

Abstract
Microglial activation is an early and common feature of almost all neuropathologies, including multiple sclerosis, Alzheimer's disease and mechanical injury. To better understand the relative contributions microglia make toward neurodegeneration and neuroprotection, we used TOGA® to identify molecules expressed by microglia and regulated by inflammatory signals. Triggering receptor expressed on myeloid cells-2 (TREM-2) was among the mRNAs identified as being expressed by unactivated microglia, but down-regulated by lipopolysaccharide/interferon γ. In the healthy CNS, not all microglia expressed TREM-2. Microglial expression of TREM-2 varied not only between brain regions but also within each brain region. Brain regions with an incomplete blood–brain barrier had the lowest percentages of TREM-2- expressing microglia, whereas the lateral entorhinal and cingulate cortex had the highest percentages. A novel form of TREM-2b that lacked a transmembrane domain was detected, perhaps indicating a soluble form of the protein. Taken together, these data suggest that (1) subsets of microglia are specialized to respond to defined extracellular signals; and (2) regional variations in TREM-2 expression may contribute to the varying sensitivities of different brain regions to similar pathological signals.
Keywords: adaptive immunity, inflammation, innate immunity, microglia, DAP12, triggering receptor expressed on myeloid cells
Tissue-specific inflammation is dependent on more than simply the presence of an antigen within a tissue and an immune response mounted against that antigen. The onset, progression and termination of inflammatory responses are largely dependent on how the resident tissue macrophage/dendritic cell interacts with both stromal tissue and tissue-infiltrating immune cells (Medzhitov and Janeway 1998; Lo et al. 1999). These interactions can shape antigen-independent and antigen-dependent immune responses toward the production of toxic molecules capable of destroying not only pathogens but also the tissues themselves. Such events have been argued to contribute to catastrophic neurodegenerative diseases such as multiple sclerosis, Alzheimer's disease and stroke (Kreutzberg 1996; Stoll and Jander 1999; Becher et al. 2000; Streit 2000; Aloisi 2001; Schwab et al. 2001; Campanella et al. 2002; Togo et al. 2002). Antigen presentation within the CNS by either microglia and/or CNS-infiltrating macrophages/dendritic cells may also mute and may potentially redirect antigen-specific immune responses toward the production of trophic factors (Ford et al. 1996; Raivich et al. 1998; Carson et al. 1999a; Kerschensteiner et al. 1999; Serpe et al. 1999). Such interactions have been suggested to contribute to the generation of immunological privilege and promotion of neuronal survival in the CNS (Schwartz et al. 1999; Streit 2000).
The tissue macrophages of the brain are the microglia (Kreutzberg 1996; Streit 2000; Aloisi 2001). They are found in all brain regions, often in close apposition with neurons, and comprise between 5 and 15% of cells in the CNS. Microglial activation is a common and early feature of nearly all CNS pathologies. In experimentally induced murine models of Alzheimer's and prion disease, microglial activation precedes more overt signs of neuropathology (Betmouni et al. 1996). The physiological consequences of microglial activation are still under debate. Among the issues in question are whether pathological signals (such as beta-amyloid, interferon (IFN) γ and endotoxin) differentially activate microglia located in different brain regions and so contribute to region-specific pathology, and whether different brain regions differentially respond to microglial activation.
Both in vivo and in vitro studies have demonstrated that activated microglia can perform many macrophage functions, including phagocytosis, cytokine production and antigen presentation (Aloisi 2001). However, several studies have shown that microglia are phenotypically distinct from the macrophage populations located in the meninges and perivascular spaces or macrophages that acutely infiltrate the CNS (Sedgwick et al. 1991; Ford et al. 1995; Carson et al. 1998, 1999b; Williams et al. 2001). In comparison to these macrophage populations, microglia express very low levels of CD45 and major histocompatibility complex (MHC). Whereas macrophages located in the meninges and perivascular regions are replenished by bone marrow-derived cells every few days, microglia in the healthy CNS are long-lived and are rarely replenished (Matsumoto and Fujiwara 1987; Hickey and Kimura 1988). When examined immediately after isolation from the adult CNS, microglia were found to be less effective stimulators of antigen-driven T-cell proliferation than other macrophage populations, but more effective stimulators of antigen-driven T-cell cytokine production (Carson et al. 1999b). Numerous in vitro studies have also demonstrated that the microglial phenotype can be dramatically altered by culture conditions and by co-culture with other cell types (Aloisi 2001). Considered together, these data indicate that microglia are more than just muted macrophages. Furthermore, local conditions are likely to tailor microglial functions to the specific needs of the healthy and diseased CNS.
To what extent and by what mechanisms the intact CNS and CNS-infiltrating immune cells separately and together drive microglial function is still being defined. Global regulation of large populations of microglia by cytokines, chemokines and other soluble products has been demonstrated in numerous studies (Aloisi 2001). More recently, several studies have examined the consequences of direct interactions between microglia and neurons or immune cells. For example, recent studies indicate that CNS neurons directly regulate microglial function in vivo. Microglia express the receptor for CD200 (Hoek et al. 2000). CD200 antibody-blocking studies and knock-out mice reveal that neuronal expression of CD200 represses microglial activation both in the healthy CNS and in the presence of neuronal degeneration or autoimmune inflammation (Hoek et al. 2000). Conversely, T lymphocytes have been demonstrated to increase the microglial production of interleukin-12 and tumour necrosis factor α via interactions between CD154, expressed on activated T cells, and CD40, the CD154 receptor expressed by activated microglia (Tan et al. 1999; Nguyen and Benveniste 2002). If microglia are stimulated by CNS-produced beta-amyloid, CD40 : CD154-triggered tumour necrosis factor α expression is greatly amplified.
To better define differences in microglial function that distinguish the healthy from the inflamed CNS, we have compared gene expression between unstimulated and lipopolysaccharide (LPS)/IFNγ-stimulated microglia. Among the molecules found to be down-regulated by inflammatory signals was the orphan receptor, triggering receptor expressed on myeloid cells-2 (TREM-2) (Bouchon et al. 2000, 2001; Daws et al. 2001). In vivo only a subset of microglia were found to express TREM-2, and the relative abundance of TREM-2-expressing microglia varied by brain region. Previous studies revealed that cross-linking this receptor on the surface of monocyte-derived dendritic cells promoted cell survival and dramatically increased the expression of molecules associated with antigen presentation to CD4 T cells (MHC class II, CD40, B7.2 and the CC-chemokine receptor CCR7) (Bouchon et al. 2001). Considered altogether, these results suggest that regional differences in TREM-2 expression may contribute to the regional variation in sensitivity and responses to pathological signals within the CNS.
Materials and methods
Microglia isolation from mixed glial cultures
Mixed glial cultures were prepared as described previously (Carson et al. 1998). Briefly, brains from newborn C57Bl/6J mice were stripped of meninges, mechanically dissociated, seeded into T-75 flasks and maintained in OM-5 media (Dulbecco's modified Eagle's medium supplemented with 30 nm SeO2, 15 nm T3, 10 ng/mL biotin, 3.7 g/mL NaHCO3, 1.5 g/mL glucose, 10% fetal bovine serum and 50 μg/mL gentamicin). After 2–4 weeks in culture, mixed glial cultures that had either been left untreated or treated for 22 h with 100 ng/mL LPS and 10 U/mL IFNγ were trypsinized and incubated as a single-cell suspension in OM-5 media without phenol red for 30 min at 37°C to allow the re-expression of trypsinized surface markers. Microglia were then purified to > 98% purity by flow cytometry using phycoerythrin (PE)-conjugated antibodies directed against Fc receptor (FcR)/CD16/CD32 (Pharmingen, San Diego, CA, USA). Cytoplasmic mRNA was prepared from isolated cells immediately after isolation as described previously (Thomas et al. 2001).
Peritoneal macrophage preparation
Peritoneal macrophages were prepared as described previously with minor modifications (Kumar et al. 1996). Briefly, 3.0 mL aged sterile thioglycolate broth was injected into the peritoneum (Difco, Detroit, MI, USA). Mice were killed 3 days after injection and peritoneal macrophages were harvested by rinsing the peritoneal cavity with two 5-mL washes of OM-5 medium supplemented with 5.0 U/mL heparin (Sigma, St Louis, MO, USA). Exudate cells were plated and allowed to adhere for 2 h. Non-adherent cells were removed by rinsing the cultures. Cells were then left untreated or treated for 22 h with 100 ng/mL LPS and 10 U/mL IFNγ. More than 90% of the remaining adherent cells were macrophages (CD11b-positive cells).
TOGA® analysis
TOGA® analysis (Digital Gene Technologies, Inc., La Jolla, CA, USA) was performed with slight modifications as described previously (Sutcliffe et al. 2000) on mRNA samples isolated from untreated and LPS/IFNγ-treated microglia and macrophages. Briefly, RNA samples were converted to cDNA using a degenerate pool of biotinylated phasing primers that initiated synthesis at the beginning of the poly(A) tail on each mRNA. The cDNA collection was digested exhaustively with the restriction endonuclease MspI and the 3′ fragments were isolated by strepavidin-bead capture. A T3 promoter-containing adapter was ligated at the 5′ end to enable in vitro transcription. Following incubation with T3 polymerase, a collection of RNA fragments was produced corresponding to the 3′ portion of each starting mRNA, from its most 3′ MspI recognition sequence to the beginning of its poly(A) tail, with each fragment flanked by linker tags of known sequence. For the initial PCR step, first-strand cDNA was prepared from the RNA pool by reverse transcription and used in four separate PCRs, in which a 5′ primer that extends by one of four possible nucleotides beyond the MspI site (N1 position) was paired with a universal 3′ primer to generate an N1-specific double-stranded DNA template. In the final step, 256 primers corresponding to all possible permutations of the four nucleotides immediately adjacent to the MspI recognition site (N1N2N3N4) were matched with the appropriate N1 template and utilized in individual robotically performed PCRs paired with a fluorescent primer to produce 256 non-overlapping pools of products; these products were separated by electrophoresis. This process assigned each product an address: an eight-nucleotide sequence (the four MspI recognition and the adjacent four parsing nucleotides) and a length, both of which are attributes of the individual mRNAs. The amplitudes of the fluorescent PCR products correspond to the initial concentrations of their parent mRNAs, and these were collated automatically into a database, indexed by the addresses, which could be queried electronically to identify mRNAs whose concentrations differed among the experimental samples.
In situ hybridization
In situ hybridization was performed on free-floating cryosections as described previously, with minor modifications (de Lecea et al. 1995). Briefly, coronal sections (25 μm) were hybridized at 55°C for 16 h with a 35S-labeled riboprobe (107 cpm/mL). Excess probe was removed by washing at room temperature (23°C) for 30 min in 0.03 m NaCl, 0.003 m sodium citrate (2 × SSC) containing 10 mm β-mercaptoethanol, followed by a 1-h incubation with 4 μg/mL ribonuclease, 0.5 m NaCl, 0.5 m EDTA, 0.05 m Tris-HCl, pH 7.5, at 37°C. Sections were then washed under high-stringency conditions for 2 h at 55°C in 0.5 × SSC, 50% formamide and 10 mm β-mercaptoethanol, followed by a 1-h incubation at 68°C in 0.1 × SSC, 5 mm β-mercaptoethanol and 5% sarkosyl. Myeloid cells and blood vessels were identified by their ability to bind biotinylated tomato lectin (Sigma). Bound biotinylated tomato lectin was visualized by standard strepavidin–horseradish peroxidase methodology (Carson et al. 1999a). Sections were mounted on to FisherBrand SuperFrost/plus slides (Fischer Scientific, Pittsburgh, PA, USA) and dehydrated with ethanol and chloroform. Slides were exposed for 2–4 days to Kodak X-AR film and dipped in Ilford K-5 emulsion (Polysciences, Warrington, PA, USA). After 6–8 weeks, slides were developed with Kodak D19 developer (Fischer Scientific), fixed and counterstained with Mayer's hematoxylin. Riboprobes were prepared from sequences (antisense to detect TREM-2 mRNA expression and sense to serve as non-specific controls) corresponding to two regions of Trem-2 (GenInfo Identifier (gi) 12746556; nucleotides 71–762 and 746–1012) and to antisense sequence corresponding to interferon response gene 2 (IRG2) (gi1401063; nucleotides 1395–1706).
Quantification of TREM-2 expression on microglia
TREM-2 expression was quantified by counting the number of silver grains from the photographic emulsion exposed to radiolabeled TREM-2 riboprobe that were directly above cell nuclei. Cells were determined to express TREM-2 when the number of grains located above the nuclei/cell body was three-fold greater than background. Within the CNS, tomato lectin labels both microglia and blood vessels. Microglia were identified as nuclei surrounded by lectin-positive processes with ramified morphology. Although blood vessels were also lectin positive, they were easily excluded from analysis based on their large tubular morphology. TREM-2 expression was quantified in sections prepared from three unmanipulated adult mice. Each set of sections was hybridized with TREM-2 antisense riboprobes in separate in situ hybridization experiments. In each experiment, TREM-2 expression was examined in three microscopic fields from three different brain sections for each brain region.
Northern blot
For northern blots, 2 μg per lane poly(A)+ or 10 μg per lane total RNA was resolved by electrophoresis in a 1.5% agarose/1.2 m formaldehyde gel, transferred to nylon membrane and hybridized with 32P-radiolabeled probes as described previously (Thomas et al. 2001). Expression of each transcript was quantitated using NIH IMAGE 1.62 software (National Institutes of Mental Health, Bethesda, MD, USA).
Cloning of TREM-2b transcripts
TREM-2b PCR products were amplified and cloned from four murine templates: (i) Marathon-cDNA library generated from BALB/c whole-brain RNA (Clontech, Palo Alto, CA, USA); (ii) cDNA prepared from unstimulated C57BL/6J mixed glial culture RNA; (iii) cDNA prepared from C57BL/6J whole-brain mRNA and; (iv) cDNA prepared from C57BL/6J cultured microglial mRNA. The complete open reading frame of TREM-2b was amplified using the primers 5′-CTGCTGGCAAAGGAAAGGT-3′ and 5′-CCTGGCTGGACTTAAGCTGT-3′ for 25 cycles with an annealing melting temperature (Tm) of 59°C. PCR primers corresponding to the 3′ regions of exon 3 (5′-AAGTGGAACACAGCACCTCC-3′) and the 5′ region of exon 4 (5′-GGGTCCAGTGAGGATCTGAA-3¢) were used to amplify an internal region of TREM-2b using an annealing Tm of 59.5°C for 35 cycles. All PCR reactions used the TaKaRa premix containing Ex Taq proof-reading polymerase (TaKaRa Biomedicals, Otsu, Japan). PCR products were cloned into the pCR II TOPO vector using the TOPO TA cloning kit according the manufacturer's protocols (Invitrogen, Carlsbad, CA, USA).
Real-time quantitative RT-PCR
The relative expression levels of each form of TREM-2b (original and variant) were determined by real-time quantitative RT–PCR using the ABI PRISM 7700 Sequence Detection System (PE Biosystems, Wellesley, MA, USA). Briefly, each reaction contained 50 pg cDNA template and 5 μm forward and reverse primers. AmpliTaq Gold polymerase was used with SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Primers for real-time quantitative RT–PCR were selected by the integrated software package accompanying the ABI PRISM 7700, and the specificity of each primer pair was confirmed by gel analysis. The primer pairs were as follows:
TREM-2b variant forward 5′-AACACAGCACCTCCAGGCA-3′;
TREM-2b variant reverse 5′-TGGTAGGCTAGAGGTGACCCA-3′;
TREM-2b dominant forward 5′-CAGCACCTCCAGGAATCAAGA-3′;
TREM-2b dominant reverse 5′-GAGAAGAATGGAGGTGGGTGG-3′.
Each sample was amplified for 40 cycles. For each cDNA template, the cycle threshold (Ct) necessary to detect the amplified product was determined. The Ct refers to the cycle number at which the fluorescence of the amplified product reached an arbitrary threshold (set by the ABI PRISM 7700) that was within the exponential phase of amplification. These values were normalized to the Ct values of a standard ‘housekeeping’ control gene, glyceraldehyde-3-phosphate dehydrogenase (forward 5′-CCCTCACAATTTCCATCCCA-3′; reverse 5′-TCCCTAGGCCCCTCCTGTTA-3′).
Mice
For all studies, C57BL/6J mice were used. LPS (100 ng/mL) and/or IFNc (10 U/mL) was injected intracerebrally into the striatum in a total volume of 5 μL after mice had been anesthetized with metofane. All animal use procedures were in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals, and were approved by the Animal Care and Use Committee at The Scripps Research Institute.
Results
TREM-2 expression by cultured microglia and peritoneal macrophages is reduced by inflammatory signals
The physiological roles of microglia remain enigmatic. To gain insight into these roles and how they may differ in the healthy and inflamed CNS, we have comprehensively compared gene expression between unstimulated and LPS/IFNγ-stimulated cultured microglia using TOGA®, an automated systematic, PCR-based mRNA display method (Sutcliffe et al. 2000). Of the more than 15 000 mRNAs detected in our microglial samples, the orphan receptor TREM-2 was one of 81 mRNAs identified by TOGA® as being expressed at three-fold or higher levels in unactivated microglia (Fig. 1a) than in activated microglia treated for 22 h with LPS and IFNγ (Fig. 1b), in duplicate TOGA® experiments. A similar decrease was observed between untreated and LPS/IFNγ-treated peritoneal macrophages (Figs 1c and d). This recently described receptor is part of the Ig superfamily, and has been detected in macrophage cell lines and immature dendritic cells, but not in peripheral blood monocytes, natural killer cells, T cells or granulocytes (Bouchon et al. 2000; Bouchon et al. 2001; Daws et al. 2001).
Fig. 1.
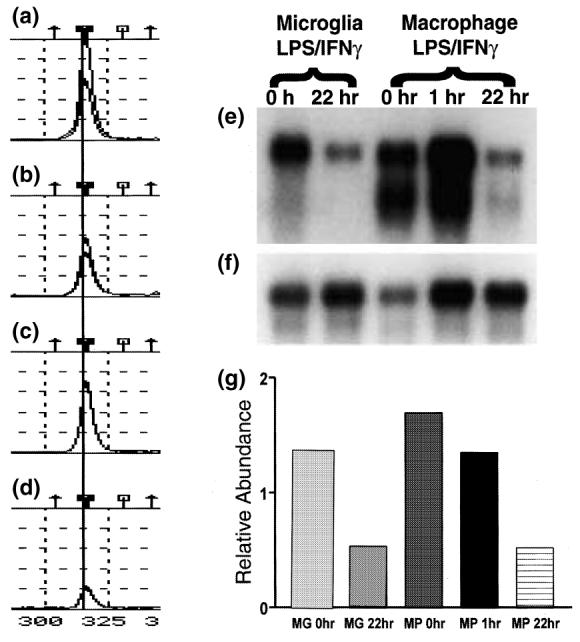
LPS/IFNγ decreases microglial (MG) and macrophage (MP) expression of TREM-2. (a–d) TOGA® profile for the product corresponding to TREM-2. The displayed TOGA® profile represents a small region of one of the 256 TOGA® electropherograms for each of the templates prepared from unstimulated cultured microglia (a), LPS/IFNγ-stimulated cultured microglia (b), unstimulated peritoneal macrophages (c) and LPS/IFNγ-stimulated peritoneal macrophages (d). The symbols on the top of (a–d) refer to the digital addresses (predicted positions) of known molecules. More details can be found in Sutcliffe et al. (2000). A line is drawn through the TOGA® PCR product corresponding to TREM-2. Peak amplitude corresponds to product abundance. (e) Northern blot with poly(A)+ RNA (2 μg/lane) prepared from unstimulated and LPS/IFNγ-stimulated cultured microglia and peritoneal macrophages was probed with a 32P-labeled TREM-2 cDNA clone. To determine the relative abundance of TREM-2 per sample, the same northern blot was re-probed with a RNA loading control, 32P-labeled cyclophilin cDNA clone (f), and the levels of TREM-2 relative to those of the loading control were quantified by densitometric analysis (g).
Northern blot analysis of mRNA generated from cultures treated in the same manner as those used for TOGA® confirmed the expression pattern seen with TOGA® (Figs 1e, f and g). Two TREM-2 transcripts, a major 1.7-kb species and a slightly smaller species, were detected. The proportions of the two transcripts differed between microglia and macrophages, with the smaller species being more readily detectable in macrophages than in microglia. The concentrations of the both TREM-2 transcripts were decreased in treated microglia and macrophages compared with untreated cultures (Figs 1e, f and g). We cannot exclude the possibility that astrocytes present in the mixed glial cultures produce factors that modify microglial expression of TREM-2. However, peritoneal macrophages cultured in the absence of other cell types also down-regulated TREM-2 expression in response to LPS/IFNγ treatment.
TREM-2 is expressed by microglia in vivo
Although previous reports had examined TREM-2 expression in a variety of purified leukocyte populations, its expression in healthy and inflamed tissues had not been described (Bouchon et al. 2001; Daws et al. 2001). Surprisingly, northern blot analysis of a panel of adult murine tissues revealed that TREM-2 was expressed at higher levels in the CNS, heart and lung than in lymph nodes or in other nonlymphoid tissue such as kidney, liver and testes (Figs 2a and c). The prolonged exposure times required to visualize the TREM-2 signal compared with the β-actin loading control (Fig. 2b) indicates that TREM-2 RNA is of low abundance, even within the CNS.
Fig. 2.
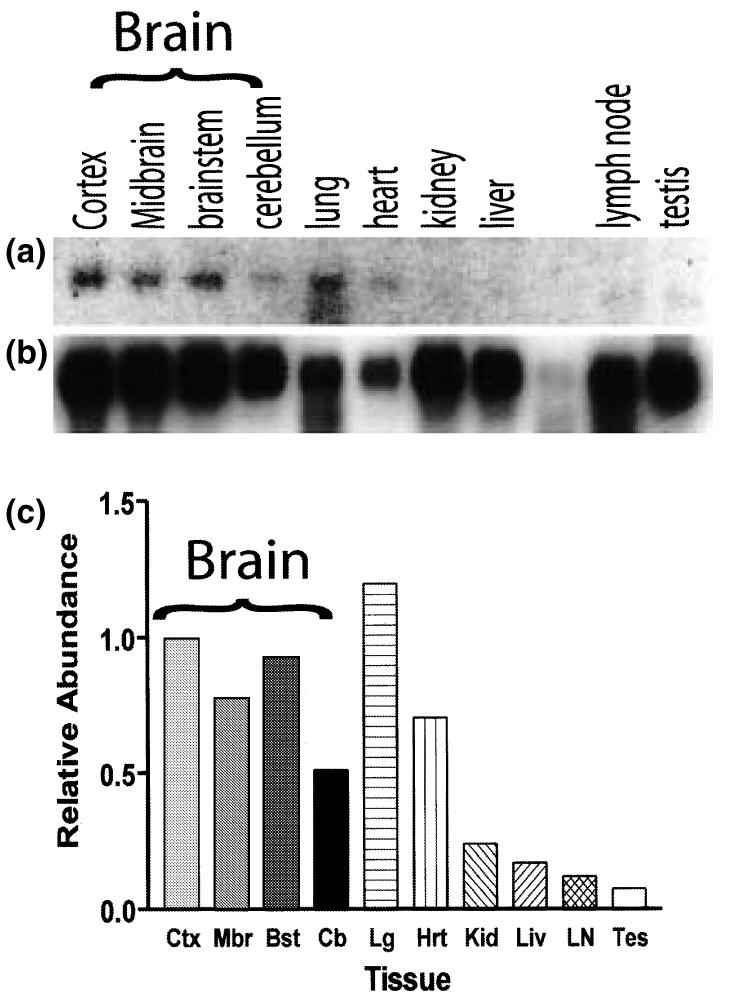
Northern blot analysis of TREM-2 expression in healthy adult mouse tissues. (a) Northern blot with poly(A)+ RNA (2 μg/lane) prepared from several adult mouse tissues was probed with a 32P-labeled TREM-2 cDNA clone. To determine the relative abundance of TREM-2 per sample, the same northern blot was re-probed with a RNA loading control, 32P-labeled β-actin cDNA clone (b), and the levels of TREM-2 relative to those of the loading control were quantified by densitometric analysis (c).
To determine the identity of the TREM-2-expressing cells within the CNS, we coupled in situ hybridization analysis with immunohistochemistry (Fig. 3). In these studies, two different 35S-labelled riboprobes, specific either for the 3′ untranslated region of TREM-2 or for the TREM-2 coding sequence, were used to detect TREM-2 expression. Tomato lectin was used to label microglia, macrophages and blood vessels. Blood vessels were easily distinguished from microglia and macrophages based on their tubular morphology. In brain sections prepared from healthy adult mice, each probe for TREM-2 labeled exclusively lectin-positive myeloid cells. Consistent with the northern blot analysis, prolonged exposure times (8 weeks) were required to visualize TREM-2 expression. Similar expression patterns were not observed even after exposure for 8 weeks using sense riboprobes (Fig. 4a) or riboprobes with similar GC content as TREM-2 riboprobes but that hybridized to mRNAs not expressed in the CNS (data not shown). Although lectin binding does not discriminate between microglia and macrophages, the lectin-positive cells expressing TREM-2 are likely to be microglia and not CNS-infiltrating macrophages owing to their location within the CNS parenchyma of healthy unmanipulated mice (Figs 3a and b) (Carson et al. 1998).
Fig. 3.
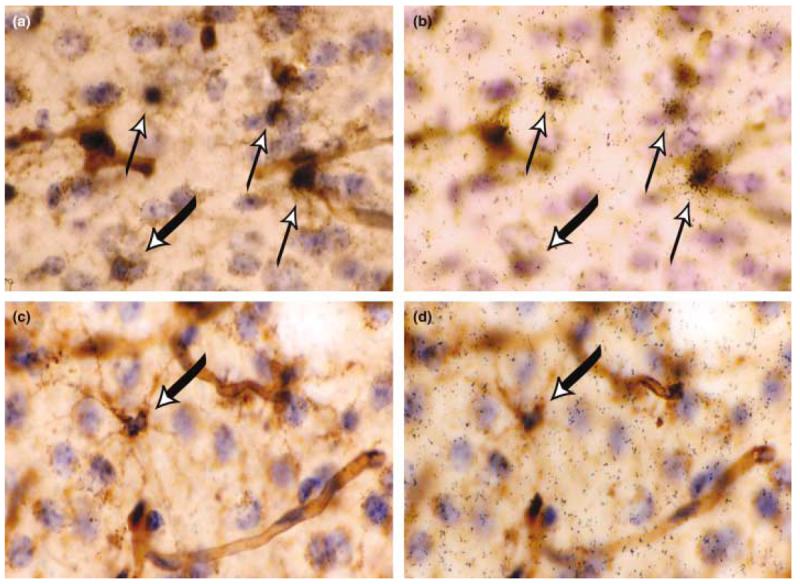
TREM-2 is expressed by lectin-positive cells in healthy adult murine brain. in situ hybridization analysis using a 35S-labeled antisense TREM-2 riboprobe (black grains) was performed on coronal sections from the CNS of untreated control mice (a and b) or from mice receiving intracerebral injections of LPS/IFNγ (c and d). In (a) and (c), the focal plane is at the level of lectin staining. In (b) and (d), the focal plane is at the level of the silver grains within the photographic emulsion. All nuclei are labeled with hematoxylin (in blue), and all myeloid cells and blood vessels are labeled with tomato lectin (in brown). Areas displayed are from the cingulate cortex. The thin upward pointing arrows in (a) and (b) indicate TREM-2-positive microglia and the thick downward pointing arrows in (a–d) indicate TREM-2-negative microglia. All cryosections were 25 μm in thickness. Magnification × 40.
Fig. 4.

Induction of IRG2 expression reveals widespread activation caused by intracerebral injection of LPS/IFNγ. (a) Coronal section taken from a control mouse hybridized with a 35S-labeled sense TREM-2 riboprobe and counterstained as detailed in Fig. 3. in situ hybridization analysis using a 35S-labeled antisense IRG2 riboprobe was performed on coronal sections taken from the same unmanipulated control (b) as depicted in Figs 3(a) and (b) and 4(a), and on coronal sections taken from the same LPS/IFNγ-injected mice (c–f) as depicted in Figs 3(c) and (d). The focal plane is at the level of silver grains in the photographic emulsion. The white arrow in Fig. 4 shows the brain region depicted in Figs 3(c) and (d). Magnification × 40.
While TREM-2 expression was limited to parenchymal microglia in the healthy CNS, not all microglia were TREM-2 positive (Figs 3a and b). Both the level of expression and the percentage of expressing cells were found to vary by brain region (Table 1). The highest percentage of TREM-2-expressing cells was found in the cingulate cortex and the lateral entorhinal cortex. The proportion of TREM-2-expressing cells was much lower in the hypothalamus and the habenula; some regions, such as the circumventricular organs, completely lacked TREM-2 expression. In all brain regions, TREM-2 expression was never detected on lectin-positive pericytes (cells completely aligned along the blood vessels in the perivascular space). However, TREM-2 expression could be detected on lectin-positive cells with processes that touched the blood vessels (Fig. 3a and b).
Table 1.
Percentage of TREM-2-expressing microglia in different brain region
| Brain region | Percentage ± SEM | |
|---|---|---|
| Lateral entorhinal cortex | 57 ± 5.8 | |
| Cingulate cortex | 45 ± 5.8 | |
| Caudate putamen | 28 ± 8.5 | |
| Medial hippocamupus | 26 ± 7.4 | |
| White matter brain regions | ||
| Fimbria | 20 ± 4.1 | |
| Corpus callosum | 28 ± 8.1 | |
| Regions with an Incomplete BBB | ||
| Anterior hypothalamic nucleus | < 10 ± N.D. | |
| Habenular nucleus | < 5 ± N.D. | |
| Circumventricular organs | ||
| Median eminence | < 1 ± N.D. | |
| Subfornical organ | < 1 ± N.D. | |
| Vascular organ | < 1 ± N.D. | |
| Medial accessory optic tract | < 1 ± N.D. | |
Inflammatory signals rapidly down-regulate in vivo expression of TREM-2
Intracerebral injection of LPS/IFNγ induces global microglial activation and macrophage infiltration within 24 h. To visualize the brain regions activated by LPS/IFNγ, the expression of IRG2 was monitored by in situ hybridization analysis. This molecule has previously been demonstrated to be induced in a wide variety of cells by LPS and IFNγ (Lee et al. 1994; Smith and Herschman 1996). IRG2 expression could not be detected in the healthy murine CNS (Fig. 4b). However, in response to intracerebral LPS/IFNγ injection, IRG2 expression was easily detected throughout the murine CNS by 24 h after injection (Figs 4c–f). As analyzed by flow cytometry, microglia isolated from the CNS of intracerebrally injected mice display an activated CD45intermediate phenotype (data not shown). Histologically, microglia become more intensely labeled by lectin (compare intensity of lectin labeling in Figs 3(a) and (c)), and their processes become more robust (Figs 3a and c). TREM-2 expression could no longer be detected in brain regions in which IRG2 expression had been induced (Figs 3c and d), consistent with the down-regulation of TREM-2 expression in vitro following LPS/IFNγ treatment (Fig. 1). Northern blot analysis of mRNA prepared from the brains of mice intracerebrally injected with LPS/IFNγ, LPS or IFNγ confirmed these results (Fig. 5). However, this experiment also showed that LPS injected alone was just as effective as simultaneous injection of LPS and IFNγ at down-regulating TREM-2 expression, and more effective than injection of IFNγ alone.
Fig. 5.
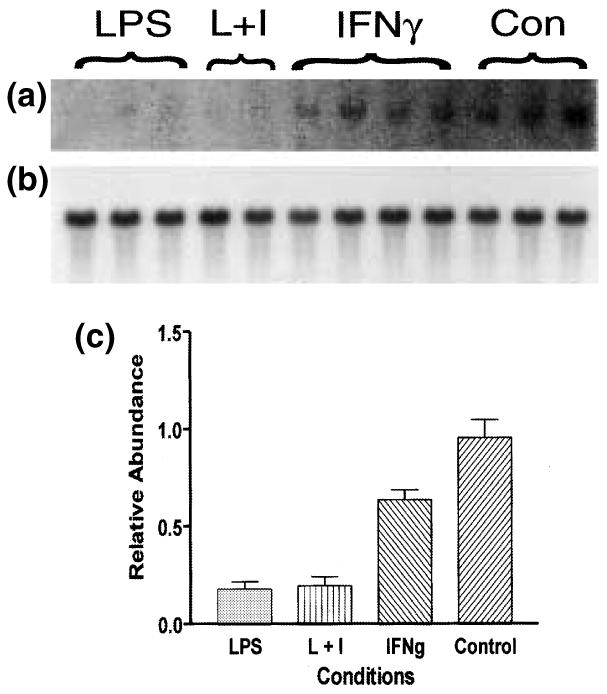
Northern blot analysis of TREM-2 expression after LPS and/or IFNγ treatment. Total RNA was prepared from three mice intracerebrally injected with LPS alone, two mice intracerebrally injected with LPS and IFNγ (L + I), four mice intracerebrally injected with IFNγ alone and three untreated control mice (Con). Total RNA (10 μg) from a single mouse brain was run in each lane. This northern blot was probed with a 32P-labeled TREM-2 cDNA clone (a). To determine the relative abundance of TREM-2 per sample, the same northern blot was re-probed with a RNA loading control, 32P-labeled β-actin cDNA clone (b), and the levels of TREM-2 with respect to those of the loading control were quantified by densitometric analysis (c).
Microglia express a novel TREM-2b transcript that does not encode a transmembrane domain
Two forms of TREM-2 (TREM-2a and TREM-2b) are expressed in the MT2 BALB/c macrophage cell line (Daws et al. 2001). Both TREM-2a and -2b transcripts encode nearly identical receptors characterized by a single V-type extracellular domain, a transmembrane domain region with a charged lysine residue, and a short cytoplasmic tail that lacks signaling motifs but that is capable of binding DAP12, a transmembrane adaptor signaling molecule. Although nucleotide differences between the two transcripts are found throughout the entire sequence, the two putative TREM-2 proteins differ by only three amino acids, all of which are located in the extracellular domain (Bouchon et al. 2000; Daws et al. 2001). As yet it is unknown whether these amino acid differences have functional consequences for ligand specificity or affinity.
To determine which form of TREM-2 was expressed by microglia, we isolated TREM-2 clones generated from cDNA templates prepared from C57BL/6J cultured microglia, C57BL/6J mixed glial cultures, C57BL/6J whole brain and BALB/c whole brain. Sequence analyses indicated that all clones encoded either of two forms of TREM-2b. In addition to the previously identified form of TREM-2b, an additional form was detected (Fig. 6). This variant differed from the original by a 55-bp insertion between exons 3 and 4. Analysis of the genomic sequence of TREM-2b suggests that the novel variant is generated by splicing exon 3 to an alternative splice site located 55 nucleotides upstream of exon 4. The frameshift caused by this insertion generates a putative TREM-2b protein that lacks a transmembrane domain and that is 22 amino acids longer than the original TREM-2b (Fig. 6).
Fig. 6.

An alternative TREM-2b splice product generates a molecule lacking a transmembrane domain and DAP12-binding domain. (a) Schematic diagram of the two forms of TREM-2b. Grey regions depict the IgV-type domain encoded by both the major (smaller membrane bound) and minor (larger splice variant) TREM-2b transcripts. The stippled region depicts the transmembrane domain encoded by the major TREM-2b transcript. The region with wavy lines depicts the frameshift in protein translation caused by the alternative splice site in the minor svTREM-2b. (b) Nucleotide sequence of the minor TREM-2b variant and the putative protein sequences of the two forms of TREM-2b from the end of exon 3 to the carboxy-terminus. The nucleotide sequence of the 55-bp insertion in the minor variant is highlighted in italic bold type. The amino acid sequence differences between the two forms of TREM-2b are displayed beneath the nucleotide sequence. The transmembrane domain present in the major transcript is shown in bold type in the upper conceptual protein translation.
The novel splice variant TREM-2b (svTREM-2b) is unlikely to be a genomic pseudogene contaminant. Both forms of TREM-2b could be PCR-amplified from cDNA templates prepared from microglia, macrophages and whole brain (Fig. 7a and data not shown). When the same primer pair was used with mouse genomic DNA as a template, neither of the TREM-2b splice products could be PCR-amplified (Fig. 7a). The only PCR product that could be detected was the ∼ 700-bp product predicted from the published murine genomic sequence.
Fig. 7.
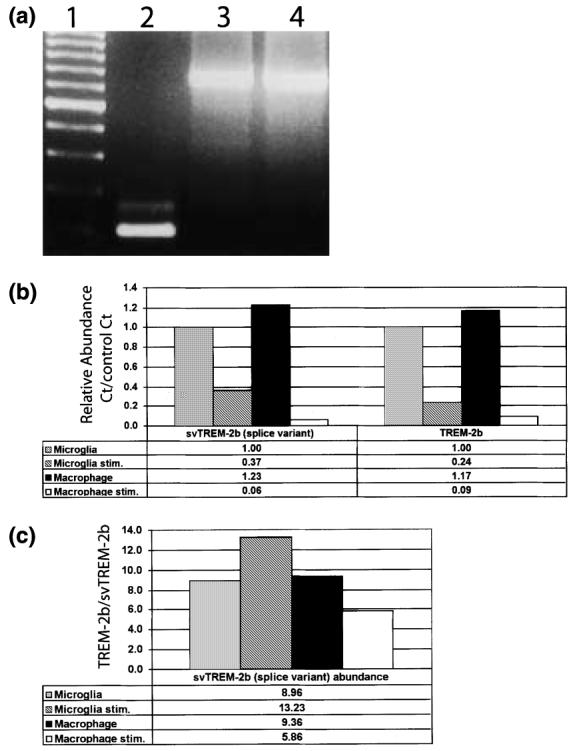
The minor svTREM-2b is not a pseudogene contaminant. (a) TREM-2b primers, corresponding to the 3′ terminal region of exon 3 and the 5′ region of exon 4, were used for RT-PCR analysis of cDNA template prepared from unstimulated microglia (lane 2) and genomic C57BL/6J DNA (lane 3, 100 ng; lane 4, 500 ng); 100-bp molecular weight ladder is shown in lane 1. (b and c) Relative expression levels of both the original membrane-bound major form (TREM-2b) and the non-membrane-bound splice variant form (svTREM-2b) were determined by real-time quantitative RT-PCR. In (b), the relative levels of svTREM-2b and TREM-2b in each sample are represented as the amplification cycle number at which the amplified product could be detected above the Ct compared with the same cycle Ct value observed in unstimulated microglia (control). (c) Relative ratio of svTREM-2b to TREM-2b in unstimulated microglia, LPS/IFNγ-stimulated microglia, unstimulated macrophages and LPS/IFNγ-stimulated macrophages.
The two forms of TREM-2b are not expressed at equivalent levels. As assayed by real-time quantitative RT–PCR, the ratio of TREM-2b to svTREM-2b was 9 : 1 in unstimulated microglia and macrophages (Figs 7b and c). Upon LPS/IFNγ stimulation, the level of expression of both forms of TREM-2b was dramatically down-regulated in microglia and macrophages (Fig. 7b). However, the ratio of TREM-2b to svTREM-2b was differentially regulated between stimulated microglia and macrophages. The ratio of TREM-2b to svTREM-2b was 14 : 1 in stimulated microglia but only 6 : 1 in stimulated macrophages.
Discussion
Microglia are found in all brain regions (Kreutzberg 1996; Becher et al. 2000; Aloisi 2001). Regional differences in cell morphology, responses to cytokines, and degree of constitutive and inducible MHC expression suggest that microglial function is likely to vary by brain region (Flaris et al. 1993; Pedersen et al. 1997; Phillips et al. 1999; Ren et al. 1999; McCluskey and Lampson 2001). Despite these observations, microglia have often been considered to be functionally similar throughout the brain. Here we have characterized TREM-2 expression in the CNS and found that microglial expression of TREM-2 is not only heterogeneous among brain regions but is also heterogeneous within single brain regions. In general, the highest percentages of TREM-2-expressing microglia were found in the cingulate cortex and lateral entorhinal cortex. By contrast, few if any microglia in the hypothalamus, circumventricular organs and the median eminence expressed TREM-2. It is important to note that even in areas with a relatively high proportion of TREM-2-expressing microglia, TREM-2-positive and TREM-2-negative microglia were found to be completely intermixed.
The significance of heterogeneous expression of TREM-2 by microglia within the healthy adult CNS is unknown. One possibility is that TREM-2-negative microglia are more activated than TREM-2-expressing microglia. Several observations provide partial support for this conclusion. In vitro, exposure to LPS/IFNγ dramatically reduced microglial expression of TREM-2. In vivo, intracerebral injection of LPS or LPS/IFNγ caused widespread microglial activation as judged morphologically, and by an increase in CD45 levels and a dramatic reduction in TREM-2 expression. Within the healthy brain, the lowest percentage of TREM-2-expressing microglia was in areas such as the hypothalamus and cirumventricular organs, which are considered to have a ‘leaky’ blood–brain barrier (BBB). Microglia in these regions display a more activated morphology, express higher levels of MHC than microglia found in areas with a complete BBB, such as the cortex, and are more responsive to cytokines (Flaris et al. 1993; Pedersen et al. 1997; Phillips et al. 1999; Ren et al. 1999; McCluskey and Lampson 2001). The apparent higher level of activation of microglia in these regions has been suggested to be due to their greater exposure to serum components.
Although TREM-2 expression is repressed by inflammatory signals, the presence of TREM-2 expression cannot be equated with quiescence, nor can the absence of TREM-2 expression be equated simply with a global state of activation. Microglia from mixed glial cultures express relatively high levels of TREM-2, yet these cells are considered semi-activated (Carson et al. 1998, 1999b; Aloisi 2001). Unlike unactivated microglia from adult CNS tissue, cultured microglia constitutively express increased levels of CD45 as well as detectable levels of MHC class I and II molecules. Similarly, unactivated monocytes do not express TREM-2, whereas peritoneal macrophages and immature dendritic cells, which develop from monocytes, do (Bouchon et al. 2001; Daws et al. 2001). Thus, TREM-2 expression may more readily correlate with a differentiation state or lineage decision than with activation state.
Recent studies on peripheral myeloid cells suggest that TREM-2 may be expressed by cells predisposed to become antigen presenters and that TREM-2-mediated cell activation may help promote this differentiation step (Bouchon et al. 2001). Although a ligand for TREM-2 has not been identified, the ligation of TREM-2 with cross-linking antibodies on the cell surface was shown to cause functional activation, as judged by the phosphorylation and activation of DAP12, the transmembrane adaptor signaling protein for TREM-2 (Bouchon et al. 2001; Daws et al. 2001).
Ligation of endogenous TREM-2 on the surface of immature myeloid dendritic cells induced a unique phenotype intermediate between an immature and fully mature dendritic cell (Bouchon et al. 2001). Immature dendritic cells express many macrophage markers (CD11b, F4/80) and are inefficient antigen-presenting cells, but are very efficient at antigen uptake and antigen processing. Conversely, mature dendritic cells are very efficient antigen-presenting cells but do not process antigen efficiently. TREM-2 ligation dramatically up-regulated molecules associated with antigen presentation to CD4-positive T cells (MHC class II, CD40, B7.2) without repressing the expression of antigen-capturing molecules (CD32, CD64, CD89). TREM-2 ligation also induced the expression of CCR7, a chemokine receptor for CCL19 (ELC) and CCL21 (SLC, 6ckine, TCA4) involved in the recruitment of antigen-presenting cells to lymph nodes and the sites of inflammation/autoimmunity (Dieu-Nosjean et al. 1999; Gunn et al. 1999; Cyster 2000; Ploix et al. 2001).
It is possible that TREM-2 activates a similar activation pathway in microglia. During experimental autoimmune encephalomyelitis (EAE), myeloid cells near and within demyelinating lesions have been reported to express DAP12 (Bakker et al. 2000). Microglia in these same regions express MHC class II, CD40 and B7.2, and have been shown to efficiently promote antigen-driven T-cell cytokine production (Bakker et al. 2000; Aloisi 2001; Juedes and Ruddle 2001). They are relatively inefficient at driving T-cell proliferation owing to their production of large quantities of nitric oxide. Interestingly, ligation of an epitope-tagged form of TREM-2 on the surface of TREM-2-transfected MT2 cells also stimulated nitric oxide release (Daws et al. 2001).
Taken together, these results may indicate that a greater percentage of microglia in cortical regions is more readily induced to become activated antigen-presenting cells than microglia in brain regions with a leaky or incomplete BBB. This relative deficit in TREM-2 expression might then be viewed as a safeguard to protect inadvertent antigen presentation. Conversely, the relatively enriched expression of TREM-2 in the entorhinal cortex and cingulate cortex may indicate the predisposition of these regions to microglial-associated pathologies or to microglial-specific protection. The recent discovery that the neurodegenerative disease Nasu-Hakola is caused by genetic deletions of either TREM-2 or DAP12 genes suggests that the TREM-2/DAP12 pathway has a necessary function within the cortex. Individuals lacking either a functional TREM-2 or DAP12 gene develop cognitive deficits in their late 20s and early 30s, coupled with the appearance of cortical plaques and death in their 40s (Paloneva et al. 2000; Paloneva et al. 2001; Paloneva et al. 2002). The absence of microglial activation resulting from the loss of this DAP12-mediated pathway has been suggested to play a key role in the onset and progression of this disease (Bakker et al. 2000).
An additional complication to the TREM-2 activation pathway is the production of a TREM-2 molecule (svTREM-2b), lacking a transmembrane domain, which could act as a soluble receptor. Regardless of the source of the TREM-2 ligand (neurons, glia or CNS-infiltrating immune cells), a soluble receptor could act as a buffer preventing microglial activation by low levels of the TREM-2 ligand. Interestingly, the ratio of svTREM-2b : TREM-2b is differentially regulated between LPS/IFNγ-stimulated microglia and macrophages. Although both forms of TREM-2b are reduced in both cell types upon LPS/IFNγ stimulation, svTREM-2b is much less abundant in treated microglia than in treated macrophages. It is possible that this differential production of svTREM-2b differentially facilitates activation of the TREM-2/DAP12 pathway in microglial-dominated compared with macrophage-dominated CNS pathologies. Such conclusions are still speculative, but the expression of TREM-2 by subsets of microglia provides a molecular basis for microglial heterogeneity within the healthy CNS. The potential of microglia to sense and to respond differentially to extracellular signals in different brain regions, and even within single brain regions, is likely to contribute to the differential sensitivities and particular responses of specific brain regions and neuronal populations to insult.
Acknowledgements
We thank Dr David Lo for critical discussion of the manuscript. This work was supported by National Institutes of Health grants NS39508 (to MJC) and GM32355 (to JGS), the Swiss National Science foundation (postdoctoral fellowship to CDS) and Digital Gene Technologies, Inc.
Abbreviations used
- BBB
blood-brain barrier
- Ct
cycle threshold
- IRG
interferon response gene
- IFN
interferon
- LPS
lipolysaccharide
- MHC
major histocompatibility complex
- SSC
saline sodium citrate buffer
- svTREM-2b
splice variant of triggering receptor expressed on myeloid cells-2b
- TREM
triggering receptor expressed on myeloid cells
References
- Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- Bakker AB, Hoek RM, Cerwenka A, Blom B, Lucian L, McNeil T, Murray R, Phillips LH, Sedgwick JD, Lanier LL. DAP12-deficient mice fail to develop autoimmunity due to impaired antigen priming. Immunity. 2000;13:345–353. doi: 10.1016/s1074-7613(00)00034-0. [DOI] [PubMed] [Google Scholar]
- Becher B, Prat A, Antel JP. Brain-immune connection: immuno-regulatory properties of CNS-resident cells. Glia. 2000;29:293–304. [PubMed] [Google Scholar]
- Betmouni S, Perry VH, Gordon JL. Evidence for an early inflammatory response in the central nervous system of mice with scrapie. Neuroscience. 1996;74:1–5. doi: 10.1016/0306-4522(96)00212-6. [DOI] [PubMed] [Google Scholar]
- Bouchon A, Dietrich J, Colonna M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J. Immunol. 2000;164:4991–4995. doi: 10.4049/jimmunol.164.10.4991. [DOI] [PubMed] [Google Scholar]
- Bouchon A, Hernandez-Munain C, Cella M, Colonna M. A dap12-mediated pathway regulates expression of cc chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 2001;194:1111–1122. doi: 10.1084/jem.194.8.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella M, Sciorati C, Tarozzo G, Beltramo M. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke. 2002;33:586–592. doi: 10.1161/hs0202.103399. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Disproportionate recruitment of CD8 (+) T cells into the central nervous system by professional antigen-presenting cells. Am. J. Pathol. 1999a;154:481–494. doi: 10.1016/S0002-9440(10)65294-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MJ, Sutcliffe JG, Campbell IL. Microglia stimulate naive T-cell differentiation without stimulating T-cell proliferation. J. Neurosci. Res. 1999b;55:127–134. doi: 10.1002/(SICI)1097-4547(19990101)55:1<127::AID-JNR14>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Cyster JG. Leukocyte migration: scent of the T zone. Curr. Biol. 2000;10:R30–R33. doi: 10.1016/s0960-9822(99)00253-5. [DOI] [PubMed] [Google Scholar]
- Daws MR, Lanier LL, Seaman WE, Ryan JC. Cloning and characterization of a novel mouse myeloid DAP12-associated receptor family. Eur. J. Immunol. 2001;31:783–791. doi: 10.1002/1521-4141(200103)31:3<783::aid-immu783>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Dieu-Nosjean MC, Vicari A, Lebecque S, Caux C. Regulation of dendritic cell trafficking: a process that involves the participation of selective chemokines. J. Leukoc Biol. 1999;66:252–262. doi: 10.1002/jlb.66.2.252. [DOI] [PubMed] [Google Scholar]
- Flaris NA, Densmore TL, Molleston MC, Hickey WF. Characterization of microglia and macrophages in the central nervous system of rats: definition of the differential expression of molecules using standard and novel monoclonal antibodies in normal CNS and in four models of parenchymal reaction. Glia. 1993;7:34–40. doi: 10.1002/glia.440070108. [DOI] [PubMed] [Google Scholar]
- Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J. Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- Ford AL, Foulcher E, Lemckert FA, Sedgwick JD. Microglia induce CD4 T lymphocyte final effector function and death. J. Exp. Med. 1996;184:1737–1745. doi: 10.1084/jem.184.5.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, Nakano H. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 1999;189:451–460. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, Blom B, Homola ME, Streit WJ, Brown MH, Barclay AN, Sedgwick JD. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290:1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- Juedes AE, Ruddle NU. Resident and infiltrating central nervous system APCs regulate the emergence and resolution of experimental autoimmune encephalomyelitis. J. Immunol. 2001;166:5168–5175. doi: 10.4049/jimmunol.166.8.5168. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M, Gallmeier E, Behrens L, Leal VV, Misgeld T, Klinkert WE, Kolbeck R, Hoppe E, Oropeza-Wekerle RL, Bartke I, Stadelmann C, Lassmann H, Wekerle H, Hohlfeld R. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J. Exp. Med. 1999;189:865–870. doi: 10.1084/jem.189.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Kumar R, Dong Z, Fidler IJ. Differential regulation of metalloelastase activity in murine peritoneal macrophages by granulocyte-macrophage colony-stimulating factor and macrophage colony-stimulating factor. J. Immunol. 1996;157:5104–5111. [PubMed] [Google Scholar]
- de Lecea L, del Rio JA, Soriano E. Developmental expression of parvalbumin mRNA in the cerebral cortex and hippocampus of the rat. Brain Res. Mol. Brain Res. 1995;32:1–13. doi: 10.1016/0169-328x(95)00056-x. [DOI] [PubMed] [Google Scholar]
- Lee CG, Demarquoy J, Jackson MJ, O'Brien WE. Molecular cloning and characterization of a murine LPS-inducible cDNA. J. Immunol. 1994;152:5758–5767. [PubMed] [Google Scholar]
- Lo D, Feng LL, Li L, Carson MJ, Crowley M, Pauza M, Nguyen A, Reilly CR. Integrating innate and adaptive immunity in the whole animal. Immunol. Rev. 1999;169:225–239. doi: 10.1111/j.1600-065x.1999.tb01318.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Fujiwara M. Absence of donor-type major histocompatibility complex class I antigen-bearing microglia in the rat central nervous system of radiation bone marrow chimeras. J. Neuroimmunol. 1987;17:71–82. doi: 10.1016/0165-5728(87)90032-4. [DOI] [PubMed] [Google Scholar]
- McCluskey LP, Lampson LA. Local immune regulation in the central nervous system by substance P vs. glutamate. J. Neuroimmunol. 2001;116:136–146. doi: 10.1016/s0165-5728(01)00295-8. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Janeway CA., Jr Innate immune recognition and control of adaptive immune responses. Semin Immunol. 1998;10:351–353. doi: 10.1006/smim.1998.0136. [DOI] [PubMed] [Google Scholar]
- Nguyen VT, Benveniste EN. Critical role of TNF-alpha and NF-kB in IFN-gamma-induced CD40 expression in microglia/macrophages. J. Biol. Chem. 2002;277:13796–13806. doi: 10.1074/jbc.M111906200. [DOI] [PubMed] [Google Scholar]
- Paloneva J, Kestila M, Wu J, Salminen A, Bohling T, Ruotsalainen V, Hakola P, Bakker AB, Phillips JH, Pekkarinen P, Lanier LL, Timonen T, Peltonen L. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 2000;25:357–361. doi: 10.1038/77153. [DOI] [PubMed] [Google Scholar]
- Paloneva J, Autti T, Raininko R, Partanen J, Salonen O, Puranen M, Hakola P, Haltia M. CNS manifestations of NasuHakola disease: a frontal dementia with bone cysts. Neurology. 2001;56:1552–1558. doi: 10.1212/wnl.56.11.1552. [DOI] [PubMed] [Google Scholar]
- Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, Bianchin M, Bird T, Miranda R, Salmaggi A, Tranebjaerg L, Konttinen Y, Peltonen L. Mutations in two genes encoding different subunits of a receptor signalling complex result in an identical disease phenotype. Am. J. Hum. Genet. 2002;71:656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen EB, McNulty JA, Castro AJ, Fox LM, Zimmer J, Finsen B. Enriched immune-environment of blood–brain barrier deficient areas of normal adult rats. J. Neuroimmunol. 1997;76:117–131. doi: 10.1016/s0165-5728(97)00038-6. [DOI] [PubMed] [Google Scholar]
- Phillips LM, Simon PJ, Lampson LA. Site-specific immune regulation in the brain: differential modulation of major histocompatibility complex (MHC) proteins in brainstem vs. hippocampus. J. Comp. Neurol. 1999;405:322–333. doi: 10.1002/(sici)1096-9861(19990315)405:3<322::aid-cne3>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Ploix C, Lo D, Carson MJ. A ligand for the chemokine receptor CCR7 can influence the homeostatic proliferation of CD4 T cells and progression of autoimmunity. J. Immunol. 2001;167:6724–6730. doi: 10.4049/jimmunol.167.12.6724. [DOI] [PubMed] [Google Scholar]
- Raivich G, Jones LL, Kloss CU, Werner A, Neumann H, Kreutzberg GW. Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J. Neurosci. 1998;18:5804–5816. doi: 10.1523/JNEUROSCI.18-15-05804.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren L, Lubrich B, Biber K, Gebicke-Haerter PJ. Differential expression of inflammatory mediators in rat microglia cultured from different brain regions. Brain Res. Mol. Brain Res. 1999;65:198–205. doi: 10.1016/s0169-328x(99)00016-9. [DOI] [PubMed] [Google Scholar]
- Schwab JM, Nguyen TD, Meyermann R, Schluesener HJ. Human focal cerebral infarctions induce differential lesional interleukin-16 (IL-16) expression confined to infiltrating granulocytes, CD8+ T-lymphocytes and activated microglia/macrophages. J. Neuroimmunol. 2001;114:232–241. doi: 10.1016/s0165-5728(00)00433-1. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Moalem G, Leibowitz Amit R, Cohen IR. Innate and adaptive immune responses can be beneficial for CNS repair. Trends Neurosci. 1999;22:295–299. doi: 10.1016/s0166-2236(99)01405-8. [DOI] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc. Natl Acad. Sci. USA. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpe CJ, Kohm AP, Huppenbauer CB, Sanders VM, Jones KJ. Exacerbation of facial motoneuron loss after facial nerve transection in severe combined immunodeficient (scid) mice. J. Neurosci. 1999;19:RC7. doi: 10.1523/JNEUROSCI.19-11-j0004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JB, Herschman HR. The glucocorticoid attenuated response genes GARG-16, GARG-39, and GARG-49/IRG2 encode inducible proteins containing multiple tetratricopeptide repeat domains. Arch. Biochem. Biophys. 1996;330:290–300. doi: 10.1006/abbi.1996.0256. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog. Neurobiol. 1999;58:233–247. doi: 10.1016/s0301-0082(98)00083-5. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglial response to brain injury: a brief synopsis. Toxicol. Pathol. 2000;28:28–30. doi: 10.1177/019262330002800104. [DOI] [PubMed] [Google Scholar]
- Sutcliffe JG, Foye PE, Erlander MG, Hilbush BS, Bodzin LJ, Durham JT, Hasel KW. TOGA: An automated parsing technology for analyzing expression of nearly all genes. Proc. Natl. Acad. Sci. USA. 2000;97:1976–1981. doi: 10.1073/pnas.040537997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Town T, Saxe M, Paris D, Wu Y, Mullan M. Ligation of microglial CD40 results in p44/42 mitogen-activated protein kinase-dependent TNF-alpha production that is opposed by TGF-beta 1 and IL-10. J. Immunol. 1999;163:6614–6621. [PubMed] [Google Scholar]
- Thomas EA, Danielson PE, Nelson PA, Pribyl TM, Hilbush BS, Hasel KW, Sutcliffe JG. Clozapine increases apolipoprotein D expression in rodent brain: towards a mechanism for neuroleptic pharmacotherapy. J. Neurochem. 2001;76:789–796. doi: 10.1046/j.1471-4159.2001.00027.x. [DOI] [PubMed] [Google Scholar]
- Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, Oda T, Tsuchiya K, Kosaka K. Occurrence of T cells in the brain of Alzheimer's disease and other neurological diseases. J. Neuroimmunol. 2002;124:83–92. doi: 10.1016/s0165-5728(01)00496-9. [DOI] [PubMed] [Google Scholar]
- Williams KC, Corey S, Westmoreland SV, Pauley D, Knight H, deBakker C, Alvarez X, Lackner AA. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J. Exp. Med. 2001;193:905–915. doi: 10.1084/jem.193.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]