

Abstract
Oncogenic mutations in the BRAF gene are detected in ∼7% of human cancer samples with a particularly high frequency of mutation in malignant melanomas. Over 40 different missense BRAF mutations have been found, but the vast majority (>90%) represent a single nucleotide change resulting in a valine → glutamate mutation at residue 600 (V600EBRAF). In cells cultured in vitro, V600EBRAF is able to stimulate endogenous MEK [MAPK (mitogen-activated protein kinase)/ERK (extracellular-signal-regulated kinase) kinase] and ERK phosphorylation leading to an increase in cell proliferation, cell survival, transformation, tumorigenicity, invasion and vascular development. Many of these hallmarks of cancer can be reversed by treatment of cells with siRNA (small interfering RNA) to BRAF or by inhibiting MEK, indicating that BRAF and MEK are attractive therapeutic targets in cancer samples with BRAF mutations. In order to fully understand the role of oncogenic BRAF in cancer development in vivo as well as to test the in vivo efficacy of anti-BRAF or anti-MEK therapies, GEMMs (genetically engineered mouse models) have been generated in which expression of oncogenic BRaf is conditionally dependent on the Cre recombinase. The delivery/activation of the Cre recombinase can be regulated in both a temporal and spatial manner and therefore these mouse models can be used to recapitulate the somatic mutation of BRAF that occurs in different tissues in the development of human cancer. The data so far obtained following Cre-mediated activation in haemopoietic tissue and the lung indicate that V600EBRAF mutation can drive tumour initiation and that its primary effect is to induce high levels of cyclin D1-mediated cell proliferation. However, hallmarks of OIS (oncogene-induced senescence) are evident that restrain further development of the tumour.
Keywords: BRAF, cancer, Cre-Lox, genetically engineered mouse model, proliferation, senescence
Introduction
Cell growth is controlled by a number of key genes and, as a general rule of thumb, some of these genes need to be mutated before a cell becomes cancerous. Although more than 350 such ‘cancer genes’ have been identified to date, many others are thought to have been overlooked [1]. With the advent of whole genome sequencing, it is now possible to provide a molecular signature of individual cancers and this has opened up the possibility of identifying the full complement of cancer genes. One of the main efforts has centred on the sequencing of protein kinase genes [2]. In the first large-scale sequencing project of this type, the BRAF protein kinase gene was identified as a key growth control gene that is frequently mutated in human cancers [3]. Although BRAF mutations are present in only ∼7% of human cancers as a whole, importantly, BRAF mutations are preponderant in malignant melanoma, being detected in up to 70% of all samples tested. They are also detected at a high frequency in papillary thyroid cancer (36-53%), serous ovarian cancer (∼30%) and colorectal cancer (5-22%). The vast majority of the BRAF mutations (>90%) represent a single nucleotide change of T to A at nucleotide 1799 resulting in a valine → glutamate mutation at residue 600 (V600E).
Over 40 different missense BRAF mutations have been detected in human cancer samples and these have been found to cluster in either the glycine-rich P loop or the activation segment of the BRAF kinase domain [4]. Wan et al. [5] analysed the kinase activity of a panel of 22 BRAF mutants and classified them as having either high, intermediate or impaired activity (Table 1). A unifying feature of the high and intermediate activity mutants is that they disrupt the hydrophobic interaction between the P loop and the activation segment of the kinase domain. This results in destabilization of the inactive conformation of BRAF, thus stimulating its kinase activity leading to increased ERK (extracellular-signal-regulated kinase) activation. However, the impaired activity mutants have less activity than the inactive WT (wild-type) kinase and thus do not fall into this neat model. Three of the four impaired activity mutants are still able to stimulate ERK and they appear to do this by activating CRAF through mechanisms that are not yet fully understood. The one remaining impaired activity mutant, D594VBRAF, behaves similarly to kinase-defective BRAF and does not stimulate ERK or CRAF activity (Table 1). How this mutant is oncogenic is currently not clear, but it is certainly intriguing and further studies of it may well lead to the unravelling of novel functions for BRAF.
Table 1. Classification of 22 missense BRAF mutations detected in human cancer samples as tested by Wan et al. [5].
Mutants were expressed as c-Myc-tagged fusion proteins in COS cells and classified as having either high, intermediate or impaired levels of BRAF kinase activity. The high and intermediate activity mutants have greater kinase activity than WTBRAF and stimulate ERK activation directly. Although these mutants also stimulate CRAF activity, siRNA (small interfering RNA) to CRAF indicates that this isoform does not account for the increased ERK activation. The impaired activity mutants have less activity than WTBRAF. In the case of three or four of these mutants, ERK activation is greater than that mediated by WTBRAF and this is thought to be mediated by CRAF. The fourth impaired mutant, D594V, does not stimulate CRAF or ERK activation and has a similar level of BRAF kinase activity as kinase-defective BRAF. The mechanism by which this mutant operates in cancer development is currently unclear
| Class of mutant | BRAF activity | CRAF activity | ERK activity | Mutant |
|---|---|---|---|---|
| High | >G12VRAS-induced BRAF; | >G12VRAS-induced CRAF | Similar levels as G12VRAS-induced | G487A |
| ∼130-700-fold greater than WTBRAF | ERK activation (∼4-fold) | E586K | ||
| V600D | ||||
| Not inhibited by CRAF siRNA | V600E | |||
| V600K | ||||
| V600R | ||||
| K601E | ||||
| Intermediate | <G12VRAS-induced BRAF but >WTBRAF; | >G12VRAS-induced CRAF | <G12VRAS-induced ERK activation | R462I |
| 1.3-64-fold more than WTBRAF | but > WTBRAF-induced ERK activation | I463S | ||
| G464E | ||||
| Not inhibited by CRAF siRNA | G464V | |||
| G466A | ||||
| G469E | ||||
| N581S | ||||
| F595L | ||||
| L597V | ||||
| T599I | ||||
| A727V | ||||
| Impaired | 0.6-0.8-fold less than WTBRAF | >G12VRAS-induced CRAF | <G12VRAS-induced ERK activation | G466E |
| but > WTBRAF-induced ERK activation | G466V | |||
| G596R | ||||
| Inhibited by CRAF siRNA | ||||
| Impaired | 0.3-fold less than WTBRAF | No activation | No activation | D594V |
Data established from sequencing human tumour samples has led to some insight into the role of BRAF in the cancer phenotype. First, the data show that oncogenic BRAF and RAS mutations are mutually exclusive, rarely being present in the same cancer samples. This has been taken as evidence that BRAF and RAS contribute to cancer progression through similar mechanisms, most notably through their shared ability to activate the downstream MEK [MAPK (mitogen-activated protein kinase)/ERK]/ERK pathway, which is known to influence many cancer hallmarks [6]. Secondly, mutations in other raf genes are extremely rare and recent studies have indicated that CRAF requires mutation within the N-region of the kinase domain in addition to V492ECRAF mutation (the equivalent of a V600EBRAF mutation) in order to be converted into an oncogene [7]. Thirdly, BRAF mutations are present in benign naevi [8] that express senescent markers and V600EBRAF can induce senescence in melanocytes [9]. These observations are consistent with the view that OIS (oncogene-induced senescence) is a bona fide tumour-suppressive mechanism in the development of human cancers [10] and suggests that inactivation of key tumour suppressor genes such as CDKN2A may be necessary to unleash the full tumorigenic effects of V600EBRAF.
Mouse models for Raf-induced cancers
As elucidated above, studies of human cancer samples have not only led to the identification of BRAF as a human oncogene but also have provided mechanistic insight into how this oncogene operates. Further progress is hampered by the limited accessibility of human cancer material, particularly from early cancer lesions that may allow assessment of the progression of the disease. A major effort therefore has focused on the generation of GEMMs (genetically engineered mouse models) for cancer. Although there are many physiological differences between laboratory mice and humans, some of which have a big impact on cancer [11], it is now possible to make the mouse more human-like and turn on/off oncogenic or tumour suppressor gene mutations at the right time in the right tissue. The availability of such accurate models is making a big impact on our understanding of the progression of cancer at the molecular level. GEMMs are also being applied to the development of anticancer therapies, including their use for target validation, toxicity studies and biomarker discovery [12].
The first mouse models for Raf-induced cancer were generated by expressing either the full-length craf gene or constitutively active craf-BxB under the control of the human surfactant protein-C promoter that directs expression to the type II epithelial cells lining the lung alveoli [13]. Both transgenics developed multifocal lung adenomas associated with high levels of phosphorylated ERK and high rates of cell proliferation that could be reduced by treatment with the MEK inhibitor CI-1040 [14]. Development of the tumours was also suppressed by bcl-2 deficiency [15] or by bag1 (Bcl-2-associated athanogene 1) haploinsufficiency [16], suggesting that suppression of apoptosis co-operates in lung tumour induction. A similar approach was used to drive V600EBRaf expression from the bovine thyroglobulin promoter that directs expression to thyroid follicular cells [17]. This oncogene was observed to initiate multifocal tumours with evidence of progression to poorly differentiated carcinomas.
More recently, conditional knockin GEMMs using the Cre-lox system have been developed by modification of the endogenous BRaf locus using homologous recombination in ES cells [18,19]. The model developed in our laboratory is illustrated in Figure 1. In this model, the endogenous BRaf locus was modified such that an LSL (Lox-Stop-Lox) cassette was placed in intron 14 consisting of three loxP sites, a mini-cDNA encoding exons 15-18 of BRaf and a neoR cassette. Thus, in the absence of Cre recombinase, the WT BRaf protein is expressed with exons 15-18 being encoded by the mini-cDNA, whereas, in the presence of the Cre recombinase, deletion of the mini-cDNA and neoR cassette occurs such that V600EBRaf is expressed. The advantage of this system over those previously described is that the mutant BRaf is expressed at physiological levels and, by controlling the activity of the Cre recombinase, it is possible to mimic the situation that occurs in human cells where one copy of the normal BRAF gene is mutated somatically to V600EBRAF. So far this system has been used to induce somatic V600EBRaf expression in MEFs (mouse embryonic fibroblasts), the haemopoietic system using the inducible Mx1-Cre strain [18] and in the lung by intranasal instillation of adenoviral Cre [19]. Current efforts are being directed towards induction of V600EBRaf expression in other tissue types, particularly melanocytes and colonocytes as well as the development of mouse models for other BRAF oncogenic mutations.
Figure 1. Schematic representation of a conditional knockin allele for V600EBRaf in mice.

In this allele, the endogenous BRaf locus was modified such that an LSL cassette was placed in intron 14. The LSL cassette consists of a mini-cDNA encoding exons 15-18 of the BRaf gene with a β-globin splice acceptor sequence at the 5′-end of the mini-cDNA and a polyadenylation (PA) sequence at the 3′-end. The cassette also has a neoR gene driven by the PGK (phosphoglycerate kinase) promoter and a PA sequence at the 3′-end. On the 3′-end of the LSL cassette, the endogenous exon 15 has been modified to include the T1799A mutation. Three loxP sites are present in the LSL cassette; the first is between exon 14 and the mini-cDNA, the second between the mini-cDNA and the neoR cassette and the third between the neoR cassette and modified exon 15. Expression of the Cre recombinase converts the BRafLSL-V600E allele into the BRafLox-V600E allele that expresses V600EBRaf. A similar model was described by Dankort et al. [19], although in this model only two loxP sites were utilized: one placed between exon 14 and the mini-cDNA and the other between the neoR cassette and the modified exon 15. Adapted from [18] with permission.
What the data currently show
In the systems so far analysed, V600EBRaf is able to induce high levels of proliferation and this is associated with the induction of high levels of phosphorylated MEK/ERK and a large increase in cyclin D1 expression. Changes in other cell cycle proteins have not yet been reported although V600EBRaf has been shown to suppress p21CIP1 expression in MEFs that may also contribute to increased S-phase progression [18]. In the haemopoietic system, V600EBRaf induces a rapidly fatal acute myeloid leukaemia reminiscent of histiocytic sarcoma in which there is amplification of circulating cells of the macrophage lineage [18]. In the lung, V600EBRAF induces the formation of mixed papillary and solid adenomas that arise from amplification of alveolar type II pneumocytes within 6-8 weeks of Cre infection [19]. This phenotype can be reversed by treatment of mice with PD0325901, a MEK inhibitor structurally similar to CI-1040. However, at later times post-infection, fewer proliferating cells are observed in the lung adenomas, even though the MEK/ERK cascade remains active, and the expression of senescent markers including p19ARF and Dec1 is induced. This result has been taken as evidence that, although V600EBRaf can promote a period of rapid cell proliferation, this is followed by OIS that restrains the further development of the tumours. Few of the adenomas spontaneously progress to adenocarcinomas although concomitant loss of TP53 or CDKN2A leads to more rapid cancer progression, presumably by overcoming OIS.
The phenotypes observed in V600EBRaf-expressing mice are similar in a number of ways to conditional knockin mice expressing oncogenic KRas but also demonstrate some differences. Expression of G12DKRas using the Mx1-Cre strain induces a different myeloproliferative disorder to V600EBRAF [20], indicating that the two oncogenes have similar proliferative effects but target different populations of haemopoietic cells. Expression of oncogenic KRas in the lung leads to the similar development of adenomas that also express senescent markers [21], but these progress more rapidly to adenocarcinomas than tumours induced by V600EBRaf [22], suggesting that additional KRas effector pathways contribute to further tumour progression.
These initial studies have led to a model of how V600EBRaf may be involved in tumour development (Figure 2). In the first stage, the oncogene is able to initiate tumour development by promoting an initial dramatic wave of cell proliferation driven by activation of the MEK/ERK cascade and high levels of cyclin D1. In the case of certain haemopoietic neoplasias such as histiocytic sarcoma, this level of proliferation may be sufficient to give severe clinical symptoms. In the case of solid cancers, however, a second stage ensues involving the induction of at least some markers of OIS that constrain the further development of the tumours. In the third stage, inactivation of key tumour suppressors such as p16INK4a or p19ARF is required to overcome the senescent phenotype to further progress the disease. How V600EBRAF can promote striking cell proliferation on the one hand and cell cycle arrest on the other is a key quandary of this model and the role of the MEK/ERK pathway in both phenotypes will require the further analysis of GEMMs combined with cell culture model systems to resolve.
Figure 2. Current model for role of V600EBRaf in tumour development and progression.
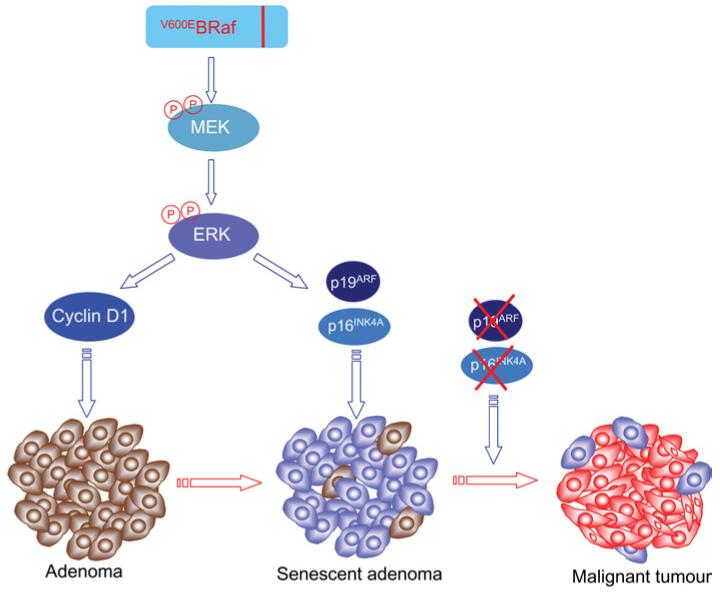
V600EBRAF is able to initiate the development of pre-malignant adenomas by inducing a rapid phase of cell proliferation through up-regulation of cyclin D1 expression. V600EBRaf can also induce the up-regulation of senescent markers including p16INK4A and p19ARF, through the MEK/ERK pathway. As a result, many of the cells within the adenoma are cell cycle arrested and the tumour cannot progress further. What controls the switch from the proliferative to senescent stage is currently not clear but there is evidence that both phenotypes are MEK/ERK-mediated. Further progression of the tumours requires a second hit within tumour suppressor genes such as CDKN2A, resulting in inactivation of p16INK4A and/or p19ARF. Cells with such an additional mutation have a proliferative advantage and allow further progression of the tumour. This model proposes that mutation of V600EBRaf is an initiating event and multiple genetic hits are required for development of the malignant disease. Proliferating V600EBRAF mutant cells are shown in brown, senescent V600EBRAF mutant cells in blue and malignant cells with V600EBRAF and CDKN2A mutations are shown in red.
Conclusion
GEMMs are being used to recapitulate BRAF-induced cancers in humans. These mice are proving vital in dissecting how oncogenic BRAF contributes to cancer and so far have shown that mutation of BRAF can be an initiating event, that oncogenic BRAF acts similarly to oncogenic RAS and that OIS may be an important step in development of the disease, at least in the lung. Further studies of these and other mouse models for BRAF-induced cancers using a wider range of Cre delivery methods to activate the oncogene in different tissues will undoubtedly help provide a complete molecular picture of events leading to metastatic disease.
Abbreviations used
- ERK
extracellular-signal-regulated kinase
- GEMM
genetically engineered mouse model
- LSL
Lox-Stop-Lox
- MAPK
mitogen-activated protein kinase
- MEF
mouse embryonic fibroblast
- MEK
MAPK/ERK kinase
- OIS
oncogene-induced senescence
- PA
polyadenylation
- siRNA
small interfering RNA
- WT
wild-type
References
- 1.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 4.Garnett MJ, Marais R. Cancer Cell. 2004;6:313–319. doi: 10.1016/j.ccr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 5.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 6.Mercer KE, Pritchard CA. Biochim. Biophys. Acta. 2003;1653:25–40. doi: 10.1016/s0304-419x(03)00016-7. [DOI] [PubMed] [Google Scholar]
- 7.Emuss V, Garnett M, Mason C, Marais R. Cancer Res. 2005;65:9719–9726. doi: 10.1158/0008-5472.CAN-05-1683. [DOI] [PubMed] [Google Scholar]
- 8.Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J, et al. Nat. Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 9.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 10.Collado M, Serrano M. Nat. Rev. Cancer. 2006;6:472–476. doi: 10.1038/nrc1884. [DOI] [PubMed] [Google Scholar]
- 11.Rangarajan A, Weinberg RA. Nat. Rev. Cancer. 2003;3:952–959. doi: 10.1038/nrc1235. [DOI] [PubMed] [Google Scholar]
- 12.Sharpless NE, Depinho RA. Nat. Rev. Drug Discov. 2006;5:741–754. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]
- 13.Kerkhoff E, Fedorov LM, Siefken R, Walter AO, Papadopoulos T, Rapp UR. Cell Growth Differ. 2000;11:185–190. [PubMed] [Google Scholar]
- 14.Kramer BW, Gotz R, Rapp UR. BMC Cancer. 2004;4:24. doi: 10.1186/1471-2407-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fedorov LM, Tyrsin OY, Papadopoulos T, Camarero G, Gotz R, Rapp UR. Cancer Res. 2002;62:6297–6303. [PubMed] [Google Scholar]
- 16.Gotz R, Kramer BW, Camarero G, Rapp UR. BMC Cancer. 2004;4:85. doi: 10.1186/1471-2407-4-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knauf JA, Ma X, Smith EP, Zhang L, Mitsutake N, Liao XH, Refetoff S, Nikiforov YE, Fagin JA. Cancer Res. 2005;65:4238–4245. doi: 10.1158/0008-5472.CAN-05-0047. [DOI] [PubMed] [Google Scholar]
- 18.Mercer K, Giblett S, Green S, Lloyd D, DaRocha Dias S, Plumb M, Marais R, Pritchard C. Cancer Res. 2005;65:11493–11500. doi: 10.1158/0008-5472.CAN-05-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. Genes Dev. 2007;21:379–384. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braun BS, Tuveson DA, Kong N, Le DT, Kogan SC, Rozmus J, Le Beau MM, Jacks TE, Shannon KM. Proc. Natl. Acad. Sci. U.S.A. 2004;101:597–602. doi: 10.1073/pnas.0307203101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, et al. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 22.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]