

Abstract
In this work, we report on a study of the structure-function relationships for three families of motor proteins, including kinesins, myosins, and F1-ATPases, by using a version of the simple elastic-network model of large-scale protein motions originally proposed by Tirion [Tirion, M. (1996) Phys. Rev. Lett. 77, 1905–1908]. We find a surprising dichotomy between kinesins and the other motor proteins (myosins and F1-ATPase). For the latter, there exist one or two dominant lowest-frequency modes (one for myosin, two for F1-ATPase) obtained from normal-mode analysis of the elastic-network model, which overlap remarkably well with the measured conformational changes derived from pairs of solved crystal structures in different states. Furthermore, we find that the computed global conformational changes induced by the measured deformation of the nucleotide-binding pocket also overlap well with the measured conformational changes, which is consistent with the “nucleotide-binding-induced power-stroke” scenario. In contrast, for kinesins, this simplicity breaks down. Multiple modes are needed to generate the measured conformational changes, and the computed displacements induced by deforming the nucleotide-binding pocket also overlap poorly with the measured conformational changes, and are insufficient to explain the large-scale motion of the relay helix and the linker region. This finding may suggest the presence of two different mechanisms for myosins and kinesins, despite their strong evolutionary ties and structural similarities.
The mechanism by which molecular motor proteins convert energy from ATP hydrolysis into mechanical work is currently an active area of research (1). The coupling of the ATP hydrolysis cycle to force generation and the determinants of motor polarity are actively being investigated by using biochemical, biophysical, and molecular approaches.
The availability of crystal structures of myosins in different nucleotide-binding states has been helpful in furthering the understanding of the working mechanism of myosin. A purely mechanical power-stroke scenario has been proposed to explain the observed conformational changes and their coupling to the binding and hydrolysis of the nucleotide. In this class of models (1, 2), the motor contains an elastic element, a spring that becomes strained as a result of the transitions between chemical states of the nucleotide: the relief of this internal strain is the driving force for the forward movement.
In the case of kinesin, the picture is more elusive, mostly due to the lack of solved structures in the ATP-binding state. Two main scenarios exist. The first is based on the mechanical power stroke that amplifies the small changes in the nucleotide and microtubule-binding sites to generate large-scale motion in the linker (1) (similar to the power-stroke scenario for myosins). The second one is based on the model of a biased Brownian ratchet where a nondeterministic random walk in a biased potential forms the basis of the mechanochemical conversion (3).
The Use of Elastic Network Models for Representing Large-Scale Motions of Motor Proteins
Advances in structure determination at atomic resolution by x-ray crystallography are making it possible to confront the observed structural changes of motor proteins, which take place during the ATP hydrolysis cycle, with models of the mechanochemical function of these remarkable machines.
Modeling the relationship of structure to function for molecular motors has been done at a variety of resolution levels ranging from all-atom simulations (4–7) to identification of rigid-body-like motions of protein subunits during the hydrolysis cycle (1).
Full-scale molecular dynamics studies of motor proteins with all atom details are still limited both by size and by time scale. The characterization of observed structural changes in terms of rigid-body motions of subunits, while being intuitively appealing, is limited to evident large-scale motions and depends on a number of assumptions (1). Here, we report the application of an alternative highly simplified approach where the large-scale motions of a protein are represented by the normal mode dynamics of an elastic network.
We use a model of the type developed by Tirion (8), and later extended by Hinsen and colleagues (9, 10), where all pairs of Cα atoms within a given cutoff distance, Rc, are joined by elastic (Hookean) springs with a single force constant. As shown by Tirion (8), the lower-frequency normal modes of models of this type correspond well to those calculated from standard semiempirical force fields. It was found that elastic-network models often succeed in representing large-scale conformational changes, such as hinge motions, which are observed in a wide range of proteins in terms of the large amplitude motion of one or two of the lowest-frequency normal modes of the model [see the application of the model to 20 proteins ranging in size from 99 to 858 residues by Tama and Sanejouand (11), and the recent review by Tama (12)]. Bahar and coworkers (13, 14) found that crystallographic temperature B factors are also well represented by the application of normal-mode analysis (NMA) to models of this type. Studies by Hinsen et al. (10) show that this characterization of observed large-scale conformational changes in terms of large-amplitude motions of a few of the lowest-frequency normal modes also maps well onto rigid-body representations of the same conformational changes. The elastic-network approach has the advantage that a single model expressed in terms of observed Cα coordinates has the capability of representing observed rigid-body-like displacements in proteins in an objective manner (15).
Here, we report a study of the observed nucleotide-induced crystallographic changes of conformation of three motor proteins, myosin, F1-ATPase, and kinesin, by using NMA of elastic-network models developed for each of these proteins.
Do Observed Nucleotide-Induced Distortions of the Binding Pocket Induce the Observed Large-Scale Conformational Changes for Motor Proteins?
One of the basic hypotheses for the relationship between the structure of a motor protein and its function as a chemical motor is the idea that the binding and subsequent hydrolysis of ATP directly induce the large-scale motions, which apply force to the substrate (actin in the case of myosin, tubulin in the case of kinesin), and thus convert the chemical energy released by the hydrolysis of ATP to ADP, to mechanical work (1).
To address this question, we apply the elastic-network model in another way. We first examine the normal-mode representation of changes in structure of a motor protein observed on adding ATP analogs. We then look at the accompanying structural changes in the nucleotide-binding pocket of the motor. From this application, we can infer an effective internal force on the Cαs constituting the binding pocket, which results from the binding of the ATP. Finally, we invert the normal-mode representation to determine whether the experimentally measured nucleotide-induced distortions of the nucleotide-binding pocket can be transmitted by means of the elastic network to account for the observed global changes in the conformation of the motor molecule.
By applying the inferred internal force that reproduces the observed nucleotide-induced changes in the binding site by means of the elastic interactions, we find that for both myosin and F1-ATPase, a remarkably simple “elastic-distortion” picture of the ATP-induced structural changes emerges, in which distortion of just one or two modes of the molecule accounts for 70–90% of the observed motions. Surprisingly, though, we do not find that the same simple picture works for kinesin.
It should be noted that none of the existing atomic resolution crystal structures for myosin or kinesin involve binding to substrate. At this stage, then, our results must be considered only as indicators of what might occur for a fully operational motor.
Methods
Elastic-Network Model. Given the Cα atomic coordinates for a protein's native structure, one can build an elastic-network model for the protein by using a harmonic potential with a single-force constant to account for pairwise interactions between all Cα atoms lying within a cutoff distance, Rc, of each other, [in practice we follow Delarue and Sanejouand (15) and set the Rc to 10 Å], as prescribed in the following equation (11):
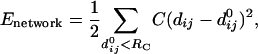 |
[1] |
where dij is the distance between the dynamical coordinates of the model representing the motions of the Cα atoms, i and j, and 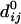 is the distance between Cα atoms i and j, as given in the crystal structure.
is the distance between Cα atoms i and j, as given in the crystal structure.
One can then perform the standard NMA on the above harmonic Hamiltonian, and, after eliminating the zero modes corresponding to three translations of the center of mass and three rotations, obtain a spectrum of frequencies (energies) for the normal modes, representing all possible elastic distortions of the model. As suggested by a number of researchers [see the review by Tama (12)], a plausible hypothesis to describe the protein's motion is that distortions of those modes with the lowest frequencies are likely to contribute importantly to the observed large-scale conformational changes that are often associated with the biological function of a protein.
To test this idea, one can compute the overlap between the conformational changes in the position of the ith Cα position, Δri, as observed between two states determined by protein crystallography and the eigenvectors, aij, of the first few lowest-frequency normal modes (generalized cosine between two vectors),
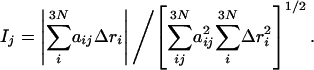 |
[2] |
Here, j denotes the index of the jth normal mode, and i denotes the index of the ith Cα.
An overlap value close to unity suggests that the crystallographically observed large-scale protein motions are well represented by the chosen set of low-frequency normal modes. This description has proved remarkably successful in describing observed large-scale motions for a range of proteins (11).
Inverting the Observed Motions: A Test of Whether the ATP-Induced Distortions of the Binding Pocket Can Account for the Observed Global Motions. For a selection of motor-protein structures and definitions of regions used in the NMA calculations, see Supporting Text, which is published as supporting information on the PNAS web site.
We suppose that the observed ATP-induced displacements of the nucleotide-binding pocket Cα positions, 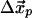 , are induced by an internal force vector,
, are induced by an internal force vector, 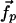 , resulting from the binding of the ATP molecule. Here, we have partitioned the complete set of Cα positions, into the Cαs of the binding pocket,
, resulting from the binding of the ATP molecule. Here, we have partitioned the complete set of Cα positions, into the Cαs of the binding pocket, 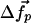 , and all of the other Cα positions,
, and all of the other Cα positions, 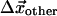 . We wish to test whether the observed displacements of all other Cα,
. We wish to test whether the observed displacements of all other Cα, 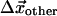 , correspond to distortions induced by
, correspond to distortions induced by 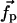 through the elastic network.
through the elastic network.
To perform this test, we need to minimize the total energy including the supposed nucleotide-induced force:
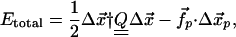 |
[3] |
where Q defines an “elastic-network matrix” resulting from the NMA after elimination of the translation and rotational modes.
Minimization of Eq. 2 with respect to the displacements 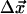 gives
gives
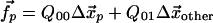 |
[4] |
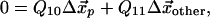 |
[5] |
where we have broken up the elasticity matrix, Qij, into the subspace of the pocket, Cα, and the subspace of all other Cα; i.e., Q00, refers to the elements of Qij, where i and j both lie in the subspace of the nucleotide-binding pocket, Q01, picks out the elements of Qij, where i lies in the pocket subspace and j in the subspace of all other Cα, and similarly for Q10 and Q11. Here, the observed pocket displacements 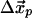 are used to determine
are used to determine 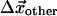 and
and 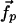 .
.
We evaluate 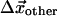 in terms of the inverse of the elasticity matrix restricted to the subspace of other residues:
in terms of the inverse of the elasticity matrix restricted to the subspace of other residues:
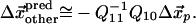 |
[6] |
Now, we can test whether the above predicted displacements compare well with the observed conformational displacements.
Results and Discussion
The Conformational Change of Myosin. A single mode dominates the ATP-induced conformational change. We consider the atomic resolution structures of scallop myosin (2, 16). We first look at the transition from the ADP-BeFx (ATP-analog) state (PDB ID code 1KK8), to the nucleotide free “near-rigor” state (PDB ID code 1KK7). To obtain the measured conformational change we superimpose 1KK7 onto 1KK8 (rms deviation = 5.2 Å). We then build an elastic-network model based on the Cα structure of 1KK8 and compute its normal modes. Finally, we project the measured conformational change from 1KK8 to 1KK7 onto the lowest 50 modes and see how well it is described by the low-frequency modes (See Fig. 1a).
Fig. 1.

The overlap of the measured conformational change with the lowest 50 normal modes for myosins and ATPases: myosin 1KK8 (with truncated lever) to 1KK7 (a); 1KK8 (with truncated lever) to 1DFL (b); and F1-ATPase PDB ID code 1H8H (chain E) to PDB ID code1H8E (chain E) (c). The thick curve is the overlap per mode, and the thin curve is the cumulative overlap using all modes below a given mode. It is observed that there exists a single mode that dominates the measured conformational change in all three cases.
It is interesting to note that a single low-frequency mode (mode 7) is able to describe the measured conformational change (rms deviation = 5.2 Å) with overlap ≈0.89, even though this is a major transition involving the rotation of the converter relative to the core domain, which drives the swinging of the long lever arm (1, 2).
We note that workers in the field generally find a better representation of large-scale motions in terms of normal modes when expanding about an “open-state” conformation than when the modes are obtained by expanding about a “closed-state” conformation (11). In the present case, we tried expanding both about the nucleotide-free state (1KK7) and about the ATP-analog state (1KK8). In the reverse direction (1KK7 to 1KK8), we found a single-dominant mode (mode 14) with higher frequency and smaller overlap (≈0.54). This finding is consistent with the above observation (11), because 1KK7 is more compact with the SH1 helix being intact and the four subdomains being closely linked. By contrast, 1KK8 is less compact and is characterized by subdomains that are loosely coupled, because the SH1 helix is unwound (16).
The observed large-scale conformational change is consistent with the ATP-induced conformational change of the nucleotide-binding pocket. To determine whether the measured conformational change may be considered as the response to the nucleotide-induced local structural change of the Cαs in the nucleotide-binding pocket, we compute the displacement induced by the pocket deformation (See Methods and Supporting Text). In Fig. 2a, we plot the amplitude of the computed displacement versus the measured one.
Fig. 2.

The amplitude of the computed displacement induced by the pocket deformation versus the measured conformational change for myosin 1KK8 (with truncated lever) to 1KK7 (a) myosin 1KK8 (with truncated lever); to 1DFL (b); and F1-ATPase 1H8H chain E to 1H8E chain E (c). Thick curve, amplitude of the measured conformational change; thin dotted curve, amplitude of the displacement induced by the nucleotide binding pocket deformation; boxes, nucleotide-binding sites.
After projecting this computed displacement for myosin onto the low-frequency normal modes, we find that it is dominated by a single mode (mode 7) with overlap ≈0.94, which is precisely the same mode that dominates the measured conformational change! Because of this coincidence, the overlap between the computed and measured displacements is 0.90.
Thus, the deformation induced in the nucleotide-binding pocket is sufficient to quantitatively account for the conformational change of the whole multidomain assembly through an elastic, or “cocked-spring” internal interaction.
We also performed NMA for the core catalytic domain of 1KK8 (results not shown), and found that a single low-frequency mode provides a decent description of the following biologically relevant conformational change, which is consistent with the power-stroke scenario (1, 2, 16). Changes in nucleotide binding deform the binding pocket, pushing (or pulling) the γ-phosphate-sensing switch II loop toward (or away from) the end of the relay helix to induce its “piston-like” motion along its axis away from (or toward) the nucleotide; this conformational change in the core domain is further transmitted and amplified by means of the SH1 helix to the converter, which then drives the swinging motion of the lever arm, which is accounted for by mode 7, identified in the above analysis.
This result gives strong support to the ability of this highly simplified elastic-network model to capture the major nucleotide-induced conformational changes observed for myosin.
To further test the above observation, we did the same calculation for the transition from 1KK8 (ATP-analog state) to the MgADP-transition state (PDB ID code 1DFL), which occurs on a larger-length scale. This state has been described as corresponding to the detached ATP state of the actomyosin cycle (16). The results are shown in Figs. 1b and 2b. Again, we found a single low-frequency mode dominates both the measured displacement and the force-induced computed displacement. The overlap between the computed and the measured displacements is 0.76.
Testing the model's robustness: Simulating the effects of actin binding and lever pulling. For calculations and results, see Supporting Text.
Conformational Change of F1-ATPase. We have performed the same elastic-network model-based calculation for an F1-ATPase and its transition from the ATP-analog state (1H8H chain E; ref. 17) to the ADP-bound state (1H8E chain E). We show the results of the overlap calculation and the pocket deformation calculation in Figs. 1c and 2c.
For this case, we found two dominant low-frequency modes with overlap equal to 0.54 (mode 8) and -0.39 (mode 9), respectively. The decomposition of the computed displacement yields the same two dominant modes with similar overlap: 0.51 for mode 8 and -0.45 for mode 9, respectively. The overlap between the computed and the measured displacements is 0.77.
This result is again consistent with the power-stroke scenario induced by nucleotide binding. As discussed by Oster and Wang (18), the deformation in the nucleotide-binding pocket triggers the β-unit to bend and push the γ-unit to rotate, thus leading to a rotary motion of the whole ATPase.
Conformational Changes of Kinesin. Based on the successful application of the elastic-network model in the above two families of motor proteins both driven by ATP hydrolysis, and the evolutionary ties and structural similarities between kinesin and myosin (19), we would naturally expect this simple model to work on kinesin as well. However, the results we find on analyzing the transition from the kinesin-like KIF1A motor-domain protein in its Mg-AMPPCP (ATP-analog)-bound state, (PDB ID code 1I6I), to the Mg-ADP-complexed state, (PDB ID code 1I5S), are surprisingly different from those found for myosin and F1-ATPase.
Instead of a single-dominant mode, we find multiple low-frequency modes (see Fig. 3a) all with overlap <0.4. Compared with myosin and ATPase, more low-frequency modes are needed to describe the measured conformational change. In other words, the nucleotide-induced distortions of the kinesin protein explore a larger configuration space with higher dimensions. We also did NMA for the transition in the reverse direction (1I5S to 1I6I) and obtained a similar result.
Fig. 3.

The overlap of the measured conformational change with the lowest 50 normal modes for kinesins:1I6I to 1I5S (a) and 1MKJ to 1BG2 (b). Thick curve, overlap per mode; thin curve, cumulative overlap using all modes below a given mode. It is observed that there does not exist a single mode that dominates the measured conformational change.
As noted above, in the case that a few normal modes can describe the overall motion, this result is more likely to show up when expanding about the open form of the protein than about the closed form. The result that many normal modes are needed to describe the motions of kinesin when expanded about either the closed or open forms suggest that the dynamics of kinesin are qualitatively different from that of myosin or ATPase. Of course, this conclusion is subject to the caveat that the elastic-network model does not break down in the case of kinesin, although apparently working well for the other two proteins examined. In view of the probable common ancestry of the motor proteins (19), this seems to be a less likely possibility (see Fig. 5 and Table 1, which are published as supporting information on the PNAS web site).
As in the myosin case, we compute the displacement induced by the pocket deformation. Here again, the measured deformation for kinesin yields a displacement composed of multiple modes, and its overlap with the measured one is only <0.2. We also note that the relatively small amplitude of the computed displacement in the relay helix and linker regions cannot account for the large motion in the measured one. It appears that the pocket deformation fails to be transmitted and amplified through the relay helix to the linker motion (Fig. 4a), which is central to the power-stroke scenario proposed in ref. 1.
Fig. 4.

The amplitude of the computed displacement induced by the pocket deformation versus the measured conformational change for kinesins: 1I6I to 1I5S (a) and 1MKJ to 1BG2 (b). Thick lines, amplitude of the measured conformational change; thin dotted lines, amplitude of the displacement induced by the nucleotide-binding pocket deformation; boxes, nucleotide-binding sites.
This result casts doubt on the applicability of the mechanical-power-stroke scenario, which works remarkably well for myosin and ATPase as shown above, to the case of kinesin. Rather than being described by a large-scale elastic deformation, the motions observed involve many local changes in the backbone conformation of the protein. Thus, in many ways, the nucleotide-induced conformational changes for kinesin are more akin to the changes occurring in protein folding (although on a lesser scale) than to a simple mechanical motion. This finding suggests that the development of mechanochemical force in kinesin may be more consistent with a Brownian ratchet-type of mechanism (3) than with the power-stroke picture.
To ensure that the above results are not caused by any particular special bias in the crystal structures used, we also did the same calculation for another pair of kinesin structures: human kinesin (PDB ID code 1BG2) and (PDB ID code 1MKJ), where the linker docking transition is observed (20) (similar to the 1I5S and 1I6I pair).
We again obtained similar results, as there are multiple low-frequency modes (see Fig. 3b), all with overlap <0.3. The pocket deformation induces a displacement whose amplitude in the relay helix region is much smaller than that of the measured one (Fig. 4b) (Although there is an anomalously large amplitude near the C-terminus, this is probably due to an artifact of the normal mode calculation, because this region in 1MKJ is a helix dangling in space without contacting the core domain, and is thus quite floppy). It is interesting to notice that the amplitude of the measured conformational change from 1MKJ to 1BG2 is similar to the one from 1I6I to 1I5S: both have large-scale motion in the relay helix and linker regions with similar details. Therefore, it does not appear that this common feature is an artifact of any particular crystal structure. In addition, the elastic-network model is, by nature, coarse-grained and robust against small variations in structural details.
The fact that, for both pairs of kinesins, single-mode dominance breaks down, and that the computed displacement induced by pocket deformation fails to account for the measured displacement, even qualitatively, suggests that the nucleotide-binding-induced power-stroke mechanism may break down for the family of kinesins. This finding may suggest that the working mechanism by which kinesin couples nucleotide binding to the large-scale motion it generates, is qualitatively different from the “cocked-spring” mechanism operating in myosin and in F1-ATPase. (However, the Mg-AMPPCP state may not represent the catalytically active state, although it contains an ATP analog. See further discussion below).
We note that, because the pocket structural details vary between human kinesin and Kif1A (see next section), this result cannot be biased by a particular bad pocket conformation in either of them. Also, whether there were to exist a common mechanism for all kinesins to amplify the pocket deformation, it cannot depend on any small structural details of those pockets, which themselves vary between different kinesins. One final comment: because the calculation is based on linear response theory, the overall scale of the pocket deformation does not affect the degree of the overlap with the normal modes.
Analysis of Differences of ATP-Induced Distortions in the Catalytic Core for Myosin Versus Kinesin. To better understand the significance of the single-mode dominance described above for the myosin catalytic cycle versus the apparent lack of single-mode dominance for the kinesin cycle, we make a detailed comparison of the relative changes observed in the available crystal structures of these two molecules. For myosin (from 1KK8 to 1KK7), both the switch I and switch II regions of the nucleotide-binding pocket move with roughly equal amplitude, whereas for kinesin (from 1I5S to 1I6I), only switch I changes significantly. The pocket conformation of 1I6I may not represent the catalytically active state although it contains an ATP analog. (Further details are given in Supporting Text.)
In a following section, we present an analysis of the robustness of the NMA against changes in the binding-pocket conformation, which suggests that, despite these important differences, the single-mode dominance of the myosin motions versus the lack of such dominance for kinesin should not be much affected by relative changes in the conformation of the nucleotide-binding regions.
Observed nucleotide-binding pocket deformation for myosin. As analyzed in ref. 21, the nucleotide-binding pocket is comprised of four conserved pieces: N1 (P loop), N2 (switch I), N3 (switch II), and N4 (base). N2 and N3 are γ-phosphate sensors that engage with phosphate through hydrogen bonding on ATP binding, but disengage on phosphate release. These regions are strongly conserved between myosin and kinesin. It was proposed that ATP binding and phosphate release trigger the most critical structural changes in the ATPase cycles (22), whereas ADP release and ATP hydrolysis may induce structural changes as well.
For myosin (from 1KK8 to 1KK7), both switch I and switch II move with roughly equal amplitude, whereas the other two pieces (N1 and N4) remain unchanged. Switch II moves into the active site, causing the distance between GswII (the conserved glycine in switch II) and Gp (the conserved glycine in the P loop) to decrease from 6.15 to 4.61 Å, suggesting that the pocket closes up to approach the ATP-like state (21).
Inferred lack of catalytic activity for available kinesin structures. For kinesin (from 1I5S to 1I6I), only switch I changes significantly from the short loop helix–loop–helix to a short pseudo-β-hairpin and it moves toward the nucleotide, while all of the other pieces remain unchanged (the distance between GswII and Gp changes little between 1I5S and 1I6I: 5.89 Å for 1I6I and 5.73 Å for I5S).
1I5S is in the ADP-binding state, while 1I6I probably represents a collision ATP complex, which is a first step in ATP binding that does not produce major conformational changes (23). Thus, the pocket conformation of 1I6I may not represent the catalytically active state, although it contains an ATP analog. A true ATP state may require stabilization by other factors not present in all those kinesin structures, such as binding with microtubules. Therefore, care must be taken to avoid drawing conclusions that rely on fine structural details of the pocket.
Very recently, Naber et al. (23) have reported EPR measurements indicating large-domain motions of the switch I region of kinesin on binding of the diphosphate complex to microtubules.
In the case of the 1BG2/MKJ pair of structures, the binding pocket changes very little (rms deviation = 0.32 Å), unlike the much larger change (rms deviation = 2.08 Å) for the KIF1A pair. In the case of 1MKJ, it appears that sulfate binding, rather than nucleotide binding, induces the observed global structural change (20). Hence, the deformation of the nucleotide-binding pocket in this case is probably not representative of a change caused by ATP binding. Nevertheless, it is interesting to notice that the amplitudes of the measured global conformational changes from 1MKJ to 1BG2 are very similar to those seen in the transition from 1I6I to 1I5S. Both have large-scale motion in the relay helix and linker regions with very similar details.
Test of Robustness of the Presence or Absence of Single-Mode Dominance. To explore whether the above differences in pocket deformations between myosin and kinesin, which might result from lack of substrate binding, could cause dramatically different responses globally, we perturb the pocket deformation in silico for both myosin and kinesin.
For myosin, we randomly perturb the displacement vectors at each pocket residue to distort the target pocket shape. We find that this distortion has little effect on the overlap between the computed and measured displacements, so that the global displacements generated by the dominant normal-mode amplitude appear to be robust against changes in fine structural details of the pocket.
For kinesin, we introduce additional motion to the switch II loop, but still could not induce a global conformational change that overlaps well with the measured one (the overlap value is always <0.3). In particular, distortions at one end of the kinesin relay helix do not appear to be transmitted to the other end of the helix, which is coupled to the motion of the neck linker.
We conclude that the presence or absence of single-mode dominance for the observed large scale global deformations of myosin versus kinesin are probably robust against minor structural distortions of the nucleotide-binding pocket and are likely to be intrinsic to their three-dimensional structure.
We note that the conformational changes that drive forward motion in myosin and kinesin occur at different steps in the ATPase cycles. ATP binding causes the forward swinging of kinesin's neck linker, but causes myosin to dissociate from actin and recock its lever arm; conversely, release of phosphate after ATP hydrolysis causes myosin to bind tightly to actin and swing its lever arm forward, although it weakens kinesin's binding with microtubules and detaches the neck linker (22). Therefore, the differences in response to pocket deformations for myosin versus kinesin may ultimately be linked to differences in their function.
Conclusions
Although the relay helices in both myosins and kinesins undergo similar conformational changes as measured by the crystallographic studies (1), our analysis reveals major underlying differences. Thus, whereas the “motions-of-a-piston” picture is consistent with a qualitative examination of the data, and, based on the more quantitative elastic-network-model-based analysis proposed here, appears to work rather well for myosin and for the corresponding hinge motion of the β-domain of F1-ATPase, it is in disagreement for the case of kinesin.
In reaching this conclusion, it is important to restate the fact that the analysis is based on structures that do not involve substrate binding. Thus, it is possible that binding to the tubulin substrate markedly affects the conformational changes for kinesin, as suggested by the electron microscopy evidence (24), whereas the binding to actin may have a less profound effect on the myosin conformational changes.
A related possibility is that the processivity of kinesin requires fundamental differences in the function of kinesin compared with that of myosin, which is more in line with a Brownian search scheme that necessarily makes the substrate binding a more essential component in the mechanochemical cycle. In contrast, the transient crossbridge action of myosin suggests that binding to the substrate may have been less critical in influencing its functional evolution.
Work on adapting the elastic-network model to conformational changes observed by cryoelectron microscopy (6) may be expected to help in highlighting the differences in the structure-function relationships of kinesin relative to myosin.
Supplementary Material
Acknowledgments
We thank Marc Delarue for bringing the NMA method to our attention and for useful comments, and Josh Shaevitz and Steve Block for helpful discussions. This work was supported by National Science Foundation Grant PHY-0140140 (to S.D.).
Abbreviation: NMA, normal-mode analysis.
References
- 1.Vale, R. & Milligan, R. (2000) Science 288, 88-95. [DOI] [PubMed] [Google Scholar]
- 2.Himmel, D., Gourinath, S., Reshetnikova, L., Shen, Y., Szent-Gyorgyi, A. & Cohen, C. (2002) Proc. Natl. Acad. Sci. USA 99, 12645-12650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cordova, N. J., Ermentrout, B. & Oster, G. F. (1992) Proc. Natl. Acad. Sci. USA 89, 339-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wriggers, W. & Schulten, K. (1998) Biophys. J. 75, 646-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xing, J., Wriggers, W., Jefferson, G. M., Stein, R., Cheung, H. C. & Rosenfeld, S. S. (2000) J. Biol. Chem. 275, 35413-35423. [DOI] [PubMed] [Google Scholar]
- 6.Chacon, P., Tama, F. & Wriggers, W. (2003) J. Mol. Biol. 326, 485-492. [DOI] [PubMed] [Google Scholar]
- 7.Minehardt, T. J., Cooke, R., Pate, E. & Kollman, P. A. (2001) Biophys. J. 80, 1151-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tirion, M. (1996) Phys. Rev. Lett. 77, 1905-1908. [DOI] [PubMed] [Google Scholar]
- 9.Hinsen, K. (1998) Proteins Struct. Funct. Genet. 33, 417-429. [DOI] [PubMed] [Google Scholar]
- 10.Hinsen, K., Thomas, A. & Field, M. J. (1999) Proteins Struct. Funct. Genet. 34, 369-382. [PubMed] [Google Scholar]
- 11.Tama F. & Sanejouand, Y. H. (2001) Protein Eng. 14, 1-6. [DOI] [PubMed] [Google Scholar]
- 12.Tama, F. (2003) Protein Pept. Lett. 10, 119-132. [DOI] [PubMed] [Google Scholar]
- 13.Bahar, I., Atligan, A. R. & Erman, B. (1997) Fold. Des. 2, 173-181. [DOI] [PubMed] [Google Scholar]
- 14.Atligan, A. R., Durell, S. D., Jernigan, R. L., Demirel, M. C., Keskin, O. & Bahar, I. (2001) Biophys. J. 80, 505-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delarue, M. & Sanejouand, Y. H. (2002) J. Mol. Biol. 320, 1011-1024. [DOI] [PubMed] [Google Scholar]
- 16.Houdusse, A., Szent-Gyorgyi, A. & Cohen, C. (2000) Proc. Natl. Acad. Sci. USA 97, 11238-11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menz, R. I., Leslie, A. G. & Walker, J. E. (2001) FEBS Lett. 494, 11-14. [DOI] [PubMed] [Google Scholar]
- 18.Oster, G. & Wang, H. (2000) Biochim. Biophys. Acta 1458, 482-510. [DOI] [PubMed] [Google Scholar]
- 19.Kull, F. J., Sablin, E. P., Lau, R., Fletterick, R. J. & Vale, R. D. (1996) Nature 380, 550-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sindelar, C. V., Budny, M. J., Rice, S., Naber, N., Fletterick, R. & Cooke, R. (2002) Nat. Struct. Biol. 9, 844-848. [DOI] [PubMed] [Google Scholar]
- 21.Sack, S., Kull, F. J. & Mandelkow, E. (1999) Eur. J. Biochem. 262, 1-11. [DOI] [PubMed] [Google Scholar]
- 22.Vale, R. D., Case, R., Sablin, E., Hart, C. & Fletterick, R. (2000) Philos. Trans. R. Soc. London B 355, 449-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naber, N., Minehardt, T. J., Rice, S., Chen, X., Grammer, J., Matuska, M., Vale, R. D., Kollman, P. A., Car, R., Yount, R. G., et al. (2003) Science 300, 798-802. [DOI] [PubMed] [Google Scholar]
- 24.Rice, S., Lin, A. W., Safer, D., Hart, C. L., Naber, N., Carragher, B. O., Cain, S. M., Pechatnikova, E., Wilson-Kubalek, E. M., Whittaker, M., et al. (1999) Nature 402, 778-784. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.