

Abstract
Cytoplasmic sequestration of wild-type p53 protein occurs in a subset of primary human tumors including breast cancer, colon cancer, and neuroblastoma (NB). The sequestered p53 localizes to punctate cytoplasmic structures that represent large protein aggregates. One functional consequence of this blocked nuclear access is impairment of the p53-mediated G1 checkpoint after DNA damage. Here we show that cytoplasmic p53 from NB cells is incompetent for specific DNA binding, probably due to its sequestration. Importantly, the C-terminal domain of sequestered p53 is masked, as indicated by the failure of a C-terminally directed antibody to detect p53 in these structures. To determine (i) which domain of p53 is involved in the aggregation and (ii) whether this phenotype is potentially reversible, we generated stable NB sublines that coexpress the soluble C-terminal mouse p53 peptide DD1 (amino acids 302–390). A dramatic phenotypic reversion occurred in five of five lines. The presence of DD1 blocked the sequestration of wild-type p53 and relocated it to the nucleus, where it accumulated. The nuclear translocation is due to shuttling of wild-type p53 by heteroligomerization to DD1, as shown by coimmunoprecipitation. As expected, the nuclear heterocomplexes were functionally inactive, since DD1 is a dominant negative inhibitor of wild-type p53. In summary, we show that nuclear access of p53 can be restored in NB cells.
Wild-type (wt) p53 protein plays a key suppressor role in cell growth and tumor formation. p53 acts as a cell cycle checkpoint in response to DNA damage, triggering G1 arrest or apoptosis, and therefore preventing genomic instability (reviewed in ref. 1). p53 protein is a nuclear transcription factor whose target genes are involved in cell cycle control, including those encoding Waf1, mdm-2, GADD45, cyclin G, bax, and insulin-like growth factor binding protein 3 (1). The p53 core domain, bound to its cognate site, reveals a novel structure, so far found only in the DNA-binding domain of NF-κB p50 homodimer (2), and the two proteins now define a novel structural class of DNA-binding domains. p53 functions as a tetramer, consisting of a dimer of dimers. The C terminus (amino acids 300–393) harbors one major and two minor nuclear localization signals (NLSs), the tetramerization domain (amino acids 324–355) and a negative regulatory domain (amino acids 363–393) (see ref. 1). The C terminus alone is a strong dominant negative inhibitor of wt p53 by generating DNA-binding-incompetent heteroligomers (3).
Disruption of p53 function strongly correlates with tumorigenesis. p53 inactivation of various types is found in over 50% of all human cancers, with p53 gene mutations being the prevalent mechanism (4). Viral oncoproteins inactivate p53 protein by complex formation as seen with simian virus 40 large T antigen (5), adenovirus type 5 E1B protein (6, 7), human papilloma virus type 16/18 E6 protein (8), and hepatitis B virus X-protein (9, 10). In 30% of soft-tissue sarcomas, p53 protein is inactivated by binding to overexpressed mdm-2 protein, which abrogates p53-mediated transactivation (11, 12). Altered cellular localization of p53 protein has emerged as an alternate mechanism of inhibiting p53 function. In a subset of primary human tumors, cytoplasmic sequestration of wt p53 protein with concomitant nuclear exclusion is found. Originally described by us in inflammatory breast cancer (13) and neuroblastoma (NB) (14), it has also been reported in colon carcinoma (15, 16) and probably occurs in malignant melanoma (17, 18). In colon carcinoma, cytoplasmic accumulation of p53 correlates with unfavorable prognosis (15, 16, 19).
Intriguingly, in NB cells the sequestered p53 localizes to punctate cytoplasmic structures representing large protein aggregates (20). While the precise mechanism of cytoplasmic p53 retention remains to be elucidated, one functional consequence of this blocked nuclear access is marked impairment of the p53-mediated G1 arrest response after low to moderate levels of DNA damage (20). In this study we determine the role of the C terminus in p53 sequestration and show that sequestration is reversible.
MATERIALS AND METHODS
Cells and Reagents.
The human NB cell lines LAN-5 and SK-N-SH (SH) were cultured in Dulbecco’s modified Eagle’s medium/10% fetal calf serum. Five independent SH sublines were generated after stable transfection with the mouse C-terminal p53 peptide, residues 302–390 (designated DD1) (ref. 3; generous gift from M. Oren), or vector alone, using the calcium phosphate method and G418 selection. Human SaOs 2 osteosarcoma cells, H 358 lung adenocarcinoma cells (both homozygously deleted for p53), and ML-1 myeloid leukemia cells (wt p53) were grown as above. Monoclonal antibodies to p53 were PAb 421 (amino acids 372–382 epitope) PAb 122 (amino acids 372–378), HR 231 (amino acids 371–380; gift of Thierry Soussi, Institute Curie, Paris) (all human and mouse specific), and PAb 1801 (amino acids 46–55, human specific) (Oncogene Science). In addition, DO-1, DO-7, and PAb 248 (see ref. 20) were used in electrophoretic mobility-shift assays (EMSAs). Waf-1 mRNA was detected by a human Waf-1 cDNA (gift of Bert Vogelstein, Johns Hopkins Oncology Center, Baltimore), and Waf-1 protein was detected by a rabbit serum against recombinant human Waf-1 (gift of David Beach, Cold Spring Harbor Laboratory, NY).
Immunofluorescence.
Immunofluorescence staining was performed as we previously described (20). The nonaqueous acetone/methanol fixative avoids artifactual shift in subcellular p53 localization (21).
Immunoblotting and Immunoprecipitation.
Precipitation from unlabeled sonicated cell lysates was performed as we previously described (20). CM-1, a rabbit polyclonal antiserum against bacterially expressed human wt p53 (Vector Laboratories), was used in ECL (enhanced chemiluminescence) detection (Amersham).
Northern (RNA) Blot Analysis.
Analyses were performed as we previously described (20).
EMSAs.
Cells were Dounce homogenized in buffer (1.5 mM Hepes, pH 7.9/0.02 mM spermine/0.05 mM spermidine/0.2 mM K·EDTA/8 mM KCl/1% glycerol/1 mM DTT/1 mM phenylmethanesulfonyl fluoride/0.7 μg/ml aprotinin/1 mM sodium orthovanadate/20 mM NaF) and centrifuged at 14,000 rpm in an Eppendorf model 5415C for 15 min at 4°C. Supernatant and pellet constituted the cytoplasmic and nuclear extracts, respectively. As controls, we used ML-1 and LAN-5 cells treated with high levels of actinomycin D (5.4 and 3.8 nM; Sigma) for 24 hr to induce functional nuclear p53. Extracts were prepared as above. The double-stranded p53 consensus oligonucleotide (p53 CON) 5′-GGGCATGTCCGGGCATGTCC-3′ was used as wt probe (22). EMSA was performed with 4 ng of 32P-labeled probe, 5 μg of cytoplasmic or nuclear extract in 30–80 μl of binding buffer [40 mM NaCl/10 mM Mops, pH 7. 0/1% Nonidet P-40/1 mM EDTA/2.5% glycerol, 0.5 μg of poly(dI-dC)·poly(dI-dC), 4 ng of pGEM nonspecific vector DNA] for 30 min at 37°C. Samples were loaded onto a nondenaturing 4.5% polyacrylamide gel and electrophoresed at 4°C and 250 V for 3 hr. When indicated, p53-specific monoclonal antibodies (0.5 μg) were included to achieve increased DNA binding activity and a supershift (23). Specific competition experiments included unlabeled p53 consensus oligonucleotide in 50-fold molar excess of radiolabeled probe. Nonspecific competition consisted of 50-fold molar excess of either p53 CON mut (5′-GGGAATTTCCGGGAATTTCC-3′) (mutated nucleotides are underlined), B1 (5′-GTTGGCTCTGACTGTACCACCATC-3′), or a 500-bp HincII/PvuI restriction fragment from pSP72 plasmid (Promega). Ni-nitrilotriacetate affinity column-purified (over 90% pure by Coomassie blue-stained gel), His-tagged human wt p53 (58 ng) from a baculovirus/insect cell translation system served as another positive control (24). Products of some unlabeled EMSA reactions, scaled up 10-fold, were denatured, transferred to a nylon membrane, and immunoblotted with DO-1 (25).
RESULTS
Sequestered wt p53 Fails to Bind to Its Specific DNA Target.
We investigated the ability of cytoplasmically sequestered p53 to sequence-specifically bind DNA. Cytoplasmic fractions of LAN-5 and SK-N-SH cells were subjected to EMSAs using the p53 CON response element. Under conditions where baculoviral p53 exhibited strong specific binding, cytoplasmic p53 failed to interact with the response element oligomer. No specific DNA·p53 complexes were seen, even when binding was stimulated by the addition of PAb 421 as previously described (23) (Fig. 1 A, lanes 2–6, and B, lanes 3–5) and 10-fold higher input of extracts (data not shown). Addition of N-terminal-specific monoclonal antibodies PAb 1801, PAb 248, DO-1, and DO-7 also failed to induce binding (data not shown). Only a nonspecific band was present, which was also seen in p53-null SaOs2 and H358 cells (Fig. 1A, lanes 10 and 11, and data not shown) and was readily displaced by both specific and nonspecific competitor DNA. A parallel immunoblot confirmed that cytoplasmic p53 had actually entered the gel and produced a distinct band above the location where the nonspecific band appeared on the corresponding EMSA gel (data not shown). In contrast, under the same conditions, purified human wt p53 yielded a specific band which was greatly enhanced and supershifted by PAb 421 (Fig. 1A, lanes 12–15; Fig. 1B, lanes 6–9 and 17–20) as had been reported previously (23). In contrast, nuclear extracts from ML-1 and LAN-5 cells that have active nuclear p53 due to extensive DNA damage after high levels of actinomycin D showed a specific DNA·p53 complex which is revealed by PAb 421 (Fig. 1B, lanes 11–15 and 21–24) and is identical to the one with recombinant DNA (lanes 6–9 and 17–20).
Figure 1.

Sequestered wt p53 fails to bind to its specific DNA target. (A) EMSA of 5 μg of cytoplasmic extract of LAN-5 cells, using the p53 CON responsive element without (lane 2) or with 50-fold excess of specific (lane 3) and various nonspecific (lanes 4–6) competitor DNA. Only a nonspecific band is seen. Lanes 2–6 contain PAb 421 for maximum stimulation of binding. In contrast, control baculoviral human wt p53 (lanes 12–15) shows a specific band which supershifts and is greatly enhanced in the presence of PAb 421. Nuclear extracts of SH DD1-10 cells (lanes 7–9) fail to show a specific shift, as do nuclear extracts from SaOs 2 p53-null cells (lanes 10 and 11). Lanes 1–10 and 12–15 are from the same gel. See text for further details. (B) EMSA of 5 μg of cytoplasmic extracts of SK-N-SH cells using p53 CON without (lanes 2 and 3) or with 50-fold excess of specific (lane 4) and mutant (lane 5) competitor DNA. As controls, 5 μg of nuclear ML-1 (lanes 11–15) and LAN-5 extracts (lanes 21–24) stimulated with actinomycin D show a specific supershifted band, identical to the one with baculoviral p53. PAb 421 was added as indicated.
p53 C-Terminal Peptide Blocks the Sequestration of Endogenous wt p53 at Cytoplasmic Sites and Reroutes It to the Nucleus.
By immunofluorescence, the N-terminal and core domains of sequestered p53 are readily detected by PAb 1801 (see Fig. 2B–D) and by PAb 248 and PAb 240 (see figure 2 of ref. 20), respectively. In contrast, the C-terminal domain of the sequestered p53 is masked. This is indicated by the failure of PAb 421, PAb 122, and HR 231 to detect p53 in these structures in situ (Fig. 2; compare A with B and C and data not shown; IgG control was identical to the one shown in figure 2 of ref. 20), suggesting the involvement of the C-terminal domain in sequestration. Using this guidance, we chose a transfection approach to determine if the C-terminal domain is involved in cytoplasmic sequestration and whether this phenotype is potentially reversible. We generated five independent stable SK-N-SH sublines that coexpress the small soluble C-terminal mouse p53 peptide DD1 (amino acids 302–390) in excess of wt p53 (Fig. 3). DD1 encompasses all three nuclear localization signals and localizes exclusively to the nucleus when transfected into transformed rat embryo fibroblasts. Furthermore, DD1 is a strong dominant negative inhibitor of wt p53 by heterocomplex formation with wt p53 (3). Upon ectopic expression, a dramatic phenotypic reversion occurred in five of five sublines. Immunofluorescence with human-specific, N-terminally directed PAb 1801 showed that the presence of DD1 peptide blocked the cytoplasmic sequestration of endogenous p53 and rerouted it into the nucleus, where it produced a moderately strong signal (compare Fig. 2 B–D with F–H and J–L). Either none (SH DD1-10) or very few p53 structures (4 remaining lines) were still detectable by PAb 1801 in the cytoplasm. In contrast, immunofluorescence of DD1 sublines with the nondiscriminatory (with respect to endogenous vs. ectopic p53), C-terminally directed PAb 421 showed exclusive nuclear staining of high intensity due to DD1 and, most likely, an additive signal from re-located endogenous p53, now recognizable by PAb 421 (Fig. 2 E and I). Currently, no human-specific C-terminal antibody is available for confirmation. As control, SK-N-SH cells transfected with vector alone showed a parental phenotype with PAb 1801 (Fig. 2D).
Figure 2.
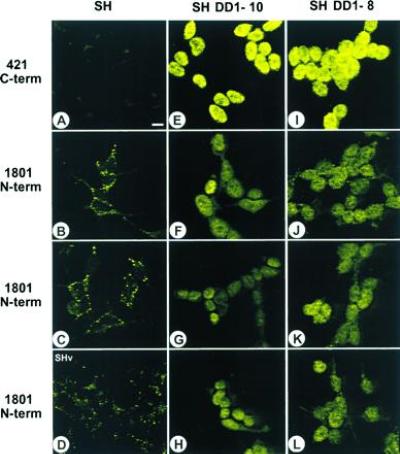
p53 C-terminal peptide (amino acids 302–390) blocks the sequestration of endogenous wt p53 at cytoplasmic sites and relocates p53 to the nucleus. The figure shows immunofluorescence of SH parental cells (A–C), SH cells transfected with vector alone (D), and two independent DD1-expressing subclones, SH DD1-10 (E–H) and SH DD1-8 (I–L). Fluorescein isothiocyanate (FITC)-based p53 localization with C-terminus-specific PAb 421 (A, E, and I) or N-terminus-specific PAb 1801 (B–D, F–H, and J–L) is shown. Note that PAb 421 fails to recognize sequestered p53 in the SH parental cells. Furthermore, PAb 1801 is specific for p53. (Scale bar = 10 μm.)
Figure 3.
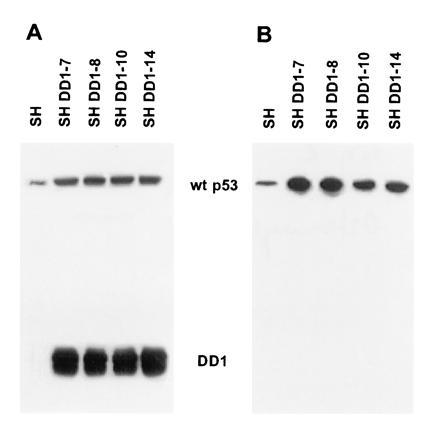
p53 C-terminal peptide (DD1) is overexpressed in SH subclones. An immunoblot of total cell lysates (50 μg of total protein per lane) was probed with PAb 421 (0.1 μg/ml) (A) or PAb 1801 (0.07 μg/ml) (B). PAb 1801 does not cross-react with DD1 peptide. Four of the five stable DD1 lines are shown here.
Wild-Type p53 Is Shuttled into the Nucleus by Means of Heterocomplex Formation with C-Terminal Peptide, a Dominant Negative Mutant.
To identify the mechanism responsible for nuclear relocation of wt p53, coimmunoprecipitation experiments were performed (Fig. 4A). When PAb 1801 was used, DD1 peptide coprecipitated with wt p53 in all sublines (i.e., lanes 5, 8, 9, and 11). In addition, unbound DD1 was apparent when cell lysates were precipitated with PAb 421 (compare increased band intensities of lanes 6, 7, 10, and 12 with lanes 5, 8, 9, and 11) and in sequential immunoprecipitations with PAb 1801 followed by PAb 421 (data not shown). Thus, heterocomplex formation between full-length protein and peptide occurs, providing a nuclear shuttle for p53. This finding is consistent with DD1 housing the tetramerization domain and all three NLS domains. Taken together, these data show that the cytoplasmic sequestration of p53 is reversible. Interestingly, the relocated wt p53 stabilized in the presence of DD1 (in Fig. 3B, compare lanes 1 with lanes 2–5, and in Fig 4A, compare lane 3 with lanes 5, 8, 9, and 11). As expected, however, nuclear heterocomplexes are functionally inactive, since DD1 also functions as a strong dominant negative inhibitor of wt p53. Heterocomplexes lacked specific DNA binding (Fig. 1A, lanes 7–9 and with PAb 421, data not shown) and failed to induce p53-response genes such as WAF-1 on the mRNA (Fig. 4B) and protein (data not shown) levels.
Figure 4.
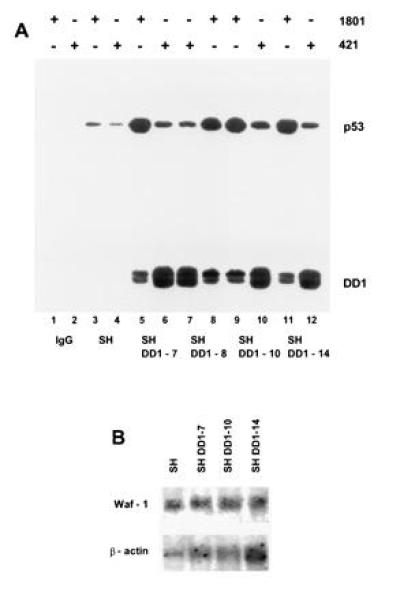
wt p53 is shuttled into the nucleus by means of heterocomplex formation with C-terminal peptide, a dominant negative mutant. (A) Coimmunoprecipitation of whole cell extracts (1.5 mg of total protein, lanes 3–12) of SH cells (lanes 3 and 4) and sublines DD1-7 (lanes 5 and 6), DD1-8 (lanes 7 and 8), DD1-10 (lanes 9 and 10), and DD1-14 (lanes 11 and 12) with either PAb 1801 (1.5 μg) (lanes 1, 3, 5, 8, 9, and 11) or PAb 421 (1.5 μg) (lanes 2, 4, 6, 7, 10, and 12) followed by immunoblotting with rabbit CM-1 antiserum. Control lanes 1 and 2 lack cell extracts. Note that PAb 421 regains partial epitope recognition after sonication of SH cell lysates (compare Fig. 2A with Fig. 3A and with Fig 4, lane 4. (B) Waf-1 mRNA analysis (10 μg per lane) comparing SH parental and several DD1 lines. Despite high levels of nuclear wt p53, no induction of p53 response genes is seen. The blot was stripped and reprobed with human β-actin cDNA as loading control.
DISCUSSION
Cytoplasmically sequestered wt p53 in human NB cells is localized to conspicuous punctate structures. These structures are not membrane enveloped and are able to pellet through a dense sucrose cushion (20). Here we show that when present in this state, this p53 species fails to bind to its specific DNA target, at least under conditions where control p53 produces a strong specific shift. These data further support a lack of function of cytoplasmic p53 (20). Cycloheximide treatment leads to a rapid disappearance of p53 aggregates within 3 hr (U.M.M., unpublished data), arguing that the p53 degradation pathway, putatively ubiquitin-mediated (26), is intact in these cells.
An important finding regarding the mechanism of sequestration is the fact that several C-terminally directed antibodies, PAb 421, PAb 122, and HR 231, fail to detect p53 in cytoplasmic structures, in contrast to antibodies directed against the N-terminal and central domains. This discordance is unexpected, since the prototypic C-terminal PAb 421 is pantropic and shows the broadest reactivity of all monoclonal p53 reagents. It reacts with all wt and mutant proteins, unless glycosylated (27), from a broad range of mammalian species (28). Similarly, several overexpression studies showed the ability of PAb 421 to detect p53 when expressed in the cytoplasm (29, 30). The 421 epitope maps to amino acids 372–382 within the negative regulatory domain and partially overlaps with NLS II (amino acids 369–375). The 421 data in conjunction with the cytoplasmic accumulation of p53 suggested that the C terminus, in particular the NLS region(s), is nonfunctional due to masking. The C terminus (amino acids 300–393) houses one major (amino acids 316–322) and two minor (amino acids 369–375 and 379–384) NLSs. Our finding that stable coexpression of the C terminus alone efficiently prevents sequestration of endogenous p53 at cytoplasmic sites and relocates it to the nucleus is completely consistent with this idea. The mouse C terminus DD1 corresponds to human residues 305–393. DD1 is small and soluble, forms heterocomplexes with human p53 (M. Oren, personal communication), and localizes exclusively to the nucleus of rat fibroblasts (3). Our data demonstrate that the C-terminal domain of p53 is involved in cytoplasmic aggregation of NB cells but that this phenotype is reversible. Heterocomplex formation with DD1 presumably provides a nuclear shuttle for p53 by providing multiple functional NLSs, thereby preventing it from being sequestered. Alternatively, nuclear relocation could be due to release of wt p53 from anchor sites by displacement by DD1, analogous to ligand swapping. We failed to detect cytoplasmically sequestered DD1 by immunofluorescence, and attempts to detect such DD1 species by sucrose ultracentrifugation of SH-DD1 cytoplasmic extracts were inconclusive. However, this possibility should be kept in mind, since the 421 epitope on DD1 could be equally masked when present in the sequestered form. An epitope-tagged fragment could test this possibility. Furthermore, there may be an additional role for a region other than the C terminus of p53 in cytoplasmic sequestration. Nevertheless, considering all current evidence, a reasonable model is that cotranslated peptide prevents C-terminal masking of p53 by driving it into a high-affinity interaction with the shuttle peptide, thus overriding those events that otherwise would lead to sequestration. NLS masking with concomitant cytoplasmic retention of transcription factors occurs physiologically to regulate nuclear access and is generally due to one of the following mechanisms: (i) intramolecular (posttranslational modification/phosphorylation or conformational); (ii) intermolecular (binding to another protein) (reviewed in ref. 31). Examples are yeast SW 15, simian virus 40 T antigen, Xenopus nuclear factor 7, and MyoD as well as the rel/NF-κB/dorsal family of transcription factors (31). Notably, however, their cytoplasmic distribution is always diffuse, in contrast to the punctate pattern seen with p53. In NB and other human malignancies, cytoplasmic p53 sequestration could be caused by an aberration of such a regulatory pathway. We showed earlier that high levels of DNA damage eventually induce levels of p53 protein that exceed the cytoplasmic retention capacity in NB cells (20). Importantly, endogenous p53 that eventually escapes retention in such a way is intrinsically functional, as shown by fluorescence-activated cell sorting analysis and induction of p53 response genes (20) as well as by specific DNA binding (Fig. 1B and ref. 32). We suggest that the interception of the cytoplasmic pathway of p53 before it engages in interactions that lead to sequestration offers hope of restoring wt p53 function in these tumors.
Acknowledgments
We thank Peter Tegtmeyer for stimulating discussions and critical reading of the manuscript. This work was supported by Public Health Service Grant CA60664 from the National Cancer Institute (to U.M.M.) and by American Cancer Society Grant JFRA-477 (to U.M.M.).
Footnotes
Abbreviations: NB, neuroblastoma; wt, wild type; NLS, nuclear localization signal; EMSA, electrophoretic mobility-shift assay.
References
- 1.Ko L J, Prives C. Genes Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 2.Muller C W, Rey F A, Sodeoka M, Verdine G L, Harrison S C. Nature (London) 1995;373:311–317. doi: 10.1038/373311a0. [DOI] [PubMed] [Google Scholar]
- 3.Shaulian E, Zauberman A, Ginsberg D, Oren M. Mol Cell Biol. 1992;12:5581–5592. doi: 10.1128/mcb.12.12.5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris C C, Hollstein M. N Engl J Med. 1993;329:1318–1327. doi: 10.1056/NEJM199310283291807. [DOI] [PubMed] [Google Scholar]
- 5.Mietz J A, Unger T, Huibregtse J M, Howley P M. EMBO J. 1992;11:5013–5020. doi: 10.1002/j.1460-2075.1992.tb05608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Debbas M, White E. Genes Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- 7.Yew P R, Berk A J. Nature (London) 1992;357:82–85. doi: 10.1038/357082a0. [DOI] [PubMed] [Google Scholar]
- 8.Scheffner M, Werness B A, Huibregtse M, Levine A J, Howley P M. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 9.Ueda H, Ullrich S J, Gangemi J D, Kappel C A, Ngo L, Feitelson M A, Jay G. Nat Genet. 1995;9:41–44. doi: 10.1038/ng0195-41. [DOI] [PubMed] [Google Scholar]
- 10.Wang X W, Forrester K, Yeh H, Feitelson M A, Gu J-R, Harris C C. Proc Natl Acad Sci USA. 1994;91:2230–2234. doi: 10.1073/pnas.91.6.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Momand J, Zambetti G P, Olson D C, George D, Levine A J. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 12.Oliner J D, Kinzler K W, Meltzer P S, George D L, Vogelstein B. Nature (London) 1992;358:80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 13.Moll U M, Riou G, Levine A J. Proc Natl Acad Sci USA. 1992;89:7262–7266. doi: 10.1073/pnas.89.15.7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moll U M, LaQuaglia M, Benard J, Riou G. Proc Natl Acad Sci USA. 1995;92:4407–4411. doi: 10.1073/pnas.92.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bosari S, Viale G, Bossi P, Maggioni M, Coggi G, Murray J J, Lee A K C. J Natl Cancer Inst. 1994;86:681–687. doi: 10.1093/jnci/86.9.681. [DOI] [PubMed] [Google Scholar]
- 16.Bosari S, Viale G, Roncalli M, Graziani D, Borsani G, Lee A K C, Coggi G. Am J Pathol. 1995;147:790–798. [PMC free article] [PubMed] [Google Scholar]
- 17.Weiss J, Heine M, Korner B, Pilch H, Jung E G. Br J Dermatol. 1995;133:23–31. doi: 10.1111/j.1365-2133.1995.tb02487.x. [DOI] [PubMed] [Google Scholar]
- 18.Castresana J S, Rubio M P, Vazquez J J, Idoate M, Sober A J, Seizinger B R, Barnhill R L. Int J Cancer. 1993;55:562–565. doi: 10.1002/ijc.2910550407. [DOI] [PubMed] [Google Scholar]
- 19.Sun X-F, Carstensen J M, Zhang H, Stal O, Wingren S, Haschek T, Nordenskjold B. Lancet. 1992;340:1369–1373. doi: 10.1016/0140-6736(92)92558-w. [DOI] [PubMed] [Google Scholar]
- 20.Moll U M, Ostermeyer A G, Haladay R, Winkfield B, Frazier M, Zambetti G. Mol Cell Biol. 1996;16:1126–1137. doi: 10.1128/mcb.16.3.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gusterson B A, Anbazhagan R, Warren W, Midgley C, Lane D P, O’Hare M, Stamps A, Carter R, Jayatilake H. Oncogene. 1991;6:1785–1789. [PubMed] [Google Scholar]
- 22.Hainaut P, Milner J. Cancer Res. 1993;53:4469–4473. [PubMed] [Google Scholar]
- 23.Hupp T R, Meek D W, Midgley C A, Lane D P. Cell. 1992;71:875–886. doi: 10.1016/0092-8674(92)90562-q. [DOI] [PubMed] [Google Scholar]
- 24.Reed M, Woelker B, Wang P, Wang Y, Anderson M E, Tegtmeyer P. Proc Natl Acad Sci USA. 1995;92:9455–9459. doi: 10.1073/pnas.92.21.9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Novak U, Paradiso L. BioTechniques. 1995;19:54–55. [PubMed] [Google Scholar]
- 26.Chowdary D R, Dermody J J, Jha K K, Ozer H L. Mol Cell Biol. 1994;14:1997–2003. doi: 10.1128/mcb.14.3.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw P, Freeman J, Bovey R, Iggo R. Oncogene. 1996;12:921–930. [PubMed] [Google Scholar]
- 28.Yewdell J W, Gannon J V, Lane D P. J Virol. 1986;59:444–452. doi: 10.1128/jvi.59.2.444-452.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez J, Georgoff I, Martinez J, Levine A J. Genes Dev. 1991;5:151–159. doi: 10.1101/gad.5.2.151. [DOI] [PubMed] [Google Scholar]
- 30.Gannon J V, Lane D P. Nature (London) 1991;349:802–806. doi: 10.1038/349802a0. [DOI] [PubMed] [Google Scholar]
- 31.Vandromme M, Gauthier-Rouviere C, Lamb N, Fernandez A. Trends Biochem Sci. 1996;21:59–64. [PubMed] [Google Scholar]
- 32.Goldman S C, Chen C-Y, Lansing T J, Gilmer T M, Kastan M B. Am J Pathol. 1996;148:1381–1385. [PMC free article] [PubMed] [Google Scholar]