

ABSTRACT
Head and neck paragangliomas (HNPs) and pheochromocytomas are rare tumors. Sporadic and hereditary forms are recognized. Four different paraganglioma syndromes (PGLs 1–4) have been described: PGL 1 is associated with mutations of the succinate dehydrogenase (SDH) subunit D (SDHD) gene; PGL 3 is caused by SDHC gene mutations; PGL 4 is caused by SDHB gene mutations; the susceptibility gene for PGL 2 is unknown. The objective of this study is to review distinct clinical features of the different PGLs. An international registry for HNPs was founded in Freiburg, Germany, in 2000. The data presented in this article have been acquired from registered HNP patients who have been screened for mutations of the genes SDHB, SDHC, and SDHD. Approximately 30% of apparent sporadic HNPs are caused by a germline mutation in one of these genes. Patients with PGL 1 or 4 have a very high lifetime risk of developing HNPs as well as thoracic and abdominal pheochromocytomas. Compared with sporadic HNPs, tumors developing in SDHB, SDHC, and SDHD mutation carriers arise at a significantly younger age. The SDHB mutations are associated with a high percentage of malignant paraganglionic tumors. We recommend molecular genetic screening of all HNP patients for SDHB, SDHC, and SDHD gene mutations. Mutation carriers must be screened for paraganglial tumors in the head, neck, thorax, and abdomen. Appropriately timed surgical intervention will minimize disease-specific morbidity and mortality. Lifelong follow-up is mandatory.
Keywords: Paraganglioma, paraganglioma syndromes, pheochromocytoma, neuroendocrine tumors, succinate dehydrogenase subunits B, C, D
Paragangliomas and pheochromocytomas are rare tumors of neural crest origin that may arise along the pathway of the paraganglial embryologic migration from the skull base to the pelvic floor.1
Several terms are used to describe paragangliomas in clinical practice. Our group uses the term pheochromocytoma for tumors located in the adrenal glands, extra-adrenal abdominal as well as in thoracic locations. Almost all these tumors are endocrinologically active. In contrast, the term paraganglioma is only used for tumors that develop in the head and neck, where most are nonfunctioning.2,3,4
Head and neck paragangliomas (HNPs) are generally named after their site of origin.5 The most common site at which they develop is the carotid body, where they are referred to as carotid body tumors (CBTs).5,6 Other common sites include the middle ear, in association with Jacobsen's tympanic plexus (glomus tympanicum tumors), the jugular bulb (glomus jugulare tumors), and paraganglia along the vagal nerve (vagal paragangliomas).5,6
Both sporadic and familial forms of HNPs and pheochromocytomas have been recognized for many years.1,5,7,8 Patients with multiple endocrine neoplasia type 2 (MEN 2), von Hippel-Lindau (vHL) disease, or neurofibromatosis type 1 (NF1) also have an increased risk of development of pheochromocytomas.9,10,11,12
In 2000, Baysal et al identified the SDHD gene as the susceptibility gene for PGL 1.13 Later that year, SDHC gene mutations were found to be associated with PGL 3 by Niemann and Müller,14 and in 2001 Astuti et al described mutations of the SDHB gene as the cause of PGL 4.15 The genetic defect leading to PGL 2 has not been identified yet.4,16 Table 1 gives an overview of the different tumor syndromes associated with HNP and pheochromocytomas. It was originally thought that the different PGL genes were exclusively associated with HNP.13,14 It soon became clear that patients with PGL 1 and PGL 4 also harbor a risk for the development of pheochromocytomas.8,15,17 A considerable number of HNP and pheochromocytoma patients with mutations of the SDHB or SDHD gene have been recorded in the last 8 years.1,2,3,4 However, only four families with SDHC gene mutations had been reported up until October 200514,18,19,20 when Schiavi et al added another five SDHC index cases.4 Interestingly, pheochromocytomas seem to be very rare in patients with PGL 3.21
Table 1.
Tumor Syndromes Associated with Pheochromocytomas and Head and Neck Paragangliomas
| Tumor Syndrome | Gene Mutation | Chromosome | Paraganglionic Tumor |
|---|---|---|---|
| MEN 2, multiple endocrine neoplasia type 2; q, short arm of a chromosome; HNP, head and neck paraganglioma; NF1, neurofibromatosis type 1; vHL, von Hippel-Lindau (disease); p, long arm of a chromosome; PGL, paraganglioma. | |||
| MEN 2 | RET | 10q11.2 | HNP* Pheochromocytoma |
| NF1 | NF1 | 17q11.2 | Pheochromocytoma |
| vHL | VHL | 3p25 | HNP Pheochromocytoma |
| PGL 1 | SDHD | 11q23 | HNP Pheochromocytoma |
| PGL 2 | Not identified | 11q13 | HNP |
| PGL 3 | SDHC | 1q21 | HNP Pheochromocytoma |
| PGL 4 | SDHB | 1p36 | HNP Pheochromocytoma |
HNP have been rarely observed in MEN 2 and vHL.4
In this article, we review the current data that are clinically relevant to paraganglioma syndromes. We also use case descriptions to illustrate distinct clinical features of PGL 1, PGL 3, and PGL 4.
CLINICAL CASE REPORTS
Patient 1: SDHB Mutation Carrier
A 34-year-old white female patient underwent resection of a right-sided CBT at another institution in February 2003. Histopathological and immunohistochemical examination of the operative specimen revealed metastatic spread to ipsilateral lymph nodes. A local recurrence developed and was resected in September 2004. During that operation, the right internal and common carotid arteries were sacrificed and replaced with Gortex bypass grafts without any subsequent neurological deficits. The ipsilateral vagus, accessory, and hypoglossal nerves were also resected. The patient first presented to our institution in February 2005. The molecular genetic screen revealed a mutation of the SDHB gene. Magnetic resonance imaging (MRI) revealed another local recurrence. An 18Fluorine L-3,4-dihydroxyphenylalanine positron emission tomography (18F DOPA PET) confirmed the tumor recurrence and showed a distant metastasis to the left femoral neck that was confirmed by MRI and computed tomography (CT) (Fig. 1). The recurrent CBT was completely resected following internal and common carotid artery balloon occlusion. Both vessels and the Gortex bypass graft were removed, and the patient did not develop any additional neurological deficit. In the meantime, the patient developed multiple bone metastases and underwent palliative external beam radiation therapy.
Figure 1.

Axial computed tomography (CT) scan with bone metastasis (arrow) in the left femoral neck of female SDHB mutation carrier (patient 1).
The patient's two sons underwent molecular genetic screening as well. A mutation of the SDHB gene was found in one son. Clinical and radiological examinations (three-body region MRIs [MRI of head and neck, abdomen, and thorax], 18F DOPA PET) did not reveal any signs of HNP or pheochromocytoma. Lifelong follow-up examinations will be mandatory for that son.
Patient 2: SDHC Mutation Carrier
This 38-year-old white female patient presented with a right-sided hearing loss in the summer of 2002. Otoscopic examination and an MRI at another institution revealed a jugular PGL. The patient had no family history of paragangliomas or pheochromocytomas. She refused surgical treatment. In February 2004, molecular genetic screening was undertaken on a blood sample. The test revealed a mutation of the SDHC gene. Magnetic resonance imaging, dynamic contrast-enhanced magnetic resonance angiography (MRA), and CT at that time revealed a large jugulotympanic paraganglioma (Fig. 2). An MRI of the thorax and abdomen as well as an 18F DOPA PET failed to detect any pheochromocytoma. The 24-hour urine assay for epinephrine, norepinephrine, and vanillylmandelic acid was unremarkable. Sadly, the patient has not consented to molecular genetic testing of her two sons. She still refuses any kind of treatment and is followed up by yearly MRI of the head and neck.
Figure 2.

Coronal T1-weighted magnetic resonance imaging (MRI) with strongly, but nonuniformly enhancing right jugulotympanic paraganglioma in female SDHC mutation carrier (patient 2).
Patient 3: SDHD Mutation Carrier
A 36-year-old white man had undergone the resection of a left CBT in January 2000. He had no postoperative cranial nerve deficits. In the months that followed, the patient developed hypertension that could not be controlled medically. A 24-hour urine catecholamine analysis was performed that revealed normal excretion of epinephrine and an elevated excretion of norepinephrine (360 μg/24 h; normal is < 100). An MRI examination revealed a pheochromocytoma of the left adrenal gland that measured 4 cm in diameter. The patient underwent complete left adrenalectomy in August 2000 with an uneventful postoperative course.
A follow-up 24-hour urine assay in December 2000 showed a persistently high norepinephrine level (219 μg/24 h). Molecular genetic screening revealed a mutation of the SDHD gene. The patient's parents and his sister were also tested for the mutation. The same SDHD mutation was detected in both his father and his sister. There was no mutation in the patient's mother.
A three-body region clinical screening including an MRI of the neck, thorax, and abdomen and an 18F DOPA PET were performed in January 2001. The 18F DOPA PET (Fig. 3) detected four lesions, with uptake of the tracer in: the right skull base, the right carotid body, the right adrenal gland, and in the atria of the heart. In addition, a remnant of the left CBT was found. All sites of uptake were confirmed as tumors by MRI. An MRA (Fig. 4) showed the right CBT and right jugular paraganglioma.
Figure 3.
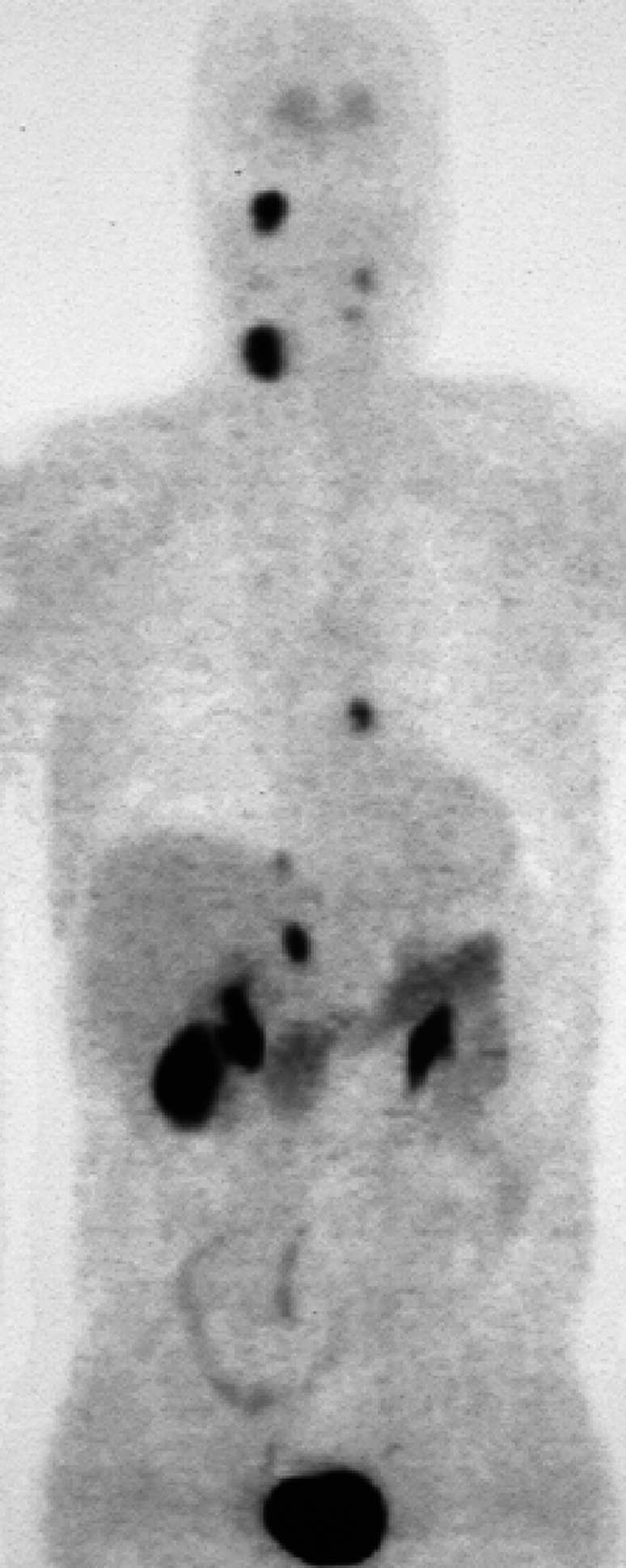
18Fluoro dopamine positron emission tomography (18F DOPA PET) in male patient with SDHD gene mutation showing CBT and jugular paraganglioma on the right, pheochromocytoma of the right adrenal gland, and pheochromocytoma in atrium of the heart. Note the remnant of the left CBT. Physiologic tracer uptake is seen in the gallbladder, the renal pelvis, and the urinary bladder (patient 3). (Reprinted from Schipper J, Boedeker CC, Maier W, Neumann HPH. Paragangliomas in the head/neck region: I. Classification and diagnosis. HNO 2004;52:569–576, with kind permission of Springer Science and Business Media.)
Figure 4.

Dynamic contrast-enhanced magnetic resonance angiography (MRA) of the SDHD mutation carrier shown in Fig. 3 confirming right CBT and jugular paraganglioma (patient 3). Contrast enhancement shows these highly vascular tumors in the early arterial phase.
Physical examination of the head and neck was unremarkable even at that stage of the illness. There were no cranial nerve deficits. Hearing was normal on both sides even though he complained of right-sided pulsatile tinnitus.
Despite genetic and clinical counseling, this patient refuses all treatment for his HNPs. A partial, laparoscopic right adrenalectomy has also been recommended, but the patient has preferred to have serial MRI studies. During the last review in January 2008, three-body region MRIs did not reveal any significant growth of the pheochromocytomas and the tumor remnant in the left neck. However, the right-sided HNPs have shown some significant growth between January 2001 and January 2008. The volume of the jugular paraganglioma has increased by 65%; the volume of the CBT has increased by 202%.
DISCUSSION
Familial HNPs have been recognized for decades.1,5,7,8 Since the identification of the SDHD gene as one of the susceptibly genes for familial HNP,13 the PGLs have attracted attention.1,2,3,4 The PGLs have been classified genetically into four entities—PGL 1, PGL 2, PGL 3, and PGL 4.13,14,15 Three of those four entities have been associated with germline mutations in genes encoding subunits of succinate dehydrogenase. The genetic mutation of the fourth syndrome, PGL 2, has not been identified yet.8
Succinate dehydrogenase (SDH), with its four subunits A, B, C and D, plays an important role in the Krebs cycle and, as part of the mitochondrial complex II, in the aerobic electron transport of the respiratory chain.8,22 Mitochondrial complex II is thought to function as a tumor suppressor because when it is defective, it results in the overexpression of several hypoxia-inducible genes that are believed to result in proliferation of paraganglia.23 Although mutations ofSDHB, SDHC, and SDHD lead to PGL 4, PGL 3, and PGL 1, respectively, germline mutations of SDHA have not been associated with HNP or pheochromocytoma. Instead, SDHA mutations are related to Leigh's syndrome, which is characterized by severe neurodegeneration.24
Malignant Head and Neck Paragangliomas and Pheochromocytomas
Approximately 10% of HNPs and pheochromocytomas have been cited to be malignant.25 Benign as well as malignant HNPs consist of the same two major cell types: epithelioid chief cells and sustentacular supporting cells. These two cell types form clusters of cells, so-called Zellballen, that are surrounded by extensive vascular sinusoids. Interestingly, no accepted pathological or immunohistochemical markers distinguish malignant from benign paraganglial tumors. Therefore, malignant HNPs and pheochromocytomas are only diagnosed when metastasis to non-neuroendocrine tissue is demonstrated.25,26 The most common site of metastatic spread for HNP is the cervical lymph nodes. Common systemic sides include bone, lung, and liver.25
In the head and neck, malignancy is most commonly found in vagal tumors (16 to 19%), followed by CBTs (~6%) and jugulotympanic tumors (2 to 4%).25 There are no internationally defined criteria concerning the preoperative search for local and distant metastases or the extent of lymphadenectomy in the removal of HNP. In HNP patients without an SDHB mutation, we do not screen for distant metastases unless clinical signs such as weight loss, fatigue, and night sweat suggest metastatic disease. All HNP patients at our department undergo B-mode sonographic examination of the neck. In patients with vagal paragangliomas and CBTs, we undertake levels 2 and 3 selective neck dissection.27 In patients with jugular paragangliomas, we remove lymph nodes immediately adjacent to the internal jugular vein at the skull base.
Imaging in Patients with Paragangliomas
Patients with SDHB (PGL 4) and SDHD (PGL 1) gene mutations have been shown to be at risk of development of multiple paraganglial tumors in the head and neck, thorax, and abdomen.1,2,3,4 In addition, PGL 4 patients' tumors can be malignant and metastasize.3,28 The SDHB and SDHD mutation carriers should undergo three-body region MRI screening. Also, PGL 4 patients should have a bone scan. As stated previously, 18F DOPA PET29,30 has been used with great success for the detection of primary and secondary tumors. Its primary advantage is that the whole body is imaged, including the skeleton. This is of great value for the detection or exclusion of multifocal or metastatic paraganglial tumors. It has also been shown to be suitable for the detection of very small tumors that may be easily missed on MRI.29 In our opinion, the 18F DOPA PET has great potential to become both a screening method and a method for clinical follow-up in PGL 1 and PGL 4 patients.
Role of Transmission and Genetic Counseling
Transmission for all PGLs is by an autosomal-dominant gene.14,31 Interestingly, there is a parent-of-origin-dependent inheritance in subjects with SDHD gene mutations (PGL 1).32 This means that the risk of manifestation of the disease phenotype is only increased if the mutation is inherited through the paternal line. Maternal transmission does not cause tumor development. Children of female SDHD mutation carriers need not undergo clinical surveillance.3 If a mutation carrier themselves, they will pass on the mutation to their offspring in an autosomal-dominant trait. This important feature has to be kept in mind when it comes to genetic counseling of affected families.
The psychological, ethical, and legal risks that often arise during genetic testing and counseling include psychological and social stress for the patient and family (e.g., survivor guilt, burden of knowledge, and social stigmatization), and threat of employment or insurance discrimination. Because of these complex issues that patients face, genetic counseling services should be offered throughout the entire testing process with follow-up when necessary.33
Distinct Clinical Features of Paraganglioma Syndromes
Only a few months after the first description of the SDHD gene as the susceptibility gene for PGL 113 in the year 2000, it became clear that those patients were also are at risk of development of adrenal and extra-adrenal pheochromocytomas.8 The same could be shown for patients with mutations of the SDHB gene in 2001.15,17 Whereas pheochromocytomas are commonly detected in patients with SDHB and SDHD gene mutations, they seem to occur very rarely in SDHC mutation carriers.21
In 2002, Neumann et al described 271 pheochromocytoma patients with apparent sporadic tumors.2 All patients underwent molecular genetic screening of the VHL gene (vHL disease), the RET gene (MEN 2), the SDHB gene (PGL 4), and the SDHD gene (PGL 1). Mutations were found in 24% of cases: 11% had vHL disease, 5% had MEN 2, 4% had PGL 1, and 4% had PGL 4. Whereas vHL disease and MEN 2 are well-documented tumor syndromes, detailed clinical information on the different PGLs, such as gene-specific clinical features and penetrance, had not been realized at that time. This information is pivotal to genetic counseling, treatment, and follow-up. In 2004, our group began to provide such information for PGL 1 and PGL 43. In that study, we encountered 53 SDHB and 47 SDHD mutation carriers. Average age at diagnosis of the first HNP or pheochromocytoma was 29.8 years in the SDHB group and 30.6 years in the SDHD group. The SDHB mutation carriers were shown to have a 50% penetrance for paraganglial tumors by the age of 35 years, increasing to 77% by the age of 50 years. The SDHD mutations conferred 50% penetrance by 31 years of age and 86% by 50 years of age. The HNP and multifocal tumors were significantly more frequent in the SDHD mutation group (p < 0.001); malignant HNP and pheochromocytomas were found in about one third of the SDHB mutation carriers. No malignant paraganglial tumors were seen in the SDHD group (p < 0.001). Additionally, renal cell cancer was observed in two young SDHB mutation carriers.
In the meantime, malignant paragangliomas have been detected in selected patients with SDHC and SDHD gene mutations.18,34,35 They are significantly more common in SDHB mutation carriers, however.3,4,28,36 Therefore, all patients presenting to our institution with an SDHB mutation are screened for local and systemic metastatic disease. This screening should include both a three-body region MRI scan as well as a bone isotope scan. An 18F DOPA PET has been shown to be an equally sensitive alternative.
Only four families with SDHC gene mutations had been described in the literature until October 2005.4,14,18,19,20 We have combined our data on 121 HNP patients with that of 371 patients from the Freiburg-Warsaw pheochromocytoma registry. All these patients were screened for mutations of the SDHB, SDHC, and SDHD gene. Prevalences in the HNP registry were 4% for SDHC, 7% for SDHB, and 17% for SDHD mutations. Altogether, ~28% of patients with apparently sporadic HNPs harbored a mutation in one of those three genes. In all three groups of mutation carriers, patients were of significantly younger age at diagnosis of their first tumor compared with patients without germline mutations. Fewer instances of multifocal tumors were found in SDHC patients when compared with SDHD mutation carriers. More CBTs where found in the SDHC group when compared with patients with sporadic HNP. Interestingly, pheochromocytomas could not been found in any SDHC patient in this study.4
The clinical cases described in the present study (patients 1, 2, and 3) highlight some of the distinct clinical features of the different PGLs.
All three patients were in their 30s when they were first diagnosed with an HNP. In patients without mutations, HNPs develop at a later age in the majority of cases.
The SDHB mutation carrier suffered from a malignant CBT. Although detected in SDHB patients in a large percentage of cases, malignant paragangliomas are rarely seen in SDHC and SDHD mutation carries.
The SDHC mutation carrier only had one HNP. Multifocal HNPs and pheochromocytomas seem to be very rare in PGL 3.
The SDHD mutation carrier presented with multiple HNPs and multiple pheochromocytomas but without any signs of metastatic spread. Multifocal paraganglial tumors are significantly more frequent in SDHD patients compared with SDHB and SDHC mutation carriers and patients with sporadic HNPs.
CONCLUSION
Approximately 30% of apparently sporadic HNPs are caused by a germline mutation in one of the SDHB, SDHC, and SDHD genes. We therefore recommend molecular genetic screening of all HNP patients for those mutations. First-degree relatives of mutation carriers have to be screened as well. There is an autosomal-dominant role of transmission for all PGLs with a parent-of-origin–dependent inheritance in SDHD mutation carriers (PGL 1). Patients with PGL 1 and 4 have a very high lifetime risk for the development of HNP as well as thoracic and abdominal paraganglionic tumors. Compared with sporadic HNP, tumors in SDHB, SDHC, and SDHD mutation carriers develop at a significantly younger age. The SDHB mutations are associated with a high percentage of malignant PGLs. Three-body region clinical screening of mutation carriers offers the chance to diagnose those tumors in an early and asymptomatic stage. Appropriately timed surgical intervention will minimize disease-specific morbidity and mortality. Genetic counseling and interdisciplinary follow-up of mutation carriers are essential. Duration of intervals is under debate, but yearly intervals may be considered.
REFERENCES
- Boedeker C C, Neumann H PH, Ridder G J, Maier W, Schipper J. Paragangliomas in patients with mutations of the SDHD gene. Otolaryngol Head Neck Surg. 2005;132:467–470. doi: 10.1016/j.otohns.2004.09.024. [DOI] [PubMed] [Google Scholar]
- Neumann H P, Bausch B, McWhinney S R, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- Neumann H PH, Pawlu C, Peçzkowska M, et al. Distinct clinical features characterize paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943–951. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- Schiavi F, Boedeker C C, Bausch B, et al. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA. 2005;294:2057–2063. doi: 10.1001/jama.294.16.2057. [DOI] [PubMed] [Google Scholar]
- Sobol S M, Dailey J C. Familial multiple cervical paragangliomas: report of a kindred and review of the literature. Otolaryngol Head Neck Surg. 1990;102:382–390. doi: 10.1177/019459989010200413. [DOI] [PubMed] [Google Scholar]
- Boedeker C C, Ridder G J, Schipper J. Paragangliomas of the head and neck: diagnosis and treatment. Fam Cancer. 2005;4:55–59. doi: 10.1007/s10689-004-2154-z. [DOI] [PubMed] [Google Scholar]
- Chase W H. Familial and bilateral tumors of the carotid body. J Pathol Bacteriol. 1933;36:1–12. [Google Scholar]
- Gimm O, Armanios M, Dziema H, Neumann H PH, Eng C. Somatic and occult germline mutations in SDHD, a mitochondrial complex II gene, in non-familial pheochromocytomas. Cancer Res. 2000;60:6822–6825. [PubMed] [Google Scholar]
- Neumann H PH, Berger D P, Siegmund G, et al. Pheochromocytomas, multiple endocrine neoplasia type 2 and von Hippel-Lindau disease. N Engl J Med. 1993;329:1531–1538. doi: 10.1056/NEJM199311183292103. [DOI] [PubMed] [Google Scholar]
- Riccardi V M. Von Recklinghausen neurofibromatosis. N Engl J Med. 1981;305:1617–1627. doi: 10.1056/NEJM198112313052704. [DOI] [PubMed] [Google Scholar]
- Eng C, Clayton D, Schuffenecker I, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA. 1996;276:1575–1579. [PubMed] [Google Scholar]
- Yip L, Lee J R, Shapiro S E, et al. Surgical management of hereditary pheochromocytoma. J Am Coll Surg. 2004;198:525–535. doi: 10.1016/j.jamcollsurg.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Baysal B E, Ferrell R E, Willett-Brozick J E, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- Niemann S, Müller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baysal B E, Willett-Brozick J E, Lawrence E C, et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet. 2002;39:178–183. doi: 10.1136/jmg.39.3.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astuti D, Douglas F, Lennard T W, et al. Germline SDHD mutation in familial phaeochromocytoma. Lancet. 2001;357:1181–1182. doi: 10.1016/S0140-6736(00)04378-6. [DOI] [PubMed] [Google Scholar]
- Niemann S, Müller U, Engelhardt D, Lohse P. Autosomal dominant malignant and catecholamine-producing paraganglioma caused by a splice donor site mutation in SDHC. Hum Genet. 2003;113:92–94. doi: 10.1007/s00439-003-0938-0. [DOI] [PubMed] [Google Scholar]
- Bauters C, Vantyghem M C, Leteutre E, et al. Hereditary phaeochromocytomas and paragangliomas: a study of five susceptibility genes [letter] J Med Genet. 2003;40:E75. doi: 10.1136/jmg.40.6.e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baysal B E, Willett-Brozick J E, Andrade Filho P A, et al. An Alu-mediated partial SDHC deletion causes familial and sporadic paraganglioma. J Med Genet. 2004;41:703–709. doi: 10.1136/jmg.2004.019224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peczkowska M, Cascon A, Prejbisz A, et al. Extra-adrenal and adrenal pheochromocytomas associated with a germline SDHC mutation. Nat Clin Pract Endocrinol Metab. 2008;4:111–115. doi: 10.1038/ncpendmet0726. [DOI] [PubMed] [Google Scholar]
- Gimenez-Roqueplo A P, Favier J, Rustin P, et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003;63:5615–5621. [PubMed] [Google Scholar]
- Baysal B E. Hereditary paraganglioma targets diverse paraganglia. J Med Genet. 2002;39:617–622. doi: 10.1136/jmg.39.9.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch-Machin M A, Taylor R W, Cochran B, Ackrell B A, Turnbull D M. Late-onset optic atrophy, ataxia, and myopathy associated with a mutation of a complex II gene. Ann Neurol. 2000;48:330–335. [PubMed] [Google Scholar]
- Lee J H, Barich F, Karnell L H, et al. National Cancer Data Base report on malignant paragangliomas of the head and neck. Cancer. 2002;94:730–737. doi: 10.1002/cncr.10252. [DOI] [PubMed] [Google Scholar]
- Persky M S, Setton A, Niimi Y, Hartman J, Frank D, Berenstein A. Combined endovascular and surgical treatment of head and neck paragangliomas: a team approach. Head Neck. 2002;24:423–431. doi: 10.1002/hed.10068. [DOI] [PubMed] [Google Scholar]
- Robbins K T, Clayman G, Levine P A, et al. American Head and Neck Society Neck dissection classification update: revisions proposed by the American Head and Neck Society and the American Academy of Otolaryngology–Head and Neck Surgery. Arch Otolaryngol Head Neck Surg. 2002;128:751–758. doi: 10.1001/archotol.128.7.751. [DOI] [PubMed] [Google Scholar]
- Vanharanta S, Buchta M, McWhinney S R, et al. Early onset renal cell carcinoma as novel extraparaganglial component of SDHB-associated hereditable paraganglioma. Am J Hum Genet. 2004;74:153–159. doi: 10.1086/381054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoegerle S, Ghanem N, Altehoefer C, et al. 18F-DOPA positron emission tomography for the detection of glomus tumours. Eur J Nucl Med Mol Imaging. 2003;30:689–694. doi: 10.1007/s00259-003-1115-3. [DOI] [PubMed] [Google Scholar]
- Brink I, Hoegerle S, Klisch J, Bley T A. Imaging of pheochromocytoma and paraganglioma. Fam Cancer. 2005;4:61–68. doi: 10.1007/s10689-004-2155-y. [DOI] [PubMed] [Google Scholar]
- Baysal B E. On the association of succinate dehydrogenase mutations with hereditary paraganglioma. Trends Endocrinol Metab. 2003;14:453–459. doi: 10.1016/j.tem.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Hensen E F, Jordanova E S, van Minderhout I J, et al. Somatic loss of maternal chromosome 11 causes parent-of-origin-dependent inheritance in SDHD-linked paraganglioma and phaeochromocytoma families. Oncogene. 2004;23:4076–4083. doi: 10.1038/sj.onc.1207591. [DOI] [PubMed] [Google Scholar]
- Shapiro S E, Cote G C, Lee J E, Gagel R F, Evans D B. The role of genetics in the surgical management of familial endocrinopathy syndromes. J Am Coll Surg. 2003;197:818–831. doi: 10.1016/j.jamcollsurg.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Timmers H J, Pacak K, Bertherat J, et al. Mutations associated with succinate dehydrogenase D-related malignant paragangliomas. Clin Endocrinol (Oxf) 2008;68:561–566. doi: 10.1111/j.1365-2265.2007.03086.x. [DOI] [PubMed] [Google Scholar]
- Papaspyrou K, Rossmann H, Fottner C, et al. Malignant paraganglioma caused by a novel germline mutation of the succinate dehydrogenase D-gene: a case report. Head Neck. 2008;30:964–969. doi: 10.1002/hed.20746. [DOI] [PubMed] [Google Scholar]
- Boedeker C C, Neumann H PH, Bausch B, Maier W, Schipper J, Ridder G J. Malignant paragangliomas in SDHB mutation carriers. Otolaryngol Head Neck Surg. 2007;137:126–129. doi: 10.1016/j.otohns.2007.01.015. [DOI] [PubMed] [Google Scholar]