

Abstract
Toll-like receptors (TLRs) play important roles in innate immunity. Several TLR family members have recently been shown to be expressed by neurons and glial cells in the adult brain, and may mediate responses of these cells to injury and infection. To address the possibility that TLRs play a functional role in development of the nervous system, we analyzed the expression of TLRs during different stages of mouse brain development and assessed the role of TLRs in cell proliferation. TLR3 protein is present in brain cells in early embryonic stages of development, and in cultured neural stem/progenitor cells (NPC). NPC from TLR3-deficient embryos formed greater numbers of neurospheres compared with neurospheres from wild-type embryos. Numbers of proliferating cells, as assessed by phospho histone H3 and proliferating cell nuclear antigen labeling, were also increased in the developing cortex of TLR3-deficient mice compared with wild-type mice in vivo. Treatment of cultured embryonic cortical neurospheres with a TLR3 ligand (polyIC) significantly reduced proliferating (BrdU-labeled) cells and neurosphere formation in wild type but not TLR3−/−-derived NPCs. Our findings reveal a novel role for TLR3 in the negative regulation of NPC proliferation in the developing brain.
Keywords: stem cells, embryo, immunity, cortex, neurogenesis, ventricle
Introduction
The family of mammalian toll-like receptors (TLRs) includes at least 12 membrane proteins that sense the invasion of pathogenic microorganisms and trigger innate immune responses through nuclear factor-κB (NF-κB)-dependent and interferon regulatory factor (IRF)-dependent pathways (Akira, 2006). TLRs were also shown to recognize endogenous ligands (Wagner, 2006). For TLR2, −3 and −4 many different endogenous ligands have been described (Rifkin et al., 2005) including heat-shock proteins or mRNA that are present after the integrity of an adjacent cell has been compromised, as well as proteins normally present in tissues such as hyaluronan or fibrinogen (Smiley et al., 2001; Termeer et al., 2002). TLRs are present in the brain, where their expression was initially believed to be limited to glial cells (Lehnardt et al., 2002; Babcock et al., 2006). However, recent evidence suggests that neurons and neural progenitor cells also express TLRs (Ma et al., 2006; Rolls et al., 2007; Tang et al., 2007). Data suggest that neurons express several TLRs responsive to viral RNA or bacterial proteins including TLR2, TLR3, TLR4 and TLR8 (Lafon et al., 2006; Ma et al., 2006; Tang et al., 2007). TLR2 and TLR4 are expressed in cerebral cortical neurons where their levels and downstream signaling via JNK are increased in response to energy deprivation in cell culture and ischemia in vivo (Tang et al., 2007). Selective elimination of TLR2 or TLR4 function suppresses activation of pro-apoptotic JNK and protects cortical neurons against death induced by energy deprivation and stroke. TLR2 expression increased rapidly in neurons within the first few hours after a stroke, well before its upregulation in microglia (Tang et al., 2007).
In contrast to their established roles in the immune and nervous systems of adult animals, the roles of mammalian TLRs in development are unknown. Several TLRs have been reported to be expressed in the developing nervous system. TLR8 is dynamically expressed during mouse brain development and localizes to neurons and axons where it may regulate neurite outgrowth and apoptosis (Ma et al., 2006). TLR2 and TLR4 are expressed in adult NPC and may influence their proliferation and differentiation (Rolls et al., 2007). TLR2 deficiency in mice impaired hippocampal neurogenesis, whereas the absence of TLR4 resulted in enhanced proliferation and neuronal differentiation (Rolls et al., 2007). However, roles for TLR3 in brain development have not yet been established. Here we show, using TLR3-deficient mice and components of the TLR3 signaling pathway, that TLR3 is a negative regulator of NPC proliferation in the developing mouse brain.
Materials and Methods
Animals.
C57BL/6 mice, TLR3 knock-out mice (B6;129S1-Tlr3tm1Flv/J) and wild type mice (TLR+/+) of the same genetic background (B6129SF1/J) were purchased from The Jackson Laboratories and were maintained in our animal facility under pathogen-free conditions on a 12 h light/12 h dark cycle with continuous access to food and water. Embryos of different gestational ages were collected from pregnant mice for tissue analysis. Neurosphere cultures were prepared using brain tissue from C57B/6, TLR3−/− and TLR+/+ mice. All procedures were approved by the National Institute on Aging Animal Care and Use Committee.
Immunostaining.
These procedures were similar to those described previously (Lathia et al., 2007). Briefly, embryonic brains were fixed in 4% paraformaldehyde in PBS overnight at 4°C before being transferred to sequential 20% and 30% solutions of sucrose (w/v) at 4°C until the brains sank to the bottom of the solution. The brains were embedded in TissueTek (Sakura) before sectioning (6 μm sections made using a Leica CM3050S cryostat) in the coronal anatomical plane. Sections were first exposed for a minimum of 30 min to PBS containing 0.1% Triton X-100 (Amresco) and 10% normal goat serum (Sigma) to block nonspecific antibody binding, followed by incubation overnight with the relevant primary antibodies at 4°C. Sections were then incubated with the appropriate species-specific Alexa Fluor 488- or 568-conjugated IgG (1:200–1:500, Invitrogen) antibodies in the dark at room temperature (RT) for 1 h, and counter-stained with Hoechst 33342 (0.05 mg/ml solution in deionized water, Invitrogen). Images were acquired using a MagnaFire camera and analyzed using Photoshop (Adobe) software. For staining of cultured cells, the cells were first fixed with 2% paraformaldehyde at RT for 30 min, followed by three washes in PBS and incubation with primary and secondary antibodies as described above. The following antibodies were used for immunohistochemistry: anti-TLR3 (1:100, CSA 790; Stressgen), anti-phospho histone H3 (PH3, 1:500, 06–570; Millipore), anti-proliferating cell nuclear antigen (PCNA, 1:500, ab29; Abcam), anti-β3-tubulin/TUJ1 (1:500, T5076; Sigma), anti-GFAP (1:200, G3893; Sigma), anti-Sox 2 (1:100, MAB2018; R & D Systems), anti-CNPase (1:250, clone 11–5B; Sigma). In vivo analysis for PH3 staining was done by quantifying the number of positive cells in the entire extent of the embryonic day (E) 13.5 dorsal telencephalon in 3 separate sections from 7 TLR+/+ and 6 TLR3 −/− mice. In vivo analysis for PCNA staining was done blindly by quantifying the amount of positive cells in a 100 by 200 μm (width by height) area from the center of the E13.5 and E16.5 dorsal telencephalon and expressed as an average based on 3 separate sections from 5 control and TLR3 −/− mice.
Immunoblot analysis.
These procedures were similar to those described previously (Tang et al., 2007). Brain tissue protein was extracted using T-PER tissue protein extraction buffer containing a protease inhibitor mixture (Sigma). Protein concentration was determined by using a BCA protein assay kit (Pierce). Protein in samples (40 μg) was separated by SDS-PAGE (8–12%) and transferred to a nitrocellulose membrane. The membrane was blocked in 5% nonfat milk for 1 h at RT, followed by an overnight incubation at 4°C with primary antibodies: TLR2 (1:1000, 2229; Cell Signaling Technology), TLR3(1:1000, 2253; Cell Signaling Technology), TLR4 (1:1000, 2246; Cell Signaling Technology), IRF3 (1:1000, 4962; Cell Signaling Technology), actin (1:1000, A5441; Sigma), synaptophysin (1:2000, SY38; Sigma), α-tubulin (1:10,000, T6074; Sigma). The membrane was then washed and incubated with a secondary antibody for 1 h at RT. Protein bands were visualized using a chemiluminescence detection kit (GE Healthcare).
Neurosphere cell culture.
NPCs from the cortex were propagated as free-floating aggregates [neurospheres (NP)], to promote proliferation of neural stem and progenitor cells before use in experiments. In brief, the dorsal telencephalon from E12.5, E13.5, or E14.5 mouse embryos were isolated, mechanically dissociated, and seeded at a density of 200,000 cells/ml in a T75 flask containing DMEM/Ham's F-12 medium, supplemented with B27 (1:50; Invitrogen), epidermal growth factor (EGF), and fibroblast growth factor (FGF)-2 (both at 20 ng/ml; R & D Systems). After neurospheres had grown for 7 d in culture, cells were dissociated using the NeuroCult cell dissociation kit (StemCell Technologies) and plated into 96 wells plates (Nunc), at 1000 cells per well. For adult neurosphere cultures, coronal sections (2-mm-thick) of tissue containing the subependymal zone were obtained from the brains of adult (2 month) C57BL/6 mice. The tissue was incubated for digestion at 37°C, 5% CO2 for 10 min in HBSS containing 2 mg/ml trypsin (Sigma). The trypsin was subsequently decanted and digestion was stopped with addition of HBSS containing 2 mg/ml trypsin inhibitor (Sigma), and the tissue was minced and mechanically dissociated by pipette trituration. Cells obtained from single cell suspensions were seeded and cultured as described above.
Neurospheres which formed were counted after 7 d in culture and the average of 8 wells was taken per experimental condition. Neurosphere forming assays for E13.5 wild-type (WT) and TLR3 −/− NPC were done in triplicate; each replicate consisted of a pool of embryos from the same litter. Neurosphere forming assays for E14.5 C57BL/6 NPC stimulated with polyIC were done a minimum of 5 times, each sample consisted of a pool of embryos from the same litter. To induce differentiation, growth factors were withdrawn, and neurospheres were plated on poly (d-lysine) and laminin-111 (10 μg/ml; Sigma)-coated eight-well chamber slides (Nunc) for 7 d before immunostaining.
Neurosphere diameter measurements.
To measure neurospheres diameter, pictures of 10 wells of each experimental condition were taken using a Magnafire camera. Images were then analyzed using NIH ImageJ software (http://rsbweb.nih.gov/ij/).
Viability assay.
NPC cultures were incubated with 10 μm Resazurin dye (Sigma) for 30 min at 37°C. Fluorescence was then measured using an HTS 7000 plus (Perkin-Elmer) plate reader with a 540 nm excitation and 595 nm emission wavelengths.
Reverse transcriptase-PCR for TLR3.
Total RNA was isolated from 600,000 cells from NPC derived from E14 embryonic and adult WT mice using the QiaZol reagent (Qiagen). Reverse transcription of RNA to cDNA was performed using SuperScript III First-Strand Synthesis SuperMix (Invitrogen). The cDNA synthesized was subjected to PCR amplification using Platinum Blue PCR SuperMix (Invitrogen) with TLR3 primers pair (forward: 5′ agctttgctgggaactttca 3′; reverse: 5′ gaaagatcgagctgggtgag 3′; 127bp) and was normalized against β-actin (forward: 5′ tggaatcctgtggcatccatgaaac 3′; reverse: 5′ taaaacgcagctcagtaacagtccg 3′, 348 bp). PCR was performed using PTC-200 (MJ Research) PCR system and visualized by ethidium bromide stained 2% TAE-agarose gel electrophoresis. PCR amplification consisted of 30 cycles of 30 s denaturation at 94°C, 1m annealing at 54°C, and 30 s of elongation at 72°C. PCR products quantified using NIH ImageJ software.
Statistical analysis.
Experimental results are expressed as mean ± SEM. Statistical comparisons were made with ANOVA, followed by Newman–Keuls post hoc analysis for pairwise comparisons.
Results
TLR3 expression is high during early brain development
To determine whether TLR expression changes during brain development, we first measured levels of several TLR proteins in mouse cortical tissues of different embryonic and early postnatal stages. In the telencephalon, neurogenesis commences at E11 and peaks between E14 and E16; during this period of time there is first a rapid expansion of NPC followed by the generation of neurons via asymmetric divisions [for review, see Götz and Huttner (2005)]. Hence, at these early stages of brain development, the majority of cells in the cortex are NPCs and over time, the number of neurons increases. Immunoblot analysis demonstrated that the TLR3 protein was present in relatively high amounts at E12.5, and its levels decreased through E17.5, matching the rate of decreased NPC expansion (Fig. 1A). However, the TLR2 protein was very low in the developing cortex at E12.5 or E13.5, but increased gradually from E15.5 to neonatal stages, corresponding to NPCs gradually differentiating as brain development proceeds (Fig. 1A). Further immunoblot analysis showed that as neuronal maturation commences, as assessed by synaptophysin expression, the expression of TLR2 protein was elevated (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), indicating that TLR2 is likely to be highly expressed in neurons. In contrast, TLR4 protein levels were detectable at E12.5 and remained at a relatively constant level through P13 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We also determined levels of the transcription factor interferon regulatory factor 3 (IRF3), which is involved in the TLR3 signaling pathway (Jiang et al., 2004) in the same brain tissue samples; IRF3 levels were highest at E12.5 and were decreased considerably after E18.5 (Fig. 1A), again matching the levels of TLR3 protein and decrease in the rate of NPC expansion. To further evaluate the dynamics of IRF3 and TLR2 and 3 protein levels in response to NPC expansion, we used proliferating neurospheres (NP) as a model system. TLR2 was not detected in NP derived from E12.5 embryos, but was expressed in cells within 2 d of culture in differentiation-promoting conditions (Fig. 1B). In contrast, cells in NP expressed TLR3 and IRF3 in high amounts, and levels of TLR3 and IRF3 decreased rapidly (within 24 h) after plating cells in differentiation-promoting conditions (Fig. 1B).
Figure 1.

Expression of TLR2, TLR3 and the TLR3-regulated transcription factor IRF3 in the developing brain. A, Immunoblot showing relative levels of TLR2 (95 kDa), TLR3 (95 kDa), and IRF3 (55 kDa) in lysates of cortical tissue collected from mice at the indicated stages of development (E, embryonic; P, postnatal). B, Immunoblot showing relative levels of TLR2 (95 kDa), TLR3 (95 kDa), and IRF3 (55 kDa) in lysates of neurospheres (NP) derived fro E12.5 mice, and dissociated cell cultures established from neurospheres (days 1–7 after plating in differentiation-promoting conditions).
The immunoblot data suggested that NPC express relatively high amounts of TLR3 and its downstream signaling effector protein IRF3, suggesting a potential role for TLR3 in regulating NPC proliferation during brain development. Notably, IRF3 expression was increased in response to PolyIC treatment in WT but not in NPC derived from TLR3-deficient mice (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). To determine the cellular localization of TLR in the developing cortex in vivo, we immunostained brain sections of different age mouse embryos with a TLR3 antibody. We chose ages that correspond to developmental periods of cortical NPC expansion (E10.5), and early (E13.5) and mid (E16.5) embryonic neurogenesis. At E10.5, when the neuroepithelium consists of dividing NPC, there was strong TLR3 immunoreactivity throughout the neuroepithelium (Fig. 2A). TLR3 was also detected in cells throughout the cortex at E13.5 (Fig. 2B), and was coexpressed in sox2-positive NPC in the cortex (data not shown). During embryonic neurogenesis, TLR3 was still detected in the NPC-containing region of the cortex and was weakly expressed in β3-tubulin positive (neuronal) cells in the outer regions of the developing cortex (Fig. 2C). TLR3-deficient embryos at both E13.5 and E16.5 were negative for staining with the TLR3 antibody, demonstrating that the antibody used for in vivo assessment was specific to TLR3 (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material).
Figure 2.
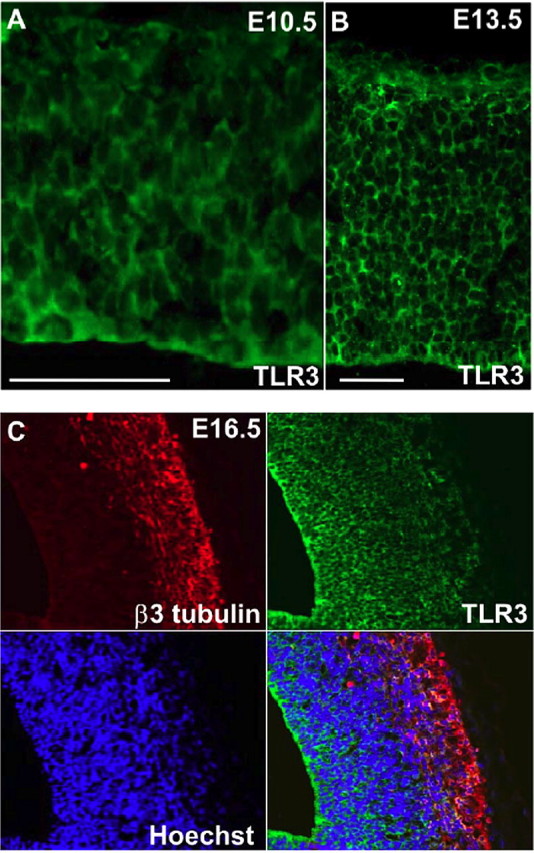
TLR3 is present in relatively high amounts in cells in the proliferative regions of the developing cerebral cortex. A, Strong TLR3 immunoreactivity (green) is present in cells throughout the neuroepithelium at E10.5. B, TLR3 immunoreactivity (green) is present in cells throughout the cortex at E13.5. C, Expression of TLR3 (green) in the E16.5 cortex; cells were counterstained with Hoechst (blue) to label all nuclei and with the neuron-specific marker β3-tubulin (red). Scale bar, 100 μm. Note that TLR3 immunoreactivity is much stronger in β3-tubulin-negative cells in the proliferative ventricular zone compared with the low TLR3 immunoreactivity in β3-tubulin-positive cells.
TLR3 negatively regulates neural progenitor cell proliferation
As we observed enriched expression of TLR3 in early stages of brain development, we next determined whether TLR3 signaling affects NPC proliferation. We used NPC-containing neurosphere cultures to assess the role of TLR3 on proliferation. Neurospheres derived from E12.5 mice were treated with the TLR3 ligand polycytidylic acid (polyIC; 10 and 20 μg/ml) and downstream signaling molecule interferon-β (200 and 500 U) for 24 h (Alexopoulou et al., 2001; Matsumoto et al., 2004). Both polyIC and interferon-β significantly reduced the number of BrdU-positive cells in cultures (Fig. 3A), suggesting that TLR3 activation inhibits NPC proliferation. We also assessed neurosphere formation in the presence of polyIC and relative to a time pregnant control at E14.5, both 2 μg/ml and 20 μg/ml of polyIC reduced the number of neurospheres which formed (Fig. 3B). We further analyzed NPC proliferation using E13.5 neurosphere cultures derived from TLR3 mutant mice, along with corresponding wild type background strain-matched controls. Neurosphere-forming assays showed significantly more neurospheres formed from TLR3-deficient NPCs (93% increase) compared with wild type controls (Fig. 3B). To assess the differentiation potential of these neurospheres, they were plated on laminin111/121 for 7 d in the absence of growth factors to promote cell differentiation. Immunofluorescence analysis showed that both the TLR3−/− (Fig. 3C) and control (data not shown) neurospheres were multipotent and capable of giving rise to neurons, astrocytes, and oligodendrocytes. There was no significant difference in the percentage of neurons, astrocytes and oligodendrocytes between WT and TLR3−/− NPC (Fig. 3D). In addition, no premature differentiation was observed in TLR3−/− NPC cultures or in the ventricular zone of TLR3 −/− embryos (data not shown).
Figure 3.
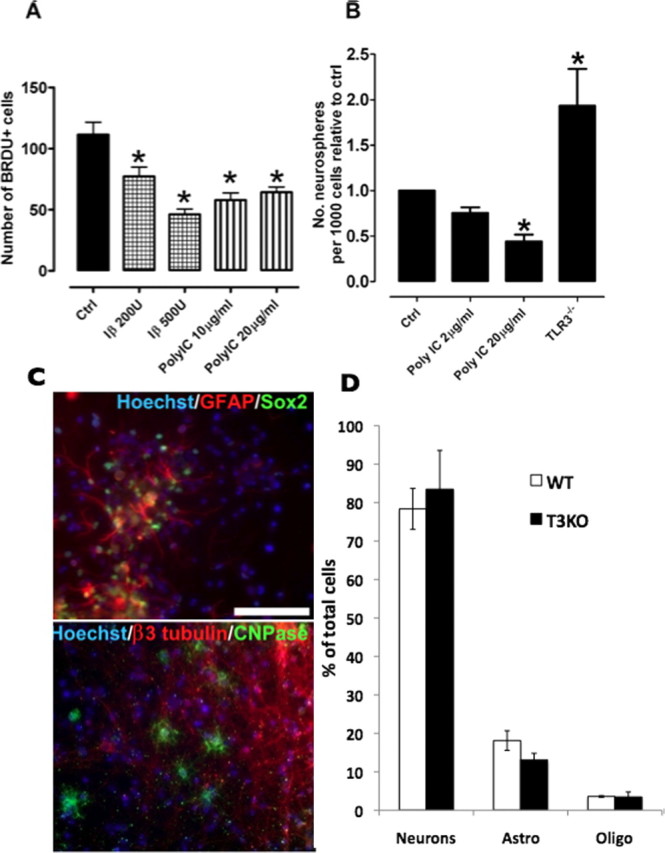
TLR3 signaling negatively regulates neural progenitor cell proliferation. A, Treatment of neurospheres with the TLR3 ligands polycytidylic acid (polyIC) (10 or 20 μg/ml) and interferon-β (200 or 500 U) significantly reduced the number of BrdU-positive (proliferating) cells. *p < 0.05 compared with the control value. B, NPC proliferation relative to control values in wild-type neurospheres treated with polyIC (2 or 20 μg/ml) and neurosphere cultures derived from TLR3−/− mice. Immunofluorescence analysis of differentiated TLR3−/− neurospheres showing their potential to remain NPCs (C, bottom, sox2, green) or differentiate into astrocytes (C, bottom, GFAP, red), oligodendrocytes (C, top, CNPase, green), or neurons (C, top, β3-tubulin, red). All images counterstained with Hoechst. Scale bar, 100 μm. D, No difference was observed in the percentage of neurons, astrocytes and oligodendrocytes derived from WT or TLR3−/− (TLR3KO) mice.
To confirm that the changes in proliferation induced by PolyIC are mediated by TLR3 signaling, we compared WT and TLR3−/− NPC proliferation following PolyIC administration. Both primary and secondary sphere formation was significantly higher in NPC derived from TLR3−/− compared with WT mice (Fig. 4A). Although 2 μg/ml PolyIC did not significantly alter neurosphere formation, NPCs treated with 10 μg/ml exhibited lower neurosphere formation than WT controls (Fig. 4B). Furthermore, 20 μg/ml polyIC significantly depressed neurosphere formation from WT NPC. In contrast, neurosphere formation by NPCs from TLR3 −/− mice remained unchanged following PolyIC administration. Whereas PolyIC did not significantly alter the diameter size of WT NPCs, TLR3−/− neurospheres were significantly larger than WT neurospheres (Fig. 4C). In addition, viability was unaltered at low concentrations of PolyIC; however, 20 μg/ml PolyIC significantly decreased viability of WT-derived neurospheres (Fig. 4D). The decrease in viability coincided with the significant decrease in total neurosphere formation observed at high PolyIC concentrations (Fig. 4B).
Figure 4.

PolyIC depresses neural progenitor cell proliferation through TLR3. A, NPC from TLR3-deficient mice form a significantly higher number of both primary and secondary neurospheres than WT controls. B, Treatment of neurospheres with PolyIC (2, 10, or 20 μg/ml) significantly depressed neurosphere formation at high concentrations, but did not alter neurosphere formation in cultures derived from TLR3-deficient mice. C, PolyIC administration did not significantly alter neurosphere diameter. However, neurospheres derived from TLR3-deficient mice are significantly larger than WT mice. D, PolyIC decreased viability of neurospheres derived from WT animals at 20 mg/ml; however, it did not alter viability of TLR3−/−-derived neurospheres. Differences are represented as ±SEM, *p < 0.05.
Given the increase in NPC proliferation in cells derived from TLR3−/− embryos, we also assessed the proliferation in vivo by using markers for different parts of the cell cycle; PCNA marking cells in early G1 to S-phase and PH3 marking cells in M-phase. Proliferation as assessed by PCNA-positive cells was significantly (p < 0.05) increased in E13.5 but not on E16.5 TLR3−/− embryos (Fig. 5A). Consistent with this, the number of PH3-positive cells was also significantly increased in TLR3−/− mice (Fig. 5B) (31.1 ± 1.96 and 37.77 ± 2.89 in WT and TLR3−/−, respectively). Collectively, these findings suggest that TLR3 signaling inhibits the proliferation of NPC at early (E13.5) embryonic stages when TLR3 is highly expressed and that this effect is abrogated at later (E16.5) embryonic stage when TLR3 expression is reduced.
Figure 5.

TLR3 signaling negatively regulates neural progenitor proliferation in vivo. A, Quantification of PCNA-positive cells from a 100 × 200 μm region of the E13.5 dorsal telencephalon of TLR3-deficient (TLR3−/−) mice showed a significant increase (*p < 0.05) when compared with control (Ctrl) mice. No difference was observed between WT and TLR3-deficient mice at E16.5. Hoechst-stained example shown on left with yellow box indicating area of analysis. V, Ventricle; D, dorsal; M, medial. B, There is a significant increase in PH3-positive cells in the dorsal telencephalon TLR3−/− mice. Scale bars, 100 μm.
Discussion
TLRs are best known as initiators of the innate immune response to pathogens and, until recently, it was believed that microglia and other glial cells were the only intrinsic types of brain cell that express TLRs and respond to TLR ligands (Bsibsi et al., 2002; Olson and Miller, 2004). However, recent reports revealed interesting roles for TLRs in the CNS. Several TLRs, including (TLR2, TLR3 and TLR4) are expressed by primary cortical neurons in culture and in the adult brain (Lafon et al., 2006; Tang et al., 2007). TLR2 and TLR4 (but not TLR3) are activated in cortical neurons in response to oxidative and metabolic stress resulting in induction of a pro-apoptotic signaling pathway involving the kinase JNK (Tang et al., 2007). The present findings suggest that TLR3 may play a particularly important role in the regulation of NPC proliferation during early stages of development of the cerebral cortex. TLR3 expression was highest in the early period of cortical development when NPC are highly proliferative, and then declined as neurogenesis and gliogenesis ensued (mid-gestation through early postnatal periods). In contrast to TLR3, TLR2 was not detected at early embryonic stages but showed strong expression in late embryonic and early postnatal brain, while TLR4 was expressed at relatively constant amounts in the brain during embryonic development. The developmental increase of TLR2 expression provides additional support for the recently reported role of TLR2 in regulating adult neurogenesis (Rolls et al., 2007). Interestingly, adult neurospheres stimulated with polyIC did not show a decrease in neurosphere formation (data not shown) as was seen with embryonic neurospheres, suggesting a context dependent role for TLR2 and 3 in regulating NPC behavior. Consistent with this adult-derived NPC express lower levels of TLR3 mRNA compared with embryonic-derived NPC (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Our data suggest that TLR3 signaling inhibits the proliferation of embryonic NPC. Consistent with this hypothesis, a TLR3 ligand and downstream signaling molecule inhibited the proliferation of embryonic mouse NPC, and NPC proliferation was increased in the developing cerebral cortex of TLR3-deficient mice. The activation of TLR signaling pathways originates from the cytoplasmic Toll/IL-1 receptor (TIR) domain; a proline residue in the TIR domain is conserved among all TLRs, except for TLR3 (Takeda and Akira, 2004). In the signaling pathway downstream of the TIR domain, an adaptor protein called myeloid differentiation factor 88 (MyD88) plays a crucial role. The TLR3 ligand dsRNA has been shown to induce activation of NF-κB in MyD88 knock-out cells (Alexopoulou et al., 2001); this MyD88-independent signaling pathway leads to the activation of the transcription factor IRF3 (Yamamoto et al., 2003). We observed a correlation between the expression levels of IRF3 and TLR3, such that both were present in highest amounts in NPC during early cortical development in vivo and in neurosphere cells in culture. These data are consistent with the possibility that IRF3 mediates the inhibition of NPC proliferation in response to TLR3 activation. Based upon previous findings showing that TLR3/IRF3 signaling induces interleukin-6 (IL-6) production (Kim et al., 2008), and that IL-6 can affect NPC proliferation (Mondal et al., 2004), IL-6 is a potential target of IRF3 that might play a role in TLR3-mediated inhibition of NPC proliferation.
Although present in higher amounts in NPC, TLR3 is also expressed in the neuronal and glial progeny of the NPC. It was recently reported that TLR3 is expressed in the mouse central and peripheral neurons where it is concentrated in the growth cones; activation of TLR3 by the synthetic ligand polyIC or by mRNA causes growth cone collapse and inhibits neurite extension (Cameron et al., 2007). The specific mechanism(s) by which TLR3 signaling inhibits the proliferation of NPC and the outgrowth of developing neurons remains to be established. However, because both cell proliferation and growth cone motility involve dynamic changes in the cytoskeleton and membranes, it is possible that TLR3 signaling inhibits such processes. Indeed, TLR signaling in dendritic cells has striking effects on endocytosis and the cytoskeleton (Watts et al., 2007). Our present findings identify a previously unknown role for TLR3 signaling in the negative regulation of NPC proliferation, a mechanism that may control the growth of the developing cerebral cortex.
Footnotes
This work was supported by the National Institute on Aging Intramural Research Program. J.D.L. is supported by the National Institutes of Health–Cambridge Graduate Partnership Program. C.ff.-C. is supported by the Wellcome Trust.
References
- Akira S. TLR signaling. Curr Top Microbiol Immunol. 2006;311:1–16. doi: 10.1007/3-540-32636-7_1. [DOI] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Babcock AA, Wirenfeldt M, Holm T, Nielsen HH, Dissing-Olesen L, Toft-Hansen H, Millward JM, Landmann R, Rivest S, Finsen B, Owens T. Toll-like receptor 2 signaling in response to brain injury: an innate bridge to neuroinflammation. J Neurosci. 2006;26:12826–12837. doi: 10.1523/JNEUROSCI.4937-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Cameron JS, Alexopoulou L, Sloane JA, DiBernardo AB, Ma Y, Kosaras B, Flavell R, Strittmatter SM, Volpe J, Sidman R, Vartanian T. Toll-like receptor 3 is a potent negative regulator of axonal growth in mammals. J Neurosci. 2007;27:13033–13041. doi: 10.1523/JNEUROSCI.4290-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz M, Huttner WB. Cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Mak TW, Sen G, Li X. Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc Natl Acad Sci U S A. 2004;101:3533–3538. doi: 10.1073/pnas.0308496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Yang E, Lee J, Kim SH, Shin JS, Park JY, Choi SJ, Kim SJ, Choi IH. Double-stranded RNA mediates interferon regulatory factor 3 activation and interleukin-6 production by engaging Toll-like receptor 3 in human brain astrocytes. Immunology. 2008;124:480–488. doi: 10.1111/j.1365-2567.2007.02799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon M, Megret F, Lafage M, Prehaud C. The innate immune facet of brain: human neurons express TLR-3 and sense viral dsRNA. J Mol Neurosci. 2006;29:185–194. doi: 10.1385/JMN:29:3:185. [DOI] [PubMed] [Google Scholar]
- Lathia JD, Patton B, Eckley DM, Magnus T, Mughal MR, Sasaki T, Caldwell MA, Rao MS, Mattson MP, ffrench-Constant C. Patterns of laminins and integrins in the embryonic ventricular zone of the CNS. J Comp Neurol. 2007;505:630–643. doi: 10.1002/cne.21520. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Li J, Chiu I, Wang Y, Sloane JA, Lü J, Kosaras B, Sidman RL, Volpe JJ, Vartanian T. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J Cell Biol. 2006;175:209–215. doi: 10.1083/jcb.200606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Funami K, Oshiumi H, Seya T. Toll-like receptor 3: a link between toll-like receptor, interferon and viruses. Microbiol Immunol. 2004;48:147–154. doi: 10.1111/j.1348-0421.2004.tb03500.x. [DOI] [PubMed] [Google Scholar]
- Mondal D, Pradhan L, LaRussa VF. Signal transduction pathways involved in the lineage-differentiation of NSCs: can the knowledge gained from blood be used in the brain? Cancer Invest. 2004;22:925–943. doi: 10.1081/cnv-200039679. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167:2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner H. Endogenous TLR ligands and autoimmunity. Adv Immunol. 2006;91:159–173. doi: 10.1016/S0065-2776(06)91004-9. [DOI] [PubMed] [Google Scholar]
- Watts C, Zaru R, Prescott AR, Wallin RP, West MA. Proximal effects of Toll-like receptor activation in dendritic cells. Curr Opin Immunol. 2007;19:73–78. doi: 10.1016/j.coi.2006.11.014. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]