

Abstract
Vessel regions with predilection to atherosclerosis have negative wall shear stress due to flow reversal. The flow reversal causes the production of superoxides (O2−), which scavenge nitric oxide (NO), leading to a decrease in NO bioavailability and endothelial dysfunction. Here, we implicate NADPH oxidase as the primary source of O2− during full flow reversal. Nitrite production and the degree of vasodilation were measured in 46 porcine common femoral arteries in an ex vivo system. Nitrite production and vasodilation were determined before and after the inhibition of NADPH oxidase, xanthine oxidase, or mitochondrial oxidase. NADPH oxidase inhibition with gp91ds-tat or apocynin restored nitrite production and vasodilation during reverse flow. Xanthine oxidase inhibition increased nitrite production at the highest flow rate, whereas mitochondrial oxidase inhibition had no effect. These findings suggest that the NADPH oxidase system can respond to directional changes of flow and is activated to generate O2− during reverse flow in a dose-dependent fashion. These findings have important clinical implications for oxidative balance and NO bioavailability in regions of flow reversal in a normal and compromised cardiovascular system.
Keywords: superoxide, nitric oxide, oxidative balance, reduced nicotinamide adenine dinucleotide phosphate
nitric oxide (NO) is released by endothelial cells to regulate blood vessel diameter acutely as well as to maintain long-term homeostatic conditions of the vessel wall (4, 30). NO is produced in the endothelium from l-arginine by the enzyme endothelial NO synthase (eNOS) (4, 41) and is released in response to endothelial shear stress and other stimuli such as acetylcholine (4). Wall shear stress (WSS) has also been found to regulate the activity and expression of eNOS (34, 40).
Oscillatory and reverse WSS have negative effects on the endothelium due to superoxide (O2−) production (14, 27, 28, 36, 52). It has been found that reverse flow in blood vessels causes reduced NO due to increased O2− production (36). Since NO is atheroprotective, regions of oscillatory and reverse WSS are more prone to atherosclerosis. Regions near branch points and curved vessels produce near-wall reverse flow (15, 16).
O2− can quickly inactivate NO, causing reduced NO bioavailability and endothelial dysfunction (9, 33). This endothelial dysfunction is implicated in atherosclerosis, hypertension, and heart failure (9, 13, 14, 18, 21). Reduced NO in the vessel wall impairs endothelium-dependent vasodilation, decreases inhibition of platelet and leukocyte adhesion, and is linked to the development of intimal hyperplasia (4, 21, 51). Our group has previously shown that NO production is reduced during reverse flow and that the addition of Tempol (an O2− scavenger) restored NO production in reverse flow to match NO production in forward flow (36). The exact enzymatic source of O2− in the endothelial cell that reduces [NO] during reverse flow, however, has not been determined.
O2− is produced in the endothelium through several pathways including NADPH oxidase (NOX), mitochondrial oxidase, xanthine oxidase (XO), uncoupled NO synthases, and cytochrome P-450 enzymes. In addition, enzymes such as lipoxygenases may also generate O2−. NOX has been implicated in several studies as the source of O2− in hypertension, atherosclerosis, and heart failure (5, 11, 17, 32, 46).
The hypothesis of the present study is that the primary source of O2− during reverse flow stems from the NOX system. We tested this hypothesis by inhibiting various pathways while assessing the nitrite production and the degree of vasodilation. Specifically, an ex vivo system was used to perfuse porcine femoral arteries over a range of positive and negative shear stresses before and after the incubation with inhibitors of the NOX, XO, or mitochondrial oxidase pathways. Nitrite was measured in the effluent of the forward and reverse flows. We found that the inhibition of NOX with gp91ds-tat or apocynin significantly restored nitrite production and a vasodilation in reverse flow, whereas the inhibition of XO increased nitrite production only for the highest flow rate. Finally, mitochondrial oxidase inhibition had no effect. The limitations and potential implications of these findings are described.
METHODS
Animal preparation and vessel harvest.
A total of 46 pigs weighing 30 ± 5 kg were used in the study. Surgical anesthesia was induced with ketamine (20 mg/kg im) and atropine (0.05 mg/kg) and maintained with isoflurane (1 to 2%). Ventilation with 100% O2 was provided with a respirator to maintain values of Po2 and Pco2 (500 and 35 mmHg, respectively). A 4 to 5 cm length of the common femoral artery was harvested. The proximal and distal ends of the vessels as well as the branches were ligated with sutures to ensure proper in vivo flow direction. Before harvest, the in vivo length of the artery was measured between the proximal and distal sutures. The vessel was also marked to indicate the proximal and distal ends. The animals were used for other acute studies and were euthanized with an overdose of pentobarbital sodium. All animal experiments were conducted in accordance with national and local ethical guidelines, including the Institute of Laboratory Animal Research guide, the Public Health Service policy, and the Animal Welfare Act, and were approved by the Insitutional Animal Use and Care Committee of the University of California, Irvine.
The freshly harvested vessel was placed in cold physiological saline solution (PSS) containing (in mmol/l) 119 NaCl, 4.7 KCl, 25 NaHCO3, 1.17 KH2PO4, 1.17 MgSO4, 1.6 CaCl, 5.5 dextrose (4°C) (all chemicals from Fisher Scientific). The vessel was then gently cleaned, and the branches were ligated in cold PSS. The two ends of the vessel were then cannulated with tubing, and an absence of leakage was confirmed. While the experimental setup was being finalized, the vessel was allowed to gradually warm up to 37°C over a 1-h period. All PSS solutions in the experiments were gassed with 95% O2-5% CO2.
Experimental setup.
The vessel placed in the chamber was stretched to the in vivo length based on the measured stretch ratio (degree of retraction from the in situ state). The proximal end of the vessel was connected to the inflow container containing PSS with bovine albumin at 37°C. The distal end of the vessel was connected to the outflow container. A manometer was used to monitor the pressure in the outflow container to maintain a pressure of 30 mmHg along the vessel. We have previously verified that a transmural pressure of 30 mmHg distends the vessel to an in vivo diameter, whereas higher pressures can overstretch the vessel and cause dysfunction of endothelium because the ex vivo vessel lacks the support from the surrounding tissue (36). Stopcocks at the inflow and outflow containers were used to collect the instantaneous sample solutions. A pressure transducer was connected to the tubing to measure the pressure at the proximal vessel. An adjustable clamp was used at the outflow tube to regulate the outflow resistance and hence control the flow rate. The manometer pressure was increased to maintain 30 mmHg along the vessel since the flow rate was increased. An ultrasonic flow probe (TS410 Transonic Systems) was placed in series with the vessel to measure the flow rate. In addition, a camera was used to continuously measure the outer diameter of the vessel.
Before every experiment, the contractile and endothelial functions of the vessel were assessed using phenylephrine (1 μM; Sigma) and acetylcholine (0.1 μM; Sigma). The flow rate was first changed in either the forward or reverse direction to 30, 60, 90, and 120 ml/min. Each flow rate was maintained for 1 min before the control and sample solutions were collected. The vessel was then rotated 180° so that the flow was reversed. The same magnitudes of flow rates were used, and the solutions were collected. In a preliminary study, we confirmed that the order of flow direction did not affect either NO production or vasodilatation. The forward and reverse flows were made randomly.
Apocynin, gp91ds-tat, oxypurinol, or rotenone was added to the exterior of the vessel in the chamber and to the lumen of the vessel with a clean syringe (n = 6 for each of the 4 groups). The vessel was incubated with either apocynin (acetovanillone; 1 mM; Fisher Scientific) for 30 min or gp91ds-tat (1 μM; EZBiolab) for 40 min or oxypurinol (1 mM; Sigma) for 1 h or rotenone (50 μm; Sigma) for 30 min. Following the incubation period, flow rates of 30, 60, 90, and 120 ml/min were imposed on the vessel in either the forward or reverse directions. The instantaneous control and sample solutions were collected at each flow rate. The vessel was then rotated to change the direction of flow, and sample solutions were collected. All solutions were frozen for storage until measurements were made.
After the completion of the experiment, vessel viability (endothelium-dependent vasodilation) was verified using acetylcholine (10−7 M) while the vessel was precontracted with phenylephrine (10−6 M). Only data from those vessels that remained viable were included in the analysis.
Vasodilation measurement.
The vasodilation of the vessel was measured as the ratio D/D0, where D0 is the diameter of the vessel without chemical treatment at zero flow rate and 30 mmHg pressure and D is diameter of the vessel at the same pressure (30 mmHg) but with flow rates of 30, 60, 90, or 120 ml/min over a 1-min period when the diameter reached a vasodilatory plateau.
NO perfusate metabolites measurement.
The sample solutions were analyzed for nitrite and nitrate (NOx) using the Eicom ENO-20 NOx Analyzer. The measurement is based on a combination of the Griess reaction and high-performance liquid chromatography. In this method, the peak of the nitrite voltage was related to nitrite concentration using a calibration solution. A dose of 10 μl of the solutions was used for the analysis. The control solution concentration was subtracted from the sample solution concentration to eliminate contamination. The NOx rate was calculated as the product of the flow rate and nitrite concentration at that flow rate.
NO vessel metabolites.
The vessel segments were used to validate the concentration of NOx increases in response to endothelial NO production. Five segments were incubated with l-arginine (1 mM), whereas five segments were incubated with NG-monomethyl-l-arginine (l-NMMA; eNOS inhibitor, 0.4 μM). Three segments were mechanically denuded with a suture attached to a metal ring (35). The segments incubated with l-arginine or l-NMMA were stimulated with acetylcholine (10−7 M). The perfusate was sampled, and NOx was measured by a NOx analyzer (Eicom, Kyoto, Japan). The vascular tissues with denuded endothelium were used to detect reactive oxygen species (ROS) with dihydroethidium (DHE).
O2− staining.
O2− staining was conducted on vessels in which forward flow, reverse flow, or reverse flow of 120 ml/min for 2 min after incubation with gp91ds-tat or apocynin was imposed. At the termination of the study, a section of the vessel was dissected and sectioned with a cryostat and mounted on microscope slides. The sections were incubated with DHE and visualized using a confocal microscope (excitation/emission, 520/610 nm; Zeiss Meta 510). The ratio of the fluorescent area to the field area was quantified for each image.
Western blot analysis.
The expression of NOX and eNOS in vascular tissue were measured with Western blot analysis. Six fresh arterial segments were exposed to forward flow at 2 ml/s for 20 min. Another six fresh arterial segments were exposed to reverse flow at −2 ml/s for 20 min. After 20 min of constant flow (2 ml/s) in either the forward or reverse direction, the respective vessel segments were immediately frozen in liquid nitrogen and stored in −80°C until Western blot analysis measurement. The artery segments were homogenized in a lysis buffer containing 50 mmol/l β-glycerophosphate, 100 μmol/l sodium orthovanadate, 2 mmol/l magnesium chloride, 1 mmol/l EGTA, 0.5% Triton X-100, 1 mmol/l dl-dithiothreitol, 20 μmol/l pepstatin, 20 μmol/l leupeptin, 0.1 U/ml aprotinin, and 1 mmol/l phenylmethylsulfonyl fluoride and then incubated on ice for 1 h. The sample was centrifuged at 1,000 g for 15 min at 1°C, and the supernatant was collected. The total protein was measured by a BCA kit (Bio-Rad). Equal amounts of protein (30 μg) were loaded and electrophoresed in 7.5% SDS-PAGE gel and transferred onto a ployvinylidene difluoride membrane. After being blocked for 2 h in 6% dried milk in TBS-Tween buffer, the membrane was incubated overnight at 4°C with specific primary antibody (either anti-eNOS, 1:1,000 dilution in blocking buffer, BD Transduction; anti-p22phox, 1:1,000, Santa Cruz Biotech; anti-p47phox and p22phox, 1:500, Santa Cruz Biotech; anti-gp91phox, 1:250, Santa Cruz Biotech; or anti-NOX1, 1:250, Santa Cruz Biotech). The membrane was then rinsed and incubated with horseradish peroxidase-conjugated secondary antibody for 2 h (1:5,000 dilution in blocking buffer, Santa Cruz Biotechnology). All samples from each group were also probed with anti-β-actin, a mouse monoclonal antibody (primary antibody 1:1,000 dilution in blocking buffer, Santa Cruz Biotechnology) to correct for sample loading.
Wall shear stress.
The flow was fully developed and at steady state, such that the WSS can be estimated as follows:
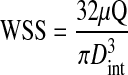 |
(1) |
where μ is fluid viscosity, Q is flow rate, and Dint is the internal diameter of the vessel. Since the outer diameter was directly measured, the inner diameter was calculated based on the incompressibility assumption; i.e.,
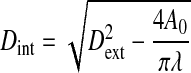 |
(2) |
where Dext is the outer diameter of the vessel at a given flow rate, λ is the stretch ratio (λ = l/l0; l and l0 represent the length of the vessel in the in vivo and no-loaded state, respectively), and A0 is the cross-sectional wall area of the vessel at zero pressure. The A0 was measured from a photograph of a transverse ring of the vessel at the conclusion of the experiment.
Statistical analysis.
The data are shown as means ± SD. Statistical significance was determined using either t-test or ANOVA (Bonferroni), which was used to determine the variation by flow rate. P < 0.05 was considered statistically significant.
RESULTS
Since nitrosylation may occur by a non-NO mechanisms in tissue, we verified the concentration of nitrite in arterial tissue to increase from 0.0021 ± 0.0008 to 0.0421 ± 0.0011 (P < 0.01) when NO production increased in response to pharmacological stimulation. When eNOS was inhibited, the concentration of nitrite did not change during acetylcholine stimulation: 0.0022 ± 0.0008 versus 0.0025 ± 0.0009 nmol/l (P > 0.1). l-Arginine enhanced the concentration of nitrite during acetylcholine stimulation: 0.0031 ± 0.0011 versus 0.0761 ± 0.016 nmol/l (P < 0.001). After the endothelium was denuded, the vasodilation and the change of ROS were not observed in the vessel segment in response to flow.
The inner diameter of the vessel was calculated from the incompressibility assumption (Eq. 2). The inner diameters of the femoral artery at flow rate of 2 ml/s and −2 ml/s were found to be 3.3 ± 1.1 and 2.7 ± 0.85 mm, respectively. The WSS was computed according to Eq. 1 for a fully developed steady-state flow. The range of WSS was ∼10–13 dyn/cm2.
The relationship between NO production and flow rate was linear for forward flow or reverse flow in both the untreated and treated (either by gp91ds-tat, apocynin, oxypurinol, or rotenone) vessels as shown in Figs. 1A or 2A, respectively. The linear least-square fit was used to determine the linear regression of NO production and flow rate (Table. 1). In the forward flow, NO production was linearly related to the flow rate in all groups. In contrast, NO production did not change in response to flow rate in the reverse flow except for the gp91ds-tat- or apocynin-treated vessels. The NO production was not affected by oxypurinol and rotenone in both the forward and reverse flow, which indicates that XO and mitochondrial oxidase are not involved in O2− anion generation induced by the reverse flow in the time scale of the present study. The vessel treated by gp91ds-tat or apocynin restored the NO production in the reverse flow, which implicates NOX in the O2− generation in the reverse flow. The slopes of the NO-production flow rate relations are summarized in Table 1, which reflects an index of NO production. The slopes in the reverse-flow group were nearly zero, except for the apocynin-treated vessels. In the forward flow, the slopes were statistically larger than those in the reverse flow (P < 0.05) (Table 1).
Fig. 1.

Nitrite production and vasodilation in forward flow (FF). A: production of nitrite in the porcine femoral artery in FFs without (control) and with the treatment of apocynin (Apo), gp91ds-tat, oxypurinol (Oxy), and rotenone (Rot). B: the degree of vasodilation is the ratio (D/D0), where D is the diameter at a given flow and D0 is the diameter at zero flow (n = 5). *P < 0.03, comparison of experimental group with control; #P < 0.02, one-way ANOVA between control and experimental group.
Fig. 2.

Nitrite production and vasodilation in reverse flow (RF). A: production of nitrite in the porcine femoral artery in RFs without control and with Apo, gp91ds-tat, Oxy, and Rot. B: the degree of vasodilation is the ratio D/D0 (n = 5). *P < 0.03, comparison of experimental group with control; #P < 0.02, one-way ANOVA between control and experimental group,.
Table 1.
The slope of relationship between nitrite production and flow rate
|
Forward Flow |
Reverse Flow
|
P | |||
|---|---|---|---|---|---|
| |Slope| | R2 | |Slope| | R2 | ||
| C | 0.0188±0.0061* | 0.811–0.939 | 0.0013±0.0011 | 0.751–0.813 | <0.01 |
| Oxy | 0.0198±0.0048* | 0.865–0.977 | 0.0043±0.0026 | 0.768–0.862 | <0.01 |
| Rot | 0.0175±0.0011 | 0.833–0.991 | 0.0022±0.0016 | 0.826–0.923 | <0.01 |
| Apo | 0.0259±0.0160 | 0.872–0.983 | 0.0205±0.0082 | 0.867–0.982 | 0.09 |
| gp91ds-tat | 0.0271±0.0180* | 0.891–0.967 | 0.0263±0.019* | 0.881–0.975 | 0.11 |
|Slope|, absolute value of slope (means ± SD); C, untreated group; Oxy, oxypurinol-treated group; Rot, rotenone-treated group; Apo, apocynin-treated group; gp91ds-tat, gp91ds-tat-treated group. P indicates the Student's t-test of the pair of forward and reverse flow.
P < 0.05, significant difference between this slope and the slope of reverse flow in untreated group.
The endothelium-dependent vasodilation, quantified as the ratio of the diameter, reflects endothelial function to produce endothelium-dependent relaxation factors, including NO, prostacyclin, and endothelium-dependent hyperpolarization factors. The vasodilation of all groups, except for the apocynin-treated group, increased similarly in response to the increase in flow rate in the forward flow. In the apocynin-treated group (in both the forward and reserve flow), the diameter at zero flow did not return to the diameter at zero flow before the apocynin treatment (D0) and resulted in a larger vasodilation with apocynin than those in control groups and the gp91ds-tat-, oxypurinol-, and rotenone-treated groups. The vessel could not generate a myogenic tone to contract to the previous diameter after the vessel was incubated with apocynin. The measurement of NOx of the apocynin-treated vessel at zero flow did not show any change in NOx concentration in the solution (data not shown). The fluorescent probe DHE did not show the suppression of O2− in apocynin-treated vessels either (Fig. 3). Those observations suggest that apocynin treatment may influence the contractility of vascular smooth muscle.
Fig. 3.

The confocal image of superoxide staining with dihydroethidium and the fractional area of staining. A: superoxide visualization with confocal microscopy of porcine common femoral artery cross-section after FF, RF, RF incubation with gp91ds-tat (RF + G), RF incubation with apocynin (RF + A), control incubation with gp91ds-tat (C + G), and control incubation with apocynin (C + A). A Zeiss LSM 510 Meta NLO multiphoton confocal microscope (excitation/emission, 514/595 nm; ×63 oil immersion objective) was used. B: the analysis of fractional area of the staining. *P < 0.05, statistical significance between groups (n = 6). NS, no statistical significance.
The fluorescent probe DHE was used to detect O2− in the vessels in the forward and reverse flow directions and in gp91ds-tat- or apocynin-treated vessels. Figure 3A shows the confocal images of vessel segments of various groups. Figure 3B represents a semiquantification of the fractional fluorescent area for each group of vessels. It can be seen that there was significantly higher O2− found in the reverse direction compared with the forward direction (P < 0.02). The incubation with gp91ds-tat or apocynin resulted in a significant decrease in O2− in the reverse direction compared with that in the control (P < 0.02). There was no significant difference between the control forward flow and the gp91ds-tat- or apocynin-treated reverse flow (P < 0.95). At zero flow, gp91ds-tat or apocynin incubation did not change the fractional area compared with that in the control.
The NOX is a complex formed when the subunits are activated. The NOX complex transfers electrons to molecular oxygen to from O2−. The expression of the subunits of NOX in vessel segments exposed to reverse flow was not statistically different to those exposed to forward flow (Fig. 4). This indicates that reverse flow activates the subunits to form more active NOX instead of upregulating the expression of NOX.
Fig. 4.

The Western blotting bands and analysis from the arterial tissue exposed to either FF or RF. A: Western blot bands. B: semiquantitative analysis of the protein content. eNOS, endothelial nitric oxide synthase; Nox-1, NADPH oxidase type 1.
DISCUSSION
We previously reported that nitrite production and vasodilation were significantly reduced in reverse flow in porcine carotid and femoral arteries due to increased O2− production in reverse flow (36). The present study shows that the major source of O2− production during full flow reversal is NOX by using the inhibitors gp91ds-tat and apocynin. This implies that the NOX can be activated by the alteration of blood flow direction, i.e., the direction of shear stress. Furthermore, when NOX is blocked by apocynin, the vessel attenuates myogenic tone and the response to flow-induced endothelium-dependent vasodilation.
The NOX complex includes transmembrane units (NOX and p22phox) and cytosolic units (p47phox, p67phox, and p40phox) (5). When the NOX is activated, p47phox and p67phox translocate to membrane and assemble with NOX and p22phox to form the NOX complex. NOX recruits NAD(P)H as an electron donor and catalyzes electron-transferring membrane to molecular oxygen to form O2− (5, 53). Several studies have demonstrated that oscillatory shear stress stimulates O2− production by NOX. Endothelial NOX activity is increased by mechanical forces such as oscillatory shear stress (33, 52). When human umbilical endothelial cells are exposed to 5 or 20 dyn/cm2, the unidirectional laminar shear stress results in transient elevation in O2− generation from NOX, but the oscillatory shear causes a sustained increase in oxidase activity (9, 27). A study of aortic endothelial cells from either normal or knockout mice that lack a component of NOX (the p47phox gene) found that a chronic exposure of endothelial cells to oscillatory shear produced O2− was inhibited by apocynin (28).
NOX can generate O2− rapidly in response to shear stress stimulation (37). In pulmonary vasculature, the membrane-localized NOX can be activated to generate ROS within several seconds (2–4 s) in response to a sudden loss of shear stress (flow cessation), which is faster than NO generation (30 s). In aortic endothelial cells, the responsive period of activation of NOX is within 30 s (37). Flow cessation causes a sudden release of forward drag force on endothelial plasma membrane, which is mechanically similar to a reverse force that retracts the membrane back.
There are several possible mechanisms for activation of NOX in response to mechanical stimuli. First, the saccharide chains that are attached to glycosylation sites found on the NOX homologue subunit which has six transmembrane helixes may be implicated (17, 50). These chains may transmit reverse shear to the NOX homologue, causing increased O2− production. Second, reverse shear causes an increase in bone morphometric protein-4 levels, which in turn can cause an increase in NOX homologue levels (11, 52). Third, the transmission of reverse shear through integrins from a focal adhesion and ECM ligand complex may cause an increase in gene expression (2, 10, 29). NOX homologue gene expression may also be increased. Finally, it is possible that the NOX directionality may lie in the membrane-bound subunits. Direct evidence for this awaits future studies where these subunits are specifically interfered with gene knockout or small-interfering RNA technique.
Apocynin inhibits the translocation of cytoplasmic units and thereafter blocks the assembly of cytosolic units and NOX (22, 40, 56). This compound requires an activation by myeloperoxidases (MPOs), which are generally expressed in the granulocyte but not in the vascular cell (25, 53). Recently, apocynin was proposed as an antioxidant in cultured endothelial and vascular smooth muscle cells but not as an inhibitor of NOX (25). Earlier reports, however, verified that apocynin decreases NADPH-stimulated O2− generation in the vasculature (9, 18, 20, 22, 28, 32, 54), where apocynin may be activated by peroxidases in vascular cells instead of MPOs (55, 56). Since there are abundant cell types in blood and in vascular wall, including granulocyte, MPOs may be present in the vessel wall (56).
Since the role of apocynin as an inhibitor is controversial, the specific NOX inhibitor gp91ds-tat was used to clarify the role of NOX in the stimulation of shear stress. The gp91ds-tat is a peptide inhibitor that interferes with the binding of gp91phox with gp47phox to inactivate NOX (42, 47). Zhou et al. (58) recently applied gp91ds-tat to block NOX in the mesenteric artery of rats. In the present study, gp91ds-tat inhibited NOX during the stimulation of shear stress, which verifies the role of NOX.
Apocynin can influence the activity of vascular smooth muscle cells through TNF-α, NF-κB, cAMP, etc. (8, 39, 45). Those signal pathways may be involved in a myogenic response of the blood vessel (38, 49). In the present study, we observed that the vessels incubated with apocynin could not develop mygenic tone in physiological loading at zero flow and remained at a significantly larger diameter than in the control, which is consistent with previous observations (49). Although the myogenic issue is too complicated to be addressed here, the present findings point to interesting future studies.
XO catalyses the oxidation of hypoxanthine to xanthine to uric acid and uses oxygen as an electron acceptor, causing O2− production (1). XO has been implicated in O2− generation in studies of congestive heart failure (CHF) and atherosclerosis and heart failure (13, 23, 43). The oxygen free radicals produced by the XO system links uric acid with endothelial dysfunction (1, 3, 48). Allopurinol treatment reduces uric acid levels in hyperuremic patients with CHF and improves the flow-dependent vasodilation (24, 26, 34). Here, we did not find XO to play a role in shear-mediated O2− in the forward or reverse flow.
Five enzyme complexes in the inner mitochondrial membrane of mammalian mitochondria compose the oxidative phosphorylation system. Complex I (NADH-quinone oxidoreductase) is made up of 46 different subunits and is inhibited by rotenone (57). Increased mitochondrial O2− generation is indicated in diabetic vasculopathy and ischemia-reperfusion (46). A study of human umbilical vein endothelial cell culture exposed to hypoxia found that rotenone eliminated increases in O2− (44). In the time scale of the present study, mitochondrial oxidase did not contribute significantly to the O2− generation in response to reverse flow. But XO and mitochondrial oxidase may be involved later, however, since they may be activated by ROS which are produced by NOX. Flow reversal and low WSS have been implicated in heart failure and atherosclerosis (19).
Diseased endothelial regions experience slow blood flow and transient variations in flow direction. However, regions that have steady blood flow are usually atheroprotected (7, 16). Endothelial dysfunction in conditions with reverse flow may be explained by the significantly increased production of O2− in the reverse flow. Higher concentrations of O2− have been found in patients with CHF and patients with atherosclerosis (6, 12). Superoxides scavenge NO, which can cause an impaired endothelium-dependent vasodilation in response to agonists or flow (34). The importance of negative WSS can be observed in studies of exercise training of patients with CHF where a negative WSS may be reduced (14). The effects of physical training on endothelium-dependent vasodilation in skeletal muscle resistance vessels were investigated in patients with heart failure. After 8 wk of training, the vasodilatory response to acetylcholine increased from pretraining values. The training appeared to specifically enhance endothelium-dependent vasodilation in the forearm skeletal muscle circulation of patients with heart failure. Vasodilation may improve by periodically increasing blood flow in the trained skeletal muscle circulation (31). In another study of patients with CHF, the peripheral blood flow improved significantly in response to acetylcholine and the effect of l-NMMA (an inhibitor of NO synthase) increased after exercise training. The increased response to l-NMMA suggests that regular exercise increases basal NO formation in resistance vessels (19) and eliminates flow reversal in larger vessels (14, 30). The present implication of the NOX system in flow reversal may serve as a foundation for a therapeutic rationale to reduce O2− and increase NO and consequently improve endothelial function.
GRANTS
This research was supported in part by National Heart, Lung, and Blood Institute Grant R01-HL-84529.
Acknowledgments
We thank Quang Dang and Carlos Linares for animal preparation.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Adachi T, Fukushima T, Usami Y, Hirano K. Binding of human xanthine oxidase to sulphated glycosaminoglycans on the endothelial-cell surface. Biochem J 289: 523–527, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alenghat FJ, Ingber DE. Mechanotransduction: all signals point to cytoskeleton, matrix, and integrins. Sci STKE 2002, PE6, 2002. [DOI] [PubMed]
- 3.Anker SD, Doehner W, Rauchhaus M, Sharma R, Francis D, Knosalla C, Davos CH, Cicoira M, Shamim W, Kemp M, Segal R, Osterziel KJ, Leyva F, Hetzer R, Ponikowski P, Coats AJ. Uric acid and survival in chronic heart failure: validation and application in metabolic, functional and hemodynamic staging. Circulation 107: 1991–1997, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Barbato JE, Tzeng E. Nitric oxide and arterial disease. J Vasc Surg 40: 187–193, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Belch JJ, Bridges AB, Scott N, Chopra M. Oxygen free radicals and congestive heart failure. Br Heart J 65: 245–248, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bharadvaj BK, Mabon RF, Giddens DP. Steady flow in a model of the human carotid bifurcation, flow visualization. J Biomech 15: 349–362, 1982. [DOI] [PubMed] [Google Scholar]
- 8.Bubolz AH, Li H, Wu Q, Liu Y. Enhanced oxidative stress impairs cAMP-mediated dilation by reducing Kv channel function in small coronary arteries of diabetic rats. Am J Physiol Heart Circ Physiol 289: H1873–H1880, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases. Circ Res 87: 840–844, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Chen KD, Li YS, Kim M, Li S, Yuan S, Chien S, Shyy JY. Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins, and Shc. J Biol Chem 274: 18393–18400, 1999. [DOI] [PubMed] [Google Scholar]
- 11.Dusting GJ, Selemidis S, Jiang F. Mechanisms for suppressing NADPH oxidase in the vessel wall. Mem Inst Oswaldo Cruz 100: 97–103, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Ellis GR, Anderson RA, Lang D, Blackman DJ, Morris RH, Morris-Thurgood J, McDowell IF, Jackson SK, Lewis MJ, Frenneaux MP. Neutrophil superoxide anion-generating capacity, endothelial function and oxidative stress in chronic heart failure: effects of short- and long-term vitamin C therapy. J Am Coll Cardiol 36: 1474–1482, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Farquharson CA, Butler R, Hill A, Belch JJ, Struthers AD. Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation 106: 221–226, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Gharib M, Beizaie M. Correlation between negative near-wall shear stress in human aorta and various stages of congestive heart failure. Ann Biomed Eng 31: 678–685, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Gimbrone MA, Topper JN, Nagel T, Anderson KR, Garcia-Cardena G. Endothelial dysfunction, hemodynamic forces and atherogenesis. Ann NY Acad Sci 902: 230–240, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Gnasso A, Irace C, Carallo C, De Franceschi MS, Motti C, Mattioli PL, Pujia A. In vivo association between low wall shear stress and plaque in subjects with asymmetrical carotid atherosclerosis. Stroke 28: 993–998, 1997. [DOI] [PubMed] [Google Scholar]
- 17.Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J 386: 401–416, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R, Channon KM. Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ Res 86: E85–E90, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Hambrecht R, Fiehn E, Weigl C, Gielen S, Hamann C, Kaiser R, Yu J, Adams V, Niebauer J, Schuler G. Regular physical exercise corrects endothelial dysfunction and improves exercise capacity in patients with chronic heart failure. Circulation 98: 2709–2715, 1998. [DOI] [PubMed] [Google Scholar]
- 20.Hamilton CA, Brosnan MJ, Al-Benna S, Berg G, Dominiczak AF. NAD(P)H oxidase inhibition improves endothelial function in rat and human blood vessels. Hypertension 40: 755–762, 2002. [DOI] [PubMed] [Google Scholar]
- 21.Harrison D, Griendling KK, Landmesser U, Horning B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol 91: 7A–11A, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi T, Juliet PA, Kano-Hayashi H, Tsunekawa T, Dingqunfang D, Sumi D, Matsui-Hirai H, Fukatsu A, Iguchi A. NADPH oxidase inhibitor, apocynin, restores the impaired endothelial-dependent and -independent responses and scavenges superoxide anion in rats with type 2 diabetes complicated by NO dysfunction. Diabetes Obes Metab 7: 334–343, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Hayden MR, Tyagi SC. Uric acid: a new look at an old risk marker for cardiovascular disease, metabolic syndrome, and type 2 diabetes mellitus: the urate redox shuttle. Nutr Metab (Lond) 1: 10, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hellsten-Westing Y Immunohistochemical localization of xanthine oxidase in human cardiac and skeletal muscle. Histochemistry 100: 215–222, 1993. [DOI] [PubMed] [Google Scholar]
- 25.Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schröder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51: 211–217, 2008. [DOI] [PubMed] [Google Scholar]
- 26.Hille R The mononuclear molybdenum enzymes. Chem Rev 96: 2757–2816, 1996. [DOI] [PubMed] [Google Scholar]
- 27.Hwang J, Ing MH, Salazar A, Lassègue B, Griendling K, Navab M, Sevanian A, Hsiai TK. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: implication for native LDL oxidation. Circ Res 93: 1225–1232, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hwang J, Saha A, Boo YC, Sorescu GP, McNally JS, Holland SM, Dikalov S, Giddens DP, Griendling KK, Harrison DG, Jo H. Oscillatory shear stress stimulates endothelial production of O2− from p47phox-dependent NAD(P)H oxidases, leading to monocyte adhesion. J Biol Chem 278: 47291–47298, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Jalali S, del Pozo MA, Chen K, Miao H, Li Y, Schwartz MA, Shyy JY, Chien S. Integrin-mediated mechanotransduction requires its dynamic interaction with specific extracellular matrix (ECM) ligands. Proc Natl Acad Sci USA 98: 1042–1046, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kassab GS, Navia JA. Biomechanical considerations in the design of graft: the mechanical homeostasis hypothesis. Annu Rev Biomed Eng 8: 499–535, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Katz SD, Yuen J, Bijou R, LeJemtel JH. Training improves endothelium-dependent vasodilation in resistance vessels of patients with heart failure. J Appl Physiol 82: 1488–1492, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 40: 477–484, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol 287: R1014–R1030, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Linke A, Recchia F, Zhang X, Hintze TH. Acute and chronic endothelial dysfunction: implications for the development of heart failure. Heart Fail Rev 8: 87–97, 2003. [DOI] [PubMed] [Google Scholar]
- 35.Lu X, Guo X, Linares C, Kassab GS. A new method to denude the endothelium without damage to media: structural, functional, and biomechanical validation. Am J Physiol Heart Circ Physiol 286: H1889–H1894, 2004. [DOI] [PubMed] [Google Scholar]
- 36.Lu X, Kassab GS. Nitric oxide is significantly reduced in ex vivo porcine arteries during reverse flow because of increased superoxide production. J Physiol 561: 575–582, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuzaki I, Chatterjee S, Debolt K, Manevich Y, Zhang Q, Fisher AB. Membrane depolarization and NADPH oxidase activation in aortic endothelium during ischemia reflect altered mechanotransduction. Am J Physiol Heart Circ Physiol 288: H336–H343, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Meininger GA, Davis MJ. Cellular mechanisms involved in the vascular myogenic response. Am J Physiol Heart Circ Physiol 263: H647–H659, 1992. [DOI] [PubMed] [Google Scholar]
- 39.Moe KT, Aulia S, Jiang F, Chua YL, Koh TH, Wong MC, Dusting GJ. Differential upregulation of NOX homologues of NADPH oxidase by tumor necrosis factor-alpha in human aortic smooth muscle and embryonic kidney cells. J Cell Mol Med 10: 231–239, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishida K, Harrison DG, Navas JP, Fisher AA, Dockery SP, Uematsu M, Nerem RM, Alexander RW, Murphy TJ. Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J Clin Invest 90: 2092–2096, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327: 524–526, 1987. [DOI] [PubMed] [Google Scholar]
- 42.Park L, Anrather J, Zhou P, Frys K, Wang G, Iadecola C. Exogenous NADPH increases cerebral blood flow through NADPH oxidase-dependent and -independent mechanisms. Arterioscler Thromb Vasc Biol 24: 1860–1865, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Patetsios P, Song M, Shutze WP, Pappas C, Rodino W, Ramirez JA, Panetta TF. Identification of uric acid and xanthine oxidase in atherosclerotic plaque. Am J Cardiol 99: 188–191, 2001. [DOI] [PubMed] [Google Scholar]
- 44.Pearlstein DP, Ali MH, Mungai PT, Hynes KL, Gewertz BL, Schumacker PT. Role of mitochondrial oxidant generation in endothelial cell responses to hypoxia. Arterioscler Thromb Vasc Biol 22: 566–573, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Piao YJ, Seo YH, Hong F, Kim JH, Kim YJ, Kang MH, Kim BS, Jo SA, Jo I, Jue DM, Kang I, Ha J, Kim SS. Nox 2 stimulates muscle differentiation via NF-kappaB/iNOS pathway. Free Radic Biol Med 38: 989–1001, 2005. [DOI] [PubMed] [Google Scholar]
- 46.Ray R, Shah AM. NADPH oxidase and endothelial cell function. Clin Sci (Lond) 109: 217–226, 2005. [DOI] [PubMed] [Google Scholar]
- 47.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2− and systolic blood pressure in mice. Circ Res 89: 408–414, 2001. [DOI] [PubMed] [Google Scholar]
- 48.Rouquette M, Page S, Bryant R, Benboubetra M, Stevens CR, Blake DR, Whish WD, Harrison R, Tosh D. Xanthine oxidoreductase is asymmetrically localised on the outer surface of human endothelial and epithelial cells in culture. FEBS Lett 426: 397–401, 1998. [DOI] [PubMed] [Google Scholar]
- 49.Schlüter T, Steinbach AC, Steffen A, Rettig R, Grisk O. Apocynin-induced vasodilation involves Rho kinase inhibition but not NADPH oxidase inhibition. Cardiovasc Res 80: 271–279, 2008. [DOI] [PubMed] [Google Scholar]
- 50.Serrander L, Jaquet V, Bedard K, Plastre O, Hartley O, Arnaudeau S, Demaurex N, Schlegel W, Krause KH. NOX5 is expressed at the plasma membrane and generates superoxide in response to protein kinase C activation. Biochimie 89: 1159–1167, 2007. [DOI] [PubMed] [Google Scholar]
- 51.Shah AM, MacCarthy PA. Paracrine and autocrine effects of nitric oxide on myocardial function. Pharmacol Ther 86: 49–86, 2000. [DOI] [PubMed] [Google Scholar]
- 52.Sorescu GP, Song H, Tressel SL, Hwang J, Dikalov S, Smith DA, Boyd NL, Platt MO, Lassègue B, Griendling KK, Jo H. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a Nox1-based NADPH oxidase. Circ Res 95: 773–779, 2004. [DOI] [PubMed] [Google Scholar]
- 53.Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol 11: 95–102, 1994. [DOI] [PubMed] [Google Scholar]
- 54.Takayama T, Wada A, Tsutamoto T, Ohnishi M, Fujii M, Isono T, Horie M. Contribution of vascular NAD(P)H oxidase to endothelial dysfunction in heart failure and the therapeutic effects of HMG-CoA reductase inhibitor. Circ J 68: 1067–1075, 2004. [DOI] [PubMed] [Google Scholar]
- 55.Touyz RM Apocynin, NADPH oxidase, and vascular cells: a complex matter. Hypertension 51: 172–174, 2008. [DOI] [PubMed] [Google Scholar]
- 56.Vejrazka M, Mícek R, Stípek S. Apocynin inhibits NADPH oxidase in phagocytes but stimulates ROS production in non-phagocytic cells. Biochim Biophys Acta 1722: 143–147, 2005. [DOI] [PubMed] [Google Scholar]
- 57.Yagi T, Seo BB, Nakamaru-Ogiso E, Marella M, Barber-Singh J, Yamashita T, Kao MC, Matsuno-Yagi A. Can a single subunit yeast NADH dehydrogenase (Ndi1) remedy disease caused by respiratory complex I defects? Rejuvenation Res 9: 191–197, 2006. [DOI] [PubMed] [Google Scholar]
- 58.Zhou X, Bohlen HG, Miller SJ, Unthank JL. NAD(P)H oxidase-derived peroxide mediates elevated basal and impaired flow-induced NO production in SHR mesenteric arteries in vivo. Am J Physiol Heart Circ Physiol 295: H1008–H1016, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]