

Abstract
The product of the c-abl protooncogene is a nonreceptor tyrosine kinase found in both the cytoplasm and the nucleus. We report herein that cell adhesion regulates the kinase activity and subcellular localization of c-Abl. When fibroblastic cells are detached from the extracellular matrix, kinase activity of both cytoplasmic and nuclear c-Abl decreases, but there is no detectable alteration in the subcellular distribution. Upon adhesion to the extracellular matrix protein fibronectin, a transient recruitment of a subset of c-Abl to early focal contacts is observed coincident with the export of c-Abl from the nucleus to the cytoplasm. The cytoplasmic pool of c-Abl is reactivated within 5 min of adhesion, but the nuclear c-Abl is reactivated after 30 min, correlating closely with its return to the nucleus and suggesting that the active nuclear c-Abl originates in the cytoplasm. In quiescent cells where nuclear c-Abl activity is low, the cytoplasmic c-Abl is similarly regulated by adhesion but the nuclear c-Abl is not activated upon cell attachment. These results show that c-Abl activation requires cell adhesion and that this tyrosine kinase can transmit integrin signals to the nucleus where it may function to integrate adhesion and cell cycle signals.
Keywords: fibronectin, signal transduction, confocal microscopy, cell cycle
Integrins are heterodimeric transmembrane receptors composed of an α subunit and a β subunit that bind extracellular matrix proteins such as fibronectin (FN) or other cell surface molecules such as the cell adhesion molecules VCAM-1 and ICAM-1 (for review, see ref. 1). Integrin cytoplasmic domains connect to the cytoskeleton to regulate cell spreading, cell migration, and cytoskeletal organization. Integrin-dependent signals are also critical for the adhesion-dependent regulation of gene expression, progression through the cell cycle, and differentiation. These biological effects are mediated through the activation of multiple signal transducers by cell adhesion, in particular tyrosine kinases such as focal adhesion kinase (2) and Src family members (3, 4), mitogen-activated protein (MAP) kinases (5, 6), protein kinase C (7, 8), phosphatidylinositol kinases (9), and the small GTPase Rho (10).
The tyrosine kinase encoded by the c-abl protooncogene is found in both the cytoplasm and the nucleus (for review, see ref. 11). It is a multidomain protein containing src homology SH2 and SH3 domains, a tyrosine kinase domain, and binding domains for actin and DNA. The kinase activity of nuclear c-Abl is regulated during cell cycle progression. In quiescent and G1 cells, nuclear c-Abl is kept in an inactive state by the retinoblastoma protein (RB) that binds to the c-Abl tyrosine kinase domain and inhibits its activity (12, 13). Phosphorylation of RB by cyclin-dependent kinases at the G1/S boundary disrupts the RB–c-Abl complex, leading to activation of the c-Abl tyrosine kinase. Activated nuclear c-Abl can phosphorylate the C-terminal repeated domain (CTD) of RNA polymerase II to modulate transcription (14, 15). Nuclear c-Abl is part of the RB–E2F complex (16); thus, the G1/S activation of c-Abl might contribute to the regulation of genes involved in S-phase entry.
Unlike nuclear c-Abl, the cytoplasmic pool of c-Abl is not regulated during cell cycle progression and it is active in resting or G1 cells (13). Previously, no physiological signals have been identified that regulate the activity of cytoplasmic c-Abl or the partitioning of c-Abl between the nucleus and cytoplasm. Constitutively activated forms of Abl such as the v-Abl tyrosine kinase of Abelson murine leukemia virus and the Bcr-Abl tyrosine kinase of human chronic myelogenous leukemia can transform cells (11). v-Abl protein is localized predominantly in the cytoplasm (17) and the Bcr-Abl tyrosine kinase is exclusively cytoplasmic, where it stably associates with actin filaments (18). In susceptible fibroblastic cells, Bcr-Abl induces anchorage-independent proliferation but does not abrogate the requirement for growth factors (19).
Because oncogenic Abl can induce anchorage-independent proliferation, we investigated whether cell adhesion to fibronectin could influence c-Abl localization or tyrosine kinase activity. We show herein that the c-Abl kinase activity is strictly dependent on integrin-mediated cell adhesion for activation in the cytoplasmic and the nuclear compartments. Thus, c-Abl may mediate effects of cell adhesion on cell cycle progression or gene expression.
MATERIALS AND METHODS
Immunofluorescence.
Glass coverslips were coated with FN at 25 μg/ml or poly(l-lysine) for 1 hr at 37°C, washed with PBS, and blocked with heat-denatured BSA at 10 mg/ml. Cells were detached with trypsin, washed in DMEM containing soybean trypsin inhibitor at 250 μg/ml, and plated on FN- or poly(l-lysine)-coated coverslips in DMEM containing either 1% nuclease- and protease-free BSA or 10% fetal calf serum. After incubations at 37°C for 5–90 min, cells were washed 1× with PBS and fixed with 3.7% formaldehyde in 0.1 M Pipes (pH 6.8), containing 1 mM MgCl2 and 1 mM EGTA. Cells were permeabilized with 0.5% Nonidet P-40 and blocked with 10% normal goat serum. Primary and secondary antibody incubations were for 90 min at 37°C, and antibodies were diluted into the blocking solution. The following antibodies were used: mouse anti-c-Abl, 8E9, 10 μg/ml; rabbit anti-α5-integrin, 1:50 (20); rabbit anti-nuclear lamins A, B and C, 8188, 1:100, from L. Gerace (Scripps Research Institute). For confocal analysis, immunofluorescent samples were scanned with a Bio-Rad MRC 600 laser confocal microscope equipped with a Zeiss ×63 objective.
Fractionation and Immunoblot Analysis.
Suspended 10T½ cells (1 × 107 cells) were replated onto 15-cm tissue culture plastic plates coated with FN or polylysine at 37°C. Cells were washed twice with ice-cold PBS and lysed in ice-cold fractionation buffer [FB = 10 mM Tris·HCl, pH 7.2/2 mM MgCl2/1 mM EGTA/1 mM phenylmethylsulfonyl fluoride/2 mM sodium orthovanadate/aprotinin (10 μg/ml)/leupeptin (10 μg/ml)/pepstatin (10 μg/ml)/phenanthroline (10 μg/ml)] containing 0.1% Triton X-100. The crude cell lysate was homogenized with 20 strokes of a Dounce homogenizer and centrifuged for 10 sec in a microfuge rotor. The nuclei-containing pellet was resuspended in 0.5 M sucrose in FB plus 1% BSA, layered over 1 M sucrose in FB containing 1% BSA and centrifuged at 14,000 rpm in a Microfuge for 10 min. Pellets were suspended in RIPA buffer (0.15 mM NaCl/0.05 M Tris·HCl, pH 7.2/1% Triton X-100/1% deoxycholate/0.1% SDS), sonicated, and examined by electrophoresis and immunoblot analysis with the anti-Abl antibody 8E9 at 1:2000 dilution or with polyclonal anti-retinoblastoma protein (antibody 851 at 1:10,000 dilution) or anti-lamin antibody. Bands were quantitated by densitometry. The amount of c-Abl in each fraction was normalized to that of RB or lamin, and the value of c-Abl in stably attached cells was set as 100.
Kinase Activity Assays.
Suspended cells were plated on FN or polylysine, rinsed in ice-cold PBS, and extracted with lysis buffer [10 mM Tris·HCl, pH 7.4/5 mM EDTA/130 mM NaCl/1% Triton X-100/1 mM phenylmethylsulfonyl fluoride/aprotinin (10 μg/ml)/leupeptin (10 μg/ml)/pepstatin (10 μg/ml)/phenanthroline (10 μg/ml)]. Samples were immunoprecipitated with the anti-Abl antibody K12 (Santa Cruz Biotechnology) as described (14). Immune complexes were washed and resuspended in 20 μl of kinase assay buffer (10 mM Tris·HCl, pH 7.4/10 mM MgCl2/1 mM DTT) containing GST–CTD (glutathione S-transferase fused with the terminal domain of RNA polymerase II) at 2 μg/ml. Kinase reactions were initiated by addition of 10 μM unlabeled ATP and 50 μCi of [γ-32P]ATP (1 Ci = 37 GBq). After incubating 30 min at room temperature, reactions were stopped by adding 3× Laemmli sample buffer. Samples were analyzed by SDS/PAGE on 8% gels and transferred to Immobilon-P. The 32P incorporation into GST–CTD was quantitated using a PhosphorImager (Molecular Dynamics). The amount of c-Abl in each reaction was determined by immunoblot analysis with anti-Abl 8E9 antibody. Kinase activity was defined as [32P]CTD/c-Abl and the activity found in detached cells was set as 1.
RESULTS AND DISCUSSION
Nuclear–Cytoplasmic Shuttling of c-Abl in Response to Adhesion.
The subcellular localization of c-Abl was examined in C3H 10T½ mouse fibroblasts during integrin α5β1-dependent adhesion and spreading on FN. Cells were fixed and stained with a monoclonal antibody (8E9) against c-Abl and a polyclonal antibody against the integrin α5 subunit. When detached cells were replated on FN-coated dishes, they adhered, began to spread within 5 min, and were fully spread by 40–60 min. Confocal immunofluorescence microscopy showed that a portion of the cytoplasmic c-Abl became colocalized with α5 integrin at the basal surface of the cell within 5 min of plating (Fig. 1A, note the coincidence of red and green colors). c-Abl also colocalized with the focal adhesion protein talin when plated on FN for 20 min (Fig. 1B Lower). Focal adhesion localization of c-Abl was maximal at 20 min and then declined. It should be noted, however, that only a fraction of the focal adhesions appeared to contain c-Abl. 8E9 stained the nuclei of stably adherent fibroblasts containing the c-Abl protein but showed no detectable staining of fibroblasts isolated from c-Abl-deficient mice (Fig. 1C), demonstrating that staining was specific. Given that c-Abl localization to focal contacts is transient, these results suggest that early but not mature focal contacts contain c-Abl. Transient localization of c-Abl to early focal contacts was also observed when cells were plated on vitronectin or collagen, where adhesion depends on integrins αvβ3 or α2β1, respectively. Colocalization was specific, as c-Abl did not show significant colocalization with integrin α2β1 when cells were plated on FN (data not shown).
Figure 1.
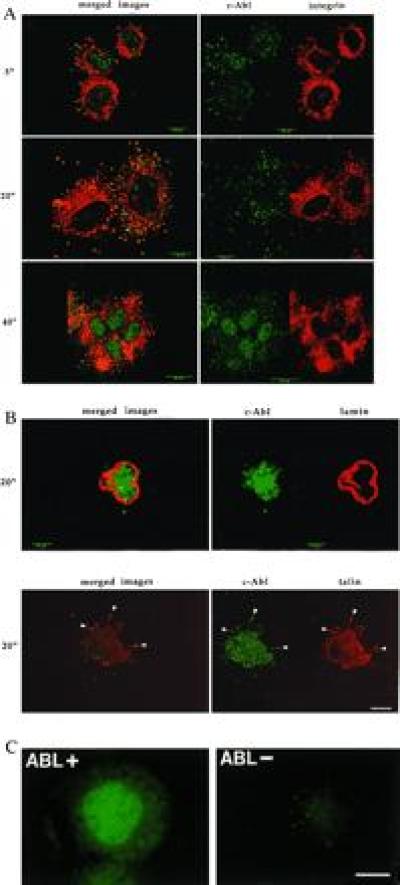
c-Abl localization during cell adhesion. (A) 10T½ cells were plated on FN in serum-free medium. Cells were fixed at the indicated times and stained with monoclonal antibody 8E9 to c-Abl (green) or a polyclonal antibody to the α5-integrin subunit (red). The images on the left show the Abl and integrin staining patterns merged. Yellow indicates codistribution. Confocal images represent the 1-μm plane nearest the substratum. (Bars: 5-min and 20-min time points, 10 μm; 40-min time point, 25 μm.) (B Upper) Cells plated on polylysine (PL) for 20 min. Cells were stained for c-Abl (green) or nuclear lamins (red) to identify the nucleus. No transport of c-Abl out of the nucleus was observed at any time between 5 and 60 min after plating (data not shown). (Lower) Colocalization with talin. Cells were stained for c-Abl (green) or talin (red). Merged images are shown on the left. Yellow indicates codistribution. (Bars: Upper, 5 μm; Lower, 10 μm.) (C) 3T3 cells isolated from abl−/− mice (Abl−) (Right) or cells that were reconstituted with c-Abl (Left) were plated for 24 hr on coverslips. Cells were stained with the 8E9 antibody for c-Abl. (Bar = 25 μm.)
Concomitant with its appearance in focal adhesions, the staining of c-Abl in the nucleus diminished, reaching a minimum at approximately 20 min and then increasing at later times (Fig. 1A). Similar movement of c-Abl between the nuclear and cytoplasmic compartments was observed in cells plated on vitronectin or collagen (data not shown), indicating that multiple integrins can trigger this process. In addition, plating of cells on an antibody directed against the integrin α5 subunit induced a transient decrease of the nuclear c-Abl (data not shown). By contrast, c-Abl did not exit the nucleus of cells adhered to poly(l-lysine), nor did it localize to contacts with the substratum (Fig. 1B Upper).
The release of c-Abl from the nuclear compartment was confirmed by fractionation (Fig. 2A). The level of nuclear c-Abl, determined by quantitative immunoblot analysis of nuclear extracts, was similar in attached or detached cells, showing that detachment alone did not cause a loss of c-Abl from the nucleus. When cells were replated on FN, however, approximately 50% of the nuclear c-Abl was lost in the first 20 min, followed by a gradual return to the level found in stably attached cells (Fig. 2A). The fact that fractionation experiments only detected a maximum of 50% loss of the nuclear c-Abl was not surprising because the export and reimport of c-Abl occurred rapidly and asynchronously in cells plated on FN. While observing single cells by immunofluorescence showed the near complete loss of nuclear c-Abl, fractionation of millions of cells could only measure the average levels of nuclear c-Abl at any time point. When detached cells were plated on polylysine, the nuclear c-Abl pool did not change, demonstrated by both immunofluorescence (Fig. 1B) and fractionation (Fig. 2B). This result further indicated that the 50% loss of nuclear c-Abl induced by plating on FN was significant. Thus, these observations demonstrate that integrin-mediated cell adhesion induces the translocation of c-Abl from the nucleus to the early focal contacts and then back to the nucleus.
Figure 2.
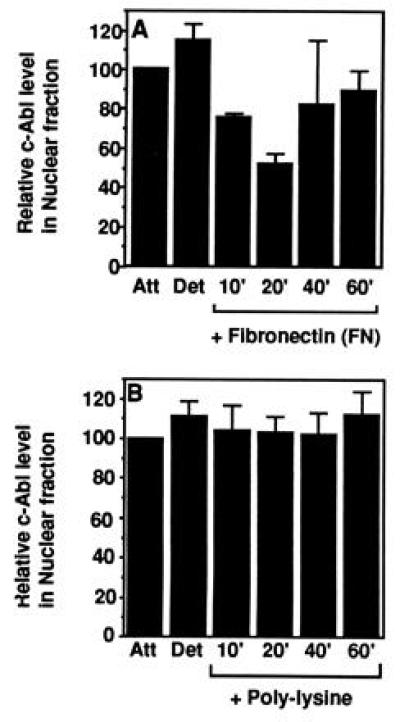
Quantitation of nuclear c-Abl. Exponentially growing 10T½ cells were stably attached (Att), detached and kept in suspension for 60 min (Det), or plated on FN (A) or poly(l-lysine) (PL) (B). At the indicated times, cells were harvested and fractionated, and the nuclear fraction was examined for c-Abl protein by immunoblot analysis. Nuclear c-Abl in stably adherent cells was set to 100%. The values shown are mean and standard deviations from three fractionation experiments.
c-Abl Kinase Activity Is Regulated by Adhesion.
Activity of nuclear c-Abl is minimal during G0/G1 phase and increases at the G1/S-phase transition (12, 13). Cytoplasmic c-Abl, however, is active in Go/G1 as well as S-phase cells (13). No physiological signals have previously been shown to regulate the cytoplasmic c-Abl kinase activity. Because adhesion altered the subcellular distribution of c-Abl, we investigated whether it also affected activity of the c-Abl tyrosine kinase. Total cellular Abl was immunoprecipitated and its kinase assayed using a GST–CTD fusion protein as a substrate (15, 21). The CTD of RNA polymerase II is a specific substrate for c-Abl, with a Km of 0.5 μM (15). Tyrosine phosphorylation of the CTD in the in vitro assay was dependent on c-Abl and could not be detected using immune complexes prepared from extracts of Abl−/− 3T3 cells (Fig. 3B). Stably attached cells contained a significant level of c-Abl tyrosine kinase activity, which decreased 3- to 5-fold upon detachment (Fig. 3 A and B). When cells were replated on FN, c-Abl kinase activity increased rapidly, reaching a peak after 10–20 min (depending on the experiment) and then decreasing to the level found in stably adherent cells. Plating on polylysine did not stimulate c-Abl activity even after 60 min (Fig. 3A). However, plating of cells on dishes coated with an antibody directed against the integrin α5 subunit led to the reactivation of c-Abl similarly to plating on FN (Fig. 3C). Although the catalytic activity of c-Abl was increased upon adhesion, we could not detect any phosphotyrosine in the c-Abl protein (data not shown). Thus, activation of c-Abl did not involve its tyrosine phosphorylation, a situation different from focal adhesion kinase or other tyrosine kinases. Thus, these results show that the c-Abl tyrosine kinase was specifically activated by integrin-mediated adhesion to an extracellular matrix protein.
Figure 3.
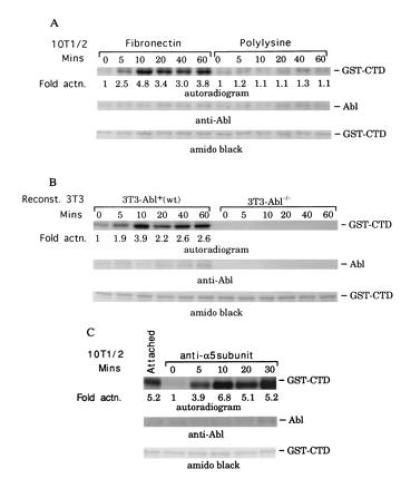
Plating on FN activates c-Abl tyrosine kinase. (A) 10T½ cells kept in suspension (time 0) or plated on FN or polylysine were extracted, c-Abl was immunoprecipitated, and kinase activity was determined. (B) Abl−/− cells stably transfected with hemagglutinin-tagged c-Abl or control transfected cells were plated on FN and analyzed as in A. (C) c-Abl tyrosine kinase activity was assayed in 10T½ cells after plating on anti-α5-integrin IgG. (Inset) Fold stimulation relative to control.
Differential Regulation of Cytoplasmic and Nuclear c-Abl.
The loss of total c-Abl activity in detached cells suggested that the cytoplasmic c-Abl was regulated by adhesion. To investigate this possibility, the cytoplasmic and nuclear fractions from asynchronous, growing cells were prepared and c-Abl tyrosine kinase activity was assayed in each fraction. Similar to the total c-Abl activity, a 3- to 5-fold reduction in the cytoplasmic Abl kinase activity was observed in detached cells (Fig. 4). Plating on FN rapidly reactivated the cytoplasmic c-Abl, which reached a peak of activity at 10–20 min, depending on the experiment, and then declined to the plateau level found in stably attached cells. These results demonstrate that cytoplasmic c-Abl tyrosine kinase is regulated by adhesion.
Figure 4.
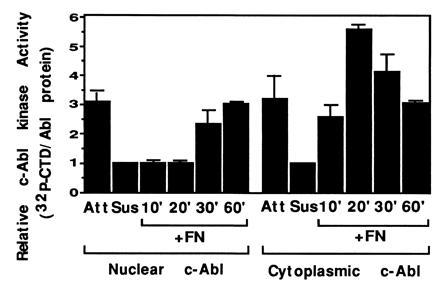
Activity of nuclear and cytoplasmic c-Abl. Cells were plated on FN and harvested at the indicated times. Cells were fractionated, and cytoplasmic and nuclear fractions were immunoprecipitated with anti-c-Abl antibody. Immunoprecipitates were analyzed for kinase activity. The mean and standard deviation from three experiments are shown.
Nuclear c-Abl, which is known to be active only after entry into S phase, also had low activity in the detached cells (Fig. 4). Upon replating on FN, nuclear c-Abl showed an increase in activity only after a distinct lag phase, regaining its activity after 30 min (Fig. 4). Interestingly, the recovery of the nuclear Abl activity closely paralleled the reentry of c-Abl to the nucleus upon reattachment to FN (Fig. 2). This suggests that the inactive nuclear c-Abl is transported to the cytoplasm where it is reactivated by integrin-dependent signals and then imported back to the nucleus.
Role of Cell Cycle.
Nuclear c-Abl activity is regulated during the cell cycle, with low activity during G0/G1 phase and high levels after entry into S phase (12, 13). To explore interactions between regulation by cell cycle and adhesion, cells were incubated in 0.1% serum for 48 hr to induce quiescence. In stably attached quiescent cells, the nuclear pool of c-Abl did not differ from that of proliferating cells, indicating that serum starvation did not alter the subcellular localization of c-Abl. Upon detachment and reattachment of quiescent cells, cytoplasmic c-Abl behaved similarly to that of growing cells, decreasing its activity upon detachment and increasing upon reattachment (Fig. 5A). Replating on FN, however, did not activate the nuclear c-Abl kinase under these conditions (Fig. 5A). Fractionation experiments showed that c-Abl was lost from the nuclear fraction upon replating on FN (Fig. 5B). Thus, the export of nuclear c-Abl could occur without an increase in kinase activity. In contrast to growing cells, the return of c-Abl protein to the nucleus was slowed in quiescent cells and was not observed within 60 min of plating on FN (Fig. 5B). The slower reimport of c-Abl was correlated with the inefficiency of quiescent cells to spread in 0.1% serum, suggesting that the nuclear translocation of c-Abl might be dependent on cell spreading. We also examined the localization of a kinase-defective c-Abl stably expressed in c-Abl deficient-fibroblasts. The kinase-defective mutant was exported from the nucleus upon attachment and returned at later times, similarly to the wild-type protein (data not shown). Thus, nuclear entry does not require activity of the c-Abl tyrosine kinase.
Figure 5.
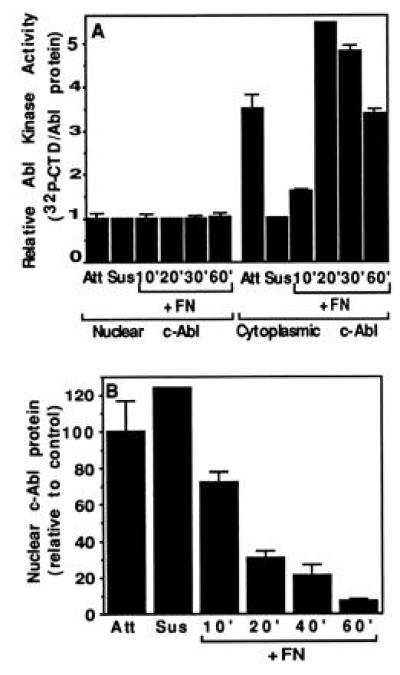
c-Abl activity and localization in quiescent cells. The 10T½ cells starved in 0.1% serum for 48 hr were trypsinized and then plated on FN in medium containing 0.1% serum. At the indicated times cells were harvested and fractionated. (A) Activity. Cells were either stably attached (Att) or trypsinized (Susp) and plated onto FN-coated plates. At the indicated times, cells were harvested and fractionated, and the c-Abl was immunoprecipitated from the lysates and the kinase activity was determined. The mean and standard deviation of three experiments are shown. (B) Nuclear c-Abl level. The relative levels of nuclear c-Abl at each time point after adhesion to FN were determined as in Fig. 2.
Thus, data shown in Figs. 2, 3, 4, 5 suggest that c-Abl is activated by integrin-dependent signals. Because c-Abl binds actin and integrins can regulate the polymerization of actin, it is possible that c-Abl is regulated by integrin-dependent reorganization of actin cytoskeleton. Consistent with this notion is the finding that c-Abl activity is inhibited if cells are treated with cytochalasin D, which disrupts the filamentous (F) actin structure (R. Baskaran, unpublished results). Thus, the integrity of F-actin may be required for c-Abl to exist in an active state. The nuclear c-Abl is additionally regulated by the cell cycle signals. The observation that nuclear c-Abl is inactivated upon detachment shows that adhesion is also required to maintain the S-phase nuclear c-Abl activity. However, the observation that nuclear c-Abl is not active in adherent G0/G1-phase cells shows that an inhibitory mechanism can superimpose on the adhesion-dependent activation to regulate the nuclear c-Abl in the cell cycle. Thus, the nuclear c-Abl tyrosine kinase can integrate at least two signals, signals of adhesion and the cell cycle.
Role of Abl Tyrosine Kinase in Integrin-Regulated Processes.
Results described herein suggests that the c-Abl tyrosine kinase is a component of integrin-regulated signaling pathways. Both c-Abl localization and activity are regulated by cell adhesion. Furthermore, the previously described cell cycle regulation of nuclear c-Abl activity depends upon cell adhesion. It is of interest to note that both c-Abl and integrins have been implicated in the regulation cell cycle progression (11, 22, 23). Thus, the fact that c-Abl can shuttle between focal contacts and the nucleus raises the possibility that c-Abl may participate in the regulation of gene expression and cell cycle progression by integrins.
These results may also be relevant to studies of human chronic myelogenous leukemia, a disease caused by the expression of the Bcr-Abl fusion protein that is a constitutively active kinase (11). Expression of Bcr-Abl in susceptible 3T3 cells leads to anchorage-independent but serum-dependent proliferation (19). This result argues that Bcr-Abl constitutively activates an integrin-dependent pathway rather than a growth factor pathway. Indeed, recent studies have identified the focal adhesion proteins paxillin and p130CAS as potential substrates of Bcr-Abl or c-Abl (refs. 24–26; W. C. Eidenmuller and J.Y.J.W., unpublished data). The identification of integrins as upstream regulators of c-Abl supports the notion that Bcr-Abl abrogates integrin signals. Identification of the integrin-dependent and c-Abl-regulated pathways may shed light on our understanding of the pathogenesis of chronic myelogenous leukemia.
Acknowledgments
We are very grateful to George Klier for confocal microscopy and to Larry Gerace for anti-lamin antibody. This work was supported by National Institutes of Health Grants RO1 CA43054 (J.Y.J.W.), RO1 GM47214 and PO1 HL48728 (M.A.S.), and F32 GM16000 (J.M.L.). This is manuscript 9981-VB from the Scripps Research Institute.
Footnotes
Abbreviations: FN, fibronectin; CTD, C-terminal repeated domain of RNA polymerase II; RB, retinoblastoma protein; GST, glutathione S-transferase.
References
- 1.Schwartz M A, Schaller M D, Ginsberg M H. Annu Rev Cell Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- 2.Schaller M D, Borgman C A, Cobb B S, Vines R R, Reynolds A B, Parsons J T. Proc Natl Acad Sci USA. 1992;89:5192–5196. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cobb B S, Schaller M D, Leu T H, Parsons J T. Mol Cell Biol. 1994;14:147–155. doi: 10.1128/mcb.14.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark E A, Brugge J S. Mol Biol Cell. 1993;3:1863–1871. doi: 10.1128/mcb.13.3.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Q, Kinch M S, Lin T H, Burridge K, Juliano R L. J Biol Chem. 1994;269:26602–26605. [PubMed] [Google Scholar]
- 6.Zhu X, Assoian R K. Mol Biol Cell. 1995;6:273–282. doi: 10.1091/mbc.6.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chun J S, Jacobson B S. Mol Biol Cell. 1993;4:271–281. doi: 10.1091/mbc.4.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vuori K, Ruoslahti E. J Biol Chem. 1993;268:21459–21462. [PubMed] [Google Scholar]
- 9.McNamee H M, Ingber D E, Schwartz M A. J Cell Biol. 1992;121:673–678. doi: 10.1083/jcb.121.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chong L D, Traynor-Kaplan A, Bokoch G M, Schwartz M A. Cell. 1994;79:507–513. doi: 10.1016/0092-8674(94)90259-3. [DOI] [PubMed] [Google Scholar]
- 11.Wang J Y J. Curr Opin Genet Dev. 1993;3:35–43. doi: 10.1016/s0959-437x(05)80338-7. [DOI] [PubMed] [Google Scholar]
- 12.Welch P J, Wang J Y J. Cell. 1993;75:779–790. doi: 10.1016/0092-8674(93)90497-e. [DOI] [PubMed] [Google Scholar]
- 13.Welch P J, Wang J Y J. Mol Cell Biol. 1995;15:5542–5551. doi: 10.1128/mcb.15.10.5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baskaran R, Dahmus M E, Wang J Y J. Proc Natl Acad Sci USA. 1993;90:11167–11171. doi: 10.1073/pnas.90.23.11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baskaran R, Chiang G G, Wang J Y J. Mol Cell Biol. 1996;16:3361–3369. doi: 10.1128/mcb.16.7.3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welch P J, Wang J Y J. Genes Dev. 1995;9:31–46. doi: 10.1101/gad.9.1.31. [DOI] [PubMed] [Google Scholar]
- 17.Van Etten R A, Jackson P, Baltimore D. Cell. 1989;58:669–678. doi: 10.1016/0092-8674(89)90102-5. [DOI] [PubMed] [Google Scholar]
- 18.McWhirter J R, Wang J Y J. Mol Biol Cell. 1991;11:1553–1565. doi: 10.1128/mcb.11.3.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Renshaw M W, McWhirter J R, Wang J Y J. Mol Cell Biol. 1995;15:1286–1293. doi: 10.1128/mcb.15.3.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giancotti F G, Ruoslahti E. Cell. 1990;60:849–859. doi: 10.1016/0092-8674(90)90098-y. [DOI] [PubMed] [Google Scholar]
- 21.Duyster J, Baskaran R, Wang J Y J. Proc Natl Acad Sci USA. 1995;92:1555–1559. doi: 10.1073/pnas.92.5.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlaepfer D D, Hanks S K, Hunter T, vanderGeer P. Nature (London) 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- 23.Fang F, Orend G, Watanabe N, Hunter T, Ruoslahti E. Science. 1996;271:499–502. doi: 10.1126/science.271.5248.499. [DOI] [PubMed] [Google Scholar]
- 24.Guadagno T M, Ohtsubo M, Roberts J M, Assoian R K. Science. 1993;262:1572–1575. doi: 10.1126/science.8248807. [DOI] [PubMed] [Google Scholar]
- 25.Salgia R, Li J L, Lo S H, Brunkhorst B, Kansas G S, Sobhanya E S, Sun Y, Pisick E, Hallek M, Ernst T, Tantravahi R, Chen L B, Griffin J D. J Biol Chem. 1995;270:5039–5047. doi: 10.1074/jbc.270.10.5039. [DOI] [PubMed] [Google Scholar]
- 26.Mayer BJ, Hirai H, Sakai R. Curr Biol. 1995;5:296–305. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]