

Abstract
Interleukin (IL)-4 has been shown to induce protection in porcine vascular endothelial cells (ECs) from killing by human complement. This protection is dependent on the PI3K/Akt signaling pathway. In this study, we investigated mechanisms downstream of Akt and found that activation of the lipid biosynthesis pathway is required for protection from complement in ECs treated with IL-4. Cells incubated with IL-4 for 48 hours contained increased fatty acids and phospholipids but cholesterol was not increased when compared with medium-treated controls. The transcription factor SREBP-1, which regulates fatty acid synthesis, was found to be activated in extracts of ECs incubated with IL-4 for 6 hours. Finally, induction of protection from complement killing with IL-4 was fully prevented by the presence of the SREBP inhibitor 25-OH cholesterol. This study showed that IL-4 induces lipid biosynthesis in porcine ECs through activation of SREBP-1 and that the activation of this pathway is critical for IL-4 to induce protection of porcine ECs from killing by human complement. Further study of these mechanisms may provide new strategies for the prevention of complement-mediated vascular injury as it occurs in xenograft rejection.
The vascular endothelium is critically important in physiologic functions such as maintaining an antithrombotic surface to blood flow, providing a selective barrier to plasma proteins, lymphocyte trafficking with the upregulation of adhesion molecules, and regulation of blood flow through control of vasomotor tone. Injury to the vascular endothelium can result in diseases such as atherosclerosis, ischemia–reperfusion injury, and vasculopathy as observed in vascularized allo- and xenograft rejection. Because it is in contact with blood, the vascular endothelium may be exposed to multiple mechanisms of injury including antibody, complement, oxygen radicals, direct cellular mediated cytotoxicity, and pore-forming toxins such as those released by bacteria. A common factor in injury induced by these mechanisms is the loss or disruption in plasma membrane integrity, which, if unchecked, can lead to inflammation, dysfunction, and cell death. Endothelial cells (ECs) possess survival mechanisms that enable limited repair and resistance to injury. Recently, it has been demonstrated that ECs are able to survive after limited exposure to streptolysin-O by resealing the lesions in the plasma membrane.1,2 It has also been demonstrated that after exposure to bacterial pore-forming toxins such as aerolysin, cells are able to survive by upregulation and activation of lipid biosynthetic pathways, which then allow for membrane synthesis.3 Antibody activation of the complement cascade plays an important role in the pathology observed in ABO-mismatch allograft and xenograft rejection. The membrane attack complex of complement may create a lesion in the plasma membrane that leads to EC activation, and in some cases ionic imbalance and even cell death. It has been observed that in some cases the vascular endothelium and grafted organ are able to resist injury despite natural reactive antibodies and an intact complement system through the up-regulation of survival signaling pathways such as HO-1,4 Bcl-2, Bcl-xL,5 and A20.6 Thus, the grafted organ is said to have achieved accommodation.
Previously, we have demonstrated that the Th2 cytokine interleukin (IL)-4 induces a cytoprotective state in porcine vascular ECs, such that they become resistant to injury mediated by human complement. Moreover the induction of protection required the activation of the PI3K/Akt signaling pathway.7,8 Now we investigated the mechanisms downstream of Akt that may participate in IL-4–induced protection from complement, in particular the role of the cell lipid synthesis pathway, which has been implicated in vascular EC survival.9
METHODS
Primary cultures of porcine ECs were incubated for 48 hours at 37°C with medium or 10 ng/mL of IL-4. The cellular lipids were extracted and analyzed by proton NMR. Lipid levels were calculated for saturated fatty acids (FA), unsaturated fatty acids (UFA), phosphatidylcholine (PC), and phosphatidylglycerol (PG). Protein expression and activation was assessed for sterol regulatory element binding proteins 1 and 2 (SREBP-1 and SREBP-2) using immunoblotting of whole cell lysates. Blocking of SREBP activation was achieved using 10 µmol/L of 25-OH cholesterol for 48 hours. 25-OH Cholesterol is a well known inhibitor of SREBP activation. Either 10 ng/mL of rIL-4 or medium was then added to the EC culture for 48 hours. Sensitivity to complement mediated killing was assessed using 15% human serum as a source of natural reactive antibodies and complement and incubating with the EC cultures for 2 hours at 37°C. Cytotoxicity was assessed with a neutral red vital dye uptake assay.
RESULTS
As indicated in Table 1, ECs incubated with IL-4 demonstrate increased lipid synthesis: Saturated FA, PC, and PG were all increased by more than 50% compared with medium controls, and UFA were increased by more than 160% compared with medium treated controls. However, IL-4 caused no change in the cellular content of cholesterol (results not shown).
Table 1.
IL-4 Induces Fatty Acid and Phospholipid Synthesis in Porcine Endothelial Cells
| % Lipid Increase Over Medium | |
|---|---|
| Treated Controls | |
| Saturated fatty acids | 67 |
| Unsaturated fatty acids | 165 |
| Phosphatidylcholine | 52 |
| Phosphatidylglycerol | 60 |
pNMR of primary culture porcine vascular EC incubated with 10 ng/mL IL-4 for 48 hours of 37°C. Percentage of lipid increase in IL-4-treated cells over medium-treated EC controls.
Immunoblotting of the ECs treated with IL-4 showed increased activation of SREBP-1 at 6 hours as denoted by the cleaved product present at 6 hours (Fig 1A); however, IL-4 did not modify SREBP-2 activation (data not shown). These results suggest that IL-4 differentially activates lipid synthesis toward fatty acid synthesis rather than sterol synthesis.
Fig. 1.
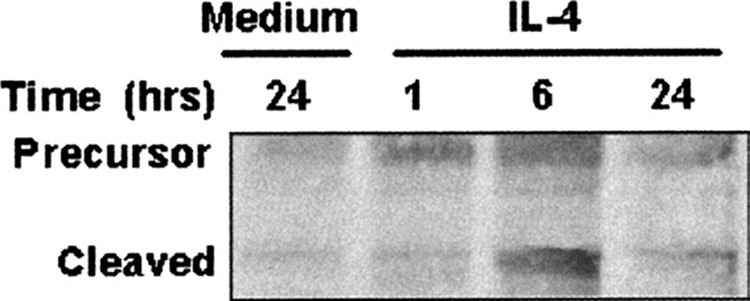
Western blot analysis of extracts from endothelial cells that were stimulated with IL-4 or medium shows that IL-4 induces activation of SREBP-1. Primary culture porcine aortic ECs incubated with 10 ng/mL IL-4 or medium at 37°C for 48 hours. The cleaved product or active portion of SREBP-1 is present at 6 hours.
Inhibition of SREBP activation with 25-OH cholesterol completely blocked IL-4–induced protection against complement-mediated killing (Table 2). IL-4 treatment reduced cellular cytotoxicity in ECs incubated with human complement from 70.5% to 12.7%. Cells that were treated with 25 OH-cholesterol and incubated with IL-4 failed to develop protection.
Table 2.
Inhibition of SREBP with 25-OH Cholesterol Blocks the Induction of Protection From Complement by IL-4 in Porcine Endothelial Cells
| % Cytotoxicity | |
|---|---|
| Medium | 70.5 ± 1.7 |
| IL-4 | 12.7 ± 4.2 |
| 25 OH-Cholesterol | 75.4 ± 1.3 |
| 25 OH-Cholesterol + IL-4 | 72.6 ± 1.9 |
Primary cultures of porcine aortic ECs were treated with either medium, 10 ng/mL IL-4, 10 µmol/L 25-OH cholesterol alone or in combination with 10 ng/mL rIL-4 for 48 hours at 37°C. ECs were then incubated with 15% human serum for 2 hours and killing assessed by a neutral red vital dye uptake assay.
DISCUSSION
The PI3K/Akt pathway has been shown in other studies to be crucial for EC survival and growth.10 It is only recently that it has been demonstrated to be linked mechanistically with the lipid synthesis pathway through SREBP,11,12 which is not surprising because membrane synthesis is required for cell growth. In this study, we investigated the induction of a cytoprotective phenotype in which ECs become resistant to injury by complement with the Th2 cytokine IL-4 and examined the mechanism of that protection. This study suggests that IL-4 induces lipogenesis of ECs, resulting in increased cellular lipid content. Induction of this pathway involves Akt phosphorylation and activation of SREBP-1, suggesting that fatty acid synthesis appears to be more important than sterol synthesis for IL-4–induced protection from complement. The role of this pathway appears to be critical because blocking SREBP activation fully prevented the development of protection. Further delineation of the mechanisms mediating IL-4–induced protection may provide new strategies for the prevention of complement-mediated vascular injury as it occurs in xenograft rejection.
Acknowledgments
Supported by grants from NIH and Minnesota Medical Foundation. SMB was supported by a NRSA from NIH.
REFERENCES
- 1.Walev I, Hombach M, Bobkiewicz W, et al. Resealing of large transmembrane pores produced by streptolysin O in nucleated cells is accompanied by NF-kappaB activation and downstream events. FASEB J. 2002;16:237. doi: 10.1096/fj.01-0572fje. [DOI] [PubMed] [Google Scholar]
- 2.Walev I, Bhakdi SC, Hofmann F, et al. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc Natl Acad Sci U SA. 2001;98:3185. doi: 10.1073/pnas.051429498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gurcel L, Abrami L, Girardin S, et al. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell. 2006;126:1135. doi: 10.1016/j.cell.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 4.Bach FH. Heme oxygenase-1: a therapeutic amplification funnel. FASEB J. 2005;19:1216. doi: 10.1096/fj.04-3485cmt. [DOI] [PubMed] [Google Scholar]
- 5.He CH, Waxman AB, Lee CG, et al. Bcl-2-related protein A1 is an endogenous and cytokine-stimulated mediator of cytoprotection in hyperoxic acute lung injury. J Clin Invest. 2005;115:1039. doi: 10.1172/JCI23004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daniel S, Arvelo MB, Patel VI, et al. A20 protects endothelial cells from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase 8 activation. Blood. 2004;104:2376. doi: 10.1182/blood-2003-02-0635. [DOI] [PubMed] [Google Scholar]
- 7.Grehan JF, Levay-Young BK, Fogelson JL, et al. IL-4 and IL-13 induce protection of porcine endothelial cells from killing by human complement and from apoptosis through activation of a phosphatidylinositide 3-kinase/Akt pathway. J Immunol. 2005;175:1903. doi: 10.4049/jimmunol.175.3.1903. [DOI] [PubMed] [Google Scholar]
- 8.Black SM, Grehan JF, Rivard AL, et al. Porcine endothelial cells and iliac arteries transduced with AdenoIL-4 are intrinsically protected, through Akt activation, against immediate injury caused by human complement. J Immunol. 2006;177:7355. doi: 10.4049/jimmunol.177.10.7355. [DOI] [PubMed] [Google Scholar]
- 9.Zhou RH, Yao M, Lee TS, et al. Vascular endothelial growth factor activation of sterol regulatory element binding protein: a potential role in angiogenesis. Circ Res. 2004;95:471. doi: 10.1161/01.RES.0000139956.42923.4A. [DOI] [PubMed] [Google Scholar]
- 10.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9:59. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porstmann T, Griffiths B, Chung YL, et al. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005;24:6465. doi: 10.1038/sj.onc.1208802. [DOI] [PubMed] [Google Scholar]
- 12.Du X, Kristiana I, Wong J, et al. Involvement of Akt in ER-to-Golgi transport of SCAP/SREBP: a link between a key cell proliferative pathway and membrane synthesis. Mol Biol Cell. 2006;17:2735. doi: 10.1091/mbc.E05-11-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]