

Abstract
Progressive multifocal leukoencephalopathy (PML) occurs most often in immunosuppressed individuals. The lesions of PML result from astrocyte and oligodendrocyte infection by the polyomavirus JC (JCV); JCV has also been shown to infect and destroy cerebellar granule cell neurons (GCNs) in 2 HIV-positive patients. To determine the prevalence and pattern of JCV infection in GCNs we immunostained formalin-fixed, paraffin-embedded cerebellar samples from 40 HIV-positive and 3 HIV-negative PML patients for JCV, glial and neuronal markers. JCV infection was detected in 30 patients (70%); 28 (93%) of these had JCV-infected cells in the granule cell layer (GCL); JCV-infected GCNs were demonstrated in 15/19 (79%) tested cases. JCV regulatory T antigen (T Ag) was expressed more frequently and abundantly in GCNs than JCV VP1 capsid protein. None of 37 HIV-negative controls but 1/35 (3%) HIV-positive subjects without PML had distinct foci of JCV-infected GCNs. Thus, JCV infection of GCNs is frequent in PML patients and may occur in the absence of cerebellar white matter demyelinating lesions. The predominance of T Ag over VP1 expression in GCNs suggests that they may be the site of early or latent central nervous system JCV infection. These results indicate that infection of GCNs is an important, previously overlooked aspect of JCV pathogenesis in immunosuppressed individuals.
Keywords: Cerebellum, Granule cell neurons, HIV, JC virus, Progressive multifocal leukoencephalopathy
INTRODUCTION
The polyomavirus JC (JCV) infects 86 % of healthy adults without causing any disease (1); it remains quiescent in the kidneys and in lymphoid organs (2, 3). In immunosuppressed individuals, JCV may reactivate and causes a productive, lytic infection of oligodendrocytes leading to a deadly demyelinating disease of the central nervous system (CNS), progressive multifocal leukoencephalopathy (PML) (4). Classical histological features of PML include multifocal areas of demyelination JCV-infected oligodendrocytes with enlarged amphophilic nuclei at the periphery of the lesions, reactive gliosis with enlarged, bizarre astrocytes, some of them sustaining JCV infection, and macrophages that phagocytose myelin and cellular debris (5). The posterior fossa is involved in up to 55% of patients with PML (6)
JCV is an encapsulated virus containing a single copy of circular double stranded genomic DNA. Early transcribed genes include two regulatory proteins, the small t and large T antigens (T Ag). T Ag is necessary for initiation of viral DNA replication; it binds to nuclear factors and JCV DNA to initiate the transcription of late genes including the VP1, VP2 and VP3 capsid protein and the agnoprotein. The viral capsid is constituted by 72 pentamers of VP1 protein, each associated with one VP2 or VP3 protein. Mature viral particles assemble in the nucleus and do not contain t or T Ag. Therefore, the detection of JCV VP1 protein by immunohistochemistry (IHC) in brain tissue indicates a full replicative cycle and the presence of mature viral particles, which also can be detected by electron microscopy (7). Detection of T Ag in the absence of VP1 suggests a restrictive, abortive infection.
Although the host cell range of JCV was thought to be limited to glial cells in the CNS, focal areas of cell loss in the cerebellar granule cell layer (GCL) were detected in up to 5% of PML patients before the AIDS epidemic (5); the reason for this has been unexplained. We reported an HIV-positive patient with PML lesions restricted to the cerebral hemispheric white matter (WM) who developed a productive and lytic JCV infection of cerebellar granule cell neurons (GCNs); this was associated with cerebellar atrophy and a clinically apparent cerebellar syndrome (7, 8). We then observed another HIV-positive patient who had cerebellar atrophy and isolated JCV infection of GCNs; we named this entity, distinct from PML, “JCV granule cell neuronopathy” (JCV GCN) (9). Other cases of JCV GCN have subsequently been reported by others (10, 11).
To determine the prevalence and the pattern of JCV infection of the GCL (including glial cells and GCNs) and the phenotype of infected cells, we studied JCV expression of T Ag and VP1 protein in cerebellar samples from a large number of PML patients and HIV-positive and -negative controls. JCV infection of GCNs was frequent in HIV-positive PML patients and the pattern of JCV protein expression in these cells differed from that in cerebellar WM.
MATERIALS AND METHODS
Origin of Brain Samples
Formalin-fixed, paraffin-embedded archival brain tissue samples from 43 patients (42 autopsies and 1 biopsy) with histologically confirmed PML were used in this study. The diagnosis of PML was established by the demonstration of JCV-infected glial cells in areas of demyelination. The diagnosis of all PML cases was confirmed in our laboratory. The materials were collected from the Departments of Pathology of the Beth Israel Deaconess Medical Center, Boston, Massachusetts, the Miriam Hospital in Providence, Rhode Island, the National NeuroAIDS Tissue Consortium, Rockville, Maryland (12) and from the UK Medical Research Council HIV Brain and Tissue Bank, Edinburgh, UK, between the years 1973 to 2007. Histological diagnoses of PML were established by a neuropathologist (JTJ, ES, JEB and members of the National NeuroAIDS Tissue Consortium). Of 43 PML patients (average age 41 years; female: male ratio 1:13), 3 were HIV-negative (2 autopsies and 1 biopsy); 2 of these had chronic lymphocytic leukemia and 1 had polymyositis as underlying diseases. Autopsy samples from 35 HIV-positive patients without PML (average age 43 years; female: male ratio 1:2.9) and 37 HIV-negative control subjects (average age 71 years; female: male ratio 1.1:1) were also investigated. The latter group included patients with stroke (n = 11), Alzheimer disease (n = 7), brain tumors (n = 7), hydrocephalus (n = 2), multiple system atrophy (n = 2) and one each with amyotrophic lateral sclerosis, chronic encephalitis, focal cerebellar sclerosis, paraneoplastic cerebellar degeneration, Parkinson disease, trauma, and trisomy 21. Samples from 2 previously reported HIV-positive patients, including 1 with PML (7) and 1 with JCV GCN (9), were used as reference for the GCN immunostaining experiments.
Immunohistochemistry and Immunofluorescence Staining Antibodies
To determine the presence of JCV-infected cells, we used the following antibodies against the simian virus 40 (SV40) that cross-react with JCV for IHC and immunofluorescence (IF) staining: mouse monoclonal anti-SV40 VP1 (PAB597 [13]), rabbit antisera against SV40 capsid proteins (Lee Biomolecular Research, San Diego, CA), rabbit polyclonal anti-SV40 VP1 (ab53977, Abcam, Cambridge, MA) and anti-VP2 and VP3 (ab53983, Abcam), mouse monoclonal anti-SV40 T Ag (PAb416; DP02-200UG, Oncogene Research Products, San Diego, CA) and rabbit polyclonal anti-SV40 T Ag (v-300; sc-20800, Santa Cruz Biotechnology, Santa Cruz, CA); The following antibodies were used to identify specific cell types: neurons: mouse monoclonal anti-MAP-2 (HM-2; M-4403; Sigma, St. Louis, MO), rabbit polyclonal anti-MAP-2 (AB5622; Chemicon, Temecula, CA) and mouse monoclonal anti-NeuN (A60; MAB377; Chemicon); astrocytes: mouse monoclonal and rabbit polyclonal anti-GFAP (6F2; M0761 and Z0334; Dako, Carpinteria, CA); oligodendrocytes and myelin: mouse monoclonal anti-CNPase (11-5B; C5922; Sigma), mouse monoclonal anti-MBP (SMI-94; SMI-94R, Covance Research Products, Denver, PA), mouse monoclonal anti-myelin/oligodendrocyte specific protein (MOSP) (CE-1; MAB328; Chemicon).
Single and Multiple IHC Staining Assays
Immunohistochemistry staining assays were performed as described in (14). For each patient, 1 to 3 cerebellar blocks were tested. The sections were either treated in an electric pressure cooker for 15 minutes in Trilogy solution (Cell Marque, Hot Springs, AR) for deparaffinization, rehydration and antigen retrieval as previously described (15) or deparaffinized in xylene and rehydrated through graded alcohols to PBS and then heated for 30 minutes at 95°C in Target Retrieval Solution, Citrate pH 6 (Dako). After incubation with primary antibodies, the Envision System/AP, Envision G/2 System/AP, Envision double stain, Envision G/2 double stain, Advance-horseradish peroxidase (HRP) kits (Dako) and PicTure Double Staining Kit (Invitrogen, Carlsbad, CA) were used according to the manufacturer's instructions. When appropriate, sections were counterstained with hematoxylin with or without with Luxol fast blue (LFB) stain for myelin before mounting. The chromogen substrates used for alkaline phosphatase were NBT/BCIP (Roche, Indianapolis, IN), Vector Blue (Vector, Burlingame, CA), Fast red or Permanent Red (Dako). For HRP, the chromogen were 3,3′-diaminobenzidine tetrahydrochloride (DAB; Dako), Vector Novared and Vector VIP (Vector).
Single and Double IF Staining Assays
Immunofluorescence staining assays were performed as described in (16). In double IF stainings, primary antibodies from different species (mouse monoclonals and rabbit polyclonals) were used with directly conjugated goat anti-mouse (IgG and/or IgM) and/or goat anti-rabbit (IgG) Alexa Fluor 350, 488 and 568 secondary antibodies (Invitrogen), according to the manufacturer's instructions.
Controls
Negative controls for IHC and IF included omission of the primary antibodies and use of sections from individuals without cerebellar abnormalities.
Qualitative Observations and Prospective Classification of the Cerebellar JCV Lesions
The selection of the cases with JCV-infected cells in the cerebellum was done on the basis of preliminary IHC experiments done on the 43 pre-selected PML patients. To be included in further analyses, a patient had to have at least 1 positive cell for either SV40 T Ag (sc-20800) or capsid proteins (PAB597 or SV40 antiserum) in 2 different IHC analyses. If after 10 tests no more than 1 positive cell was found, the patient was excluded from further analyses. For the included cases, the following experiments were done:
Characterization of the Demyelinated Areas
Double IHC for CNPase and JCV VP1 (PAB597) were performed with the Dako Envision double stain kit to estimate the extent and pattern of the demyelination in the WM and GCL of the whole section and correlate it to the location of JCV-infected cells.
Characterization of Cell Loss in the GCL
Double IHC for JCV VP1 (PAB597) and the neuronal markers NeuN or MAP-2 were performed with the Dako Envision double stain kit to determine the presence of focal GCN loss on whole sections and to correlate it with JCV-infection of the GCL. In cases with a partial or total loss of antigenicity for neuronal markers, focal loss of GCNs was estimated on hematoxylin and eosin sections.
Phenotype of the JCV-Infected Cells in the GCL
The presence of JCV-infected GCNs expressing T Ag was investigated with double IF staining assays for rabbit anti-T Ag and mouse anti-NeuN or mouse anti-MAP-2. The presence of JCV-infected GCNs expressing capsid proteins was investigated with double IF staining assays for mouse anti-NeuN and either rabbit antisera against SV40 capsid proteins, rabbit polyclonal anti-SV40 VP1 or rabbit polyclonal anti-VP2 and -VP3. Double IF assays stainings for rabbit anti-MAP-2 and mouse anti-VP1 (PAB597), mouse anti-MAP2 and either rabbit antisera, against SV40 capsid proteins, rabbit polyclonal anti-SV40 VP1 or rabbit polyclonal anti-VP2 and -VP3 were also done.
The presence of glial cells (astrocytes and oligodendrocytes) was also investigated in a series of preliminary experiments by double IHC and IF stainings for viral capsids or T Ag and oligodendrocytes (MOSP, CNPase) or astrocytes markers (GFAP).
Quantitative Image Analysis and Cell Counting
For all image analyses, light and fluorescence microscopy images were acquired with a Leica DFC300 FX Firewire camera mounted on a Leica DM5000 B microscope and connected to an Apple Mac Pro Dual Xeon 2.66GHz Dual Core Desktop. Since preliminary qualitative observations showed that JCV-infected glial cells and/or GCNs were significantly present in the GCL of some PML patients, quantification by image analysis was performed on a selection of sections that were double-stained for JCV-infected cells (VP1 or T Ag) and for neurons (NeuN or MAP-2) or double-stained for VP1 and T Ag (see below).
Quantification of VP1 and T Ag-Expressing Cells in the GCL
The numbers of JCV-infected GCNs and glial cells expressing VP1 and T Ag were determined in whole sections of 9 PML patients who had both proteins detectable in the GCL. We counted the JCV-infected cells expressing VP1 on selected sections subjected to double IHC for NeuN (DAB chromogen) and VP1 (PAB597, Vector Blue chromogen) using the Dako Envision double stain kit. Contiguous, non-compressed TIFF images encompassing the whole section or a 1 to 4 cm2 of a representative area were acquired, calibrated (with Image J, http://rsb.info.nih.gov/ij/) and merged (Photoshop CS), as previously described (14). The resulting picture was printed and used as a reference during counting performed at a magnification of 20x. During the cell counting, the area of the GCL used was reported on the printed picture and then on the file of the computer. Then, a mask (black and white picture) of the GCL was created with Image J and the area of the GCL was determined automatically. The area of the GCL analyzed was comprised between 20 and 150 mm2.
Similarly, we counted JCV-infected cells expressing T Ag on sections contiguous to those used for VP1. These sections were subjected to double IF using rabbit polyclonal anti-SV40 T Ag and either mouse anti-NeuN or mouse anti-MAP-2. To ensure that the T Ag+ cells were counted in the same areas as the VP1+ cells, the same reference pictures were used. The density of JCV-infected cells expressing the VP1 or T Ag per mm2 of GCL were calculated, as well as the ratio of T Ag+/VP1+ cells for each case.
Ratio of T Ag+ Cells/ VP1+ Cells in the GCL and Adjacent WM
To determine the ratio of T Ag and VP1 expression in the WM and GCL of the same section, the proportion of JCV-infected cells expressing VP1 or T Ag in the GCL and in the nearby WM was determined on double IFA staining (rabbit anti-T Ag and mouse anti-PAB597). This analysis was also done to determine whether there was an association between the local pattern of demyelination and the T Ag/VP1 ratio. We analyzed 4 to 8 representative pictures (20x magnification) of double IFA staining for rabbit anti-T Ag (Alexa 568, red) and mouse anti-VP1 (PAB597, Alexa 488, green) and myelin autofluorescence (blue) encompassing the GCL and WM. For each picture of the 5 patients analyzed, the area of the WM and GCL were delineated and all the cells expressing T Ag (red channel) and VP1 (green channel) of each area were counted. The local extent and pattern of demyelination were determined on the blue channel (autofluorescence) of the picture and classified as follow: 1 = no significant demyelination; 2 = medium demyelination; 3= high demyelination without cavitation; and 4 = cavitation.
Semiquantitative Analysis and Cell Counting
In light of the results of the quantitative analyses, a semiquantitative analysis was performed to estimate the relative frequency of T Ag (v-300) and VP1 (PAB597) expressing cells in all patients who had JCV-infected glial cells and/or GCNs in the GCL or in the contiguous WM. Since we sought to estimate the proportion of VP1 versus T Ag in all available patients, between 3 to 10 sections stained for capsid proteins and the neuronal markers NeuN and MAP-2 were analyzed. Similarly, we analyzed 3 to 10 sections per patient, which were stained for T Ag and the neuronal markers NeuN and MAP-2.
For each section investigated, the GCL and WM of the whole section were screened and the following semiquantitative scale was applied: 0 = no infected cell; r or rare = 1 to 10 infected cells; 1 = 11 infected cells −1% infected cells; 2 = 1% to 5% infected cells; 3 = 5% to 25% infected cells; 4 = 25% to 75% infected cells; 5 = 75% to 100% infected cells (17).
RESULTS
Frequency of JCV-Infected Cells in the Cerebellar WM and GCL
Single immunostaining for JCV T Ag or capsid proteins were performed on cerebellar samples to determine the frequency of JCV-infected cells. Of 43 PML patients studied, 27 (63%) had PML lesions in the cerebellum, consistent with a rate reported in other clinical series (6). In addition, 3 patients (7%) had JCV-infected glial cells and/or GCNs in the GCL only but had no PML lesions in the cerebellar WM. Therefore, a total of 30/43 (70%) PML patients had JCV-infected cells in the cerebellum. Of those, 28/30 (93%) had JCV-infected cells in the GCL (27 autopsies and 1 biopsy).
Patterns of Demyelination in Cerebellar WM and Correlation with and Focal GCN Loss in GCL
The double IHC, for JCV VP1 and the myelin marker CNPase or the neuronal marker NeuN, done to characterize the demyelinated areas and the cellular loss in the GCL, allowed us to classify the 27 autopsy patients with detectable JCV+ cells in the cerebellum in 4 distinct types:
Type I
Isolated JCV-infected cells in the GCL and no focal area of GCN loss in GCL (6 cases, Fig. 1A, B). Of these 6 cases, 3 had no PML lesions in the cerebellum and 3 had PML lesions in the deep cerebellar WM only.
Figure 1.
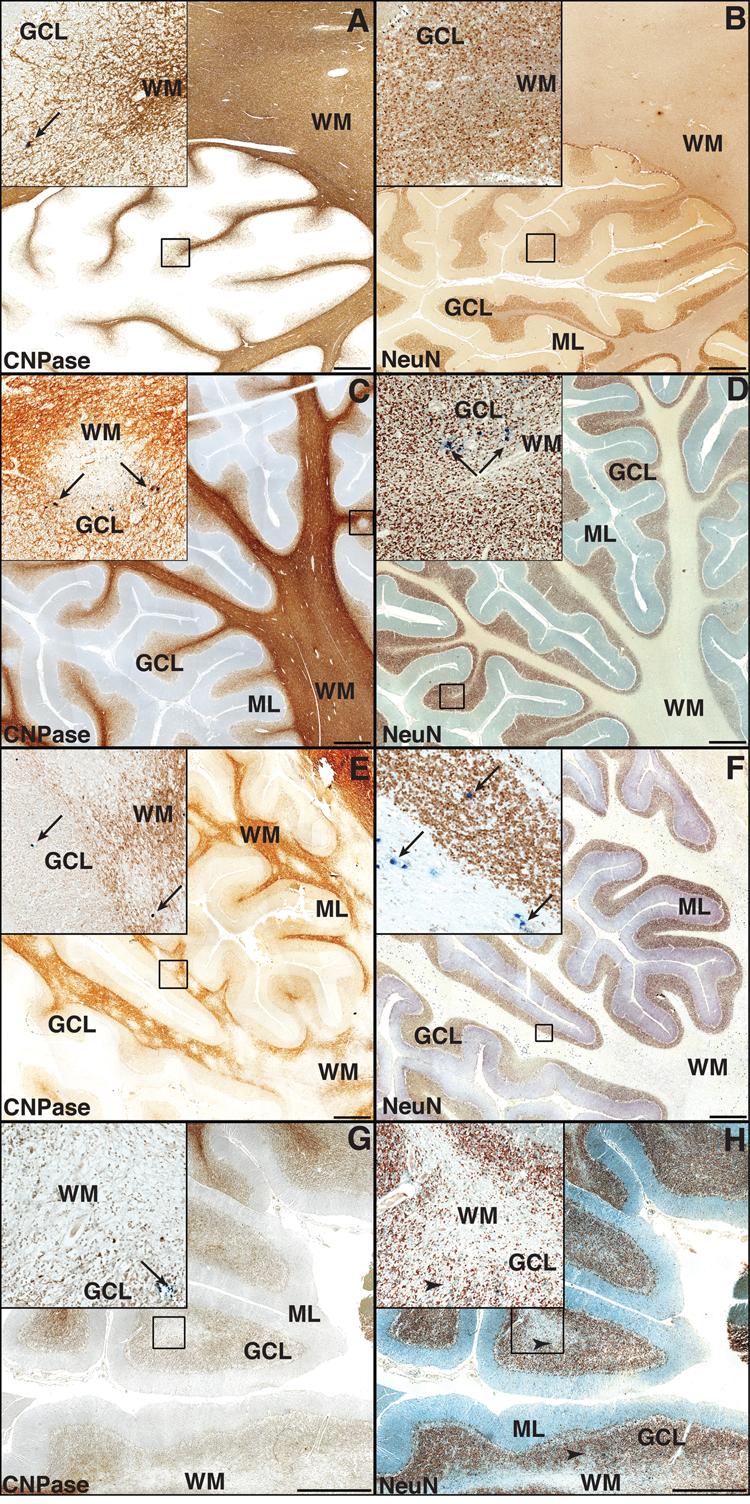
Types of demyelinating lesions in the cerebellar white matter (WM) and focal cell loss of granule cell neurons (GCNs) in the granule cells layer (GCL) of PML patients. (A, C, E, G) Double immunohistochemistry (IHC) for myelin and oligodendrocytes (CNPase, brown) and JCV VP1 capsid (PAB597, dark blue), displayed in 25,000,000 pixels pictures of 1cm2 representative portions of cerebellum, show areas of demyelination and JCV-infected cells (dark blue, marked by arrows in all insets). (B, D, F, H) Double IHC for neurons (NeuN brown, and JCV VP1 capsid (PAB597, dark blue). (A) Type I: an isolated JCV-infected cell is present in the GCL (arrow, inset) in the absence of a PML lesion in the cerebellar WM. This cell is not visible in the adjacent section (B), which shows an intact GCL. (C) Type II: a small demyelinating lesion at the GCL/WM junction contains JCV-infected cells at the periphery (arrows, inset). (D) JCV-infected cells are detected in a preserved GCL, in the same area. (E) Type III. Numerous confluent demyelinating lesions penetrating the GCL harbor JCV-infected (arrows, inset), whereas rare JCV-infected cells (arrows, inset) are present in the GCL, which has no to limited focal GCN loss (F). (G) Type IV. A cavitated PML lesion shows almost complete disappearance of myelin and oligodendrocytes in both the folia and the GCL. Almost all JCV-infected cells disappeared and only rare apoptotic JCV-infected cells or debris of JCV-infected cells (arrows, inset) are still present at the border of the demyelinated areas. These cells and debris are not visible in the inset of the adjacent section (H), which shows some areas of focal GCN loss (arrowheads, inset). (ML, molecular layer.)
Type II
Isolated lesions with JCV-infected cells mostly at the GCL-WM junction distant from PML lesions in the cerebellar WM and no focal area of GCN loss in GCL (5 cases, Fig. 1C, D)
Type III
JCV-infected cells and areas of demyelination in GCL confluent with PML lesions extending into the cerebellar WM and low to moderate areas of GCN loss in GCL (9 cases, Fig. 1E, F).
Type IV
JCV-infected cells in GCL adjacent to cavitated PML lesions in the cerebellar WM and moderate to high areas of focal GCN loss in GCL (7 cases, Fig. 1G, H).
Phenotype of JCV-Infected Cells in the GCL
To determine whether JCV-infected cells in the cerebellar GCL were neurons (GCNs), we performed double staining for JCV T Ag or capsid proteins and the neuronal markers NeuN and MAP-2 in samples that had preserved antigenicity for at least one of these neuronal marker and JCV-infected cells in the GCL. Of 28 samples from PML patients who had JCV-infected cells in the GCL, 19 (18 HIV-positive and 1 HIV-negative) could be tested by double IF staining assays for the presence of JCV-infected GCNs expressing T Ag and VP1. Interestingly, we found a variable amount of T Ag+ GCNs in the GCL of 15/19 (79%) of the tested patients (Fig. 2A, B), whereas we could detect very rare VP1+ GCNs in the GCL of 7/19 (37%) of the tested patients only (Fig. 2C, D) (Fisher exact test 2-tail p = 0.02). The one HIV-negative PML patient had both T Ag and VP1-expressing cells in the GCL and T Ag+ GCNs were found in HIV-positive PML patients, including individuals with and without highly active antiretroviral therapy (HAART). The 7 patients who had VP1-expressing GCNs always had T Ag-expressing cells as well, whereas 8 patients had GCNs expressing T Ag only. The 9 remaining samples could not be tested because of total loss of antigenicity of both neuronal markers NeuN and MAP-2.
Figure 2.
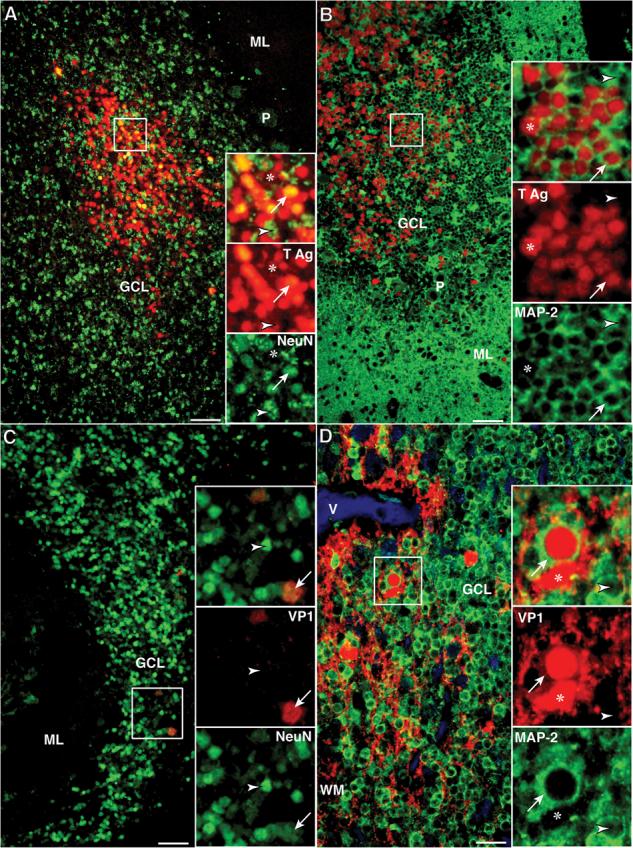
Presence of JCV T Ag and /or VP1 capsid protein in JCV-infected granule cell neurons (GCNs) of HIV+ PML patients (Bars: A–C = 50 μm; D = 25 μm). (A) Double IFA with rabbit polyclonal anti-SV40 T Ag (Alexa 568, red) and the neuronal marker mouse monoclonal anti-NeuN (Alexa 488, green) shows a cluster of hundreds of JCV-infected cells that express T Ag. Higher magnification views of cells in this cluster (square) shown in the upper inset, and viewed separately with the red (middle inset) and green channel (lower inset), show that most of the GCN, recognizable by their nuclear staining with NeuN, express T Ag in their nuclei (NeuN+/T Ag+ cells, arrows). In this cluster there are also a few JCV-infected glial cells, (NeuN−/T Ag+ cells, asterisk) and a few GCNs lacking T Ag (NeuN+/T Ag− cells, arrowhead). (B) The T Ag expressing GCNs can also be seen using double IFA with rabbit polyclonal anti-SV40 T Ag (Alexa 568, red) and a second neuronal marker mouse monoclonal anti-MAP-2 (Alexa 488, green). Because MAP-2 is mainly present in the cytoplasm, numerous MAP-2+/T Ag+ GCN are recognized by their red nuclei surrounded by a band of green cytoplasm (arrow, inset). Few MAP-2−/T Ag+ glial cells (red nuclei only, asterisk) and few non-infected GCNs (green cytoplasm only, arrowhead) are present. (C) Double IFA against JCV VP1 (SV40 antiserum, Alexa 568, red) and mouse monoclonal anti-NeuN (Alexa 488, green) reveals only isolated JCV-infected GCNs expressing VP1 capsid protein (NeuN+/VP1+, arrows), whereas the majority of GCNs do not express this protein (NeuN+/VP1−, arrowheads). (D) Similar results are seen with double IFA against VP1 (PAB597, Alexa 568, red) and rabbit polyclonal anti-MAP-2 (Alexa 488, green). Many MAP-2− glial cells displaying VP1 expression in their cytoplasm and nuclei (MAP-2−/VP1+, asterisk) are present in white mater (WM) with some extension in the granule cell layer (GCL). Rare GCNs (MAP-2+/VP1+, arrows) harboring nuclear expression of VP1 and cytoplasmic staining for MAP-2 can be found whereas most GCNs do not express VP1 (Map-2+/VP1−, arrowheads). (Asterisk, JCV-infected glial cell; Arrow, JCV-infected GCN; Arrowhead, GCN not expressing either T Ag or VP1. ML, molecular layer; P, Purkinje cells.)
Taken together, 70% (30/43) of the PML patients had JCV-infected cells in the cerebellum, 65% (28/43) in the GCL and 35% (15/43) had JCV-infected GCNs. We could only perform double staining for JCV proteins and neuronal markers on a subset of 19 samples with detectable JCV+ cells in GCL, showing JCV-positive GCNs in up to 79% (15/19). Rare JCV-infected astrocytes and oligodendrocytes that expressed T Ag and/or capsid proteins were regularly present in the GCL (not shown) and were sometimes present within the aggregates of hundreds of GCNs expressing T Ag. We found no aggregates of JCV-infected glial cells expressing T Ag in the GCL.
Relative Frequency of T Ag and VP1 Expression in the Cerebellum
Qualitative observations of sections stained by double IF assays with rabbit anti-T Ag and mouse anti-VP1 (PAB597) showed “normal active PML lesions” in the WM, some containing equivalent high numbers of cells expressing either the T Ag, the VP1 protein, or both, distributed throughout the lesion (Fig. 3A, C). Other, mostly cavitated PML lesions, showed lesser amounts of infected glial cells with dual expression of both T Ag and VP1 protein or preferential expression of VP1 at the edges of the lesions (Fig. 3B, D). The same sections showed, however, that sometimes a different type of lesion could be present in the GCL with a different pattern of JCV protein expression. Whereas few cases had limited numbers of either T, VP1, or dual-positive cells (Fig. 3A, C), most had a predominance of cells expressing T Ag only, often present in large numbers. For example, in the case shown in Figure 3 B and D, the number of T Ag-expressing cells was more than 1000 times higher than that of VP1-expressing cells.
Figure 3.
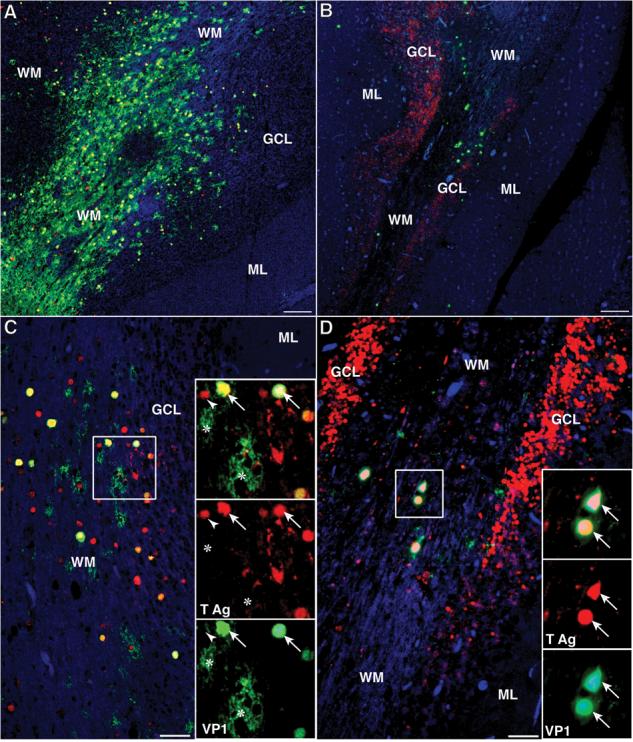
Comparison of JCV T Ag and VP1 protein expression in the cerebellum of 2 HIV-positive patients with PML. (A–D) Double IFA against the regulatory T Ag (rabbit polyclonal v-300. Alexa Fluor 568, red) and the VP1 capsid protein (mouse monoclonal PAB597, Alexa Fluor 488, green). (A, C) The partially demyelinated white matter (WM) of this patient contains numerous JCV-infected glial cells (A, bar = 250 μm). A higher magnification in (C) (bar = 50 μm) and in the inset show that many cells co-express both proteins, (T Ag+/VP1+, yellow, arrows), whereas separate channels show localization of T Ag in their nuclei (red, arrows) and VP1 in their nuclei and cytoplasm (green, arrows). In addition, some cells express VP1 only (T Ag−/VP1+, green, asterisks) whereas others express T Ag only (T Ag+/VP1−, red, arrowheads). The same pattern can be observed in the smaller number of JCV-infected cells in the granule cell layer (GCL). (B, D). The cavitating PML lesion in the WM of this patient contains rare JCV-infected glial cells (B, bar = 250 μm) that all express VP1 either without T Ag (T Ag−/VP1+, green) or, as seen in a higher magnification in (D) (bar = 50 μm) and in the inset, with co-expression of T Ag in their nuclei (T Ag+/VP1+, yellow, arrows). Dual protein expression is clearly demonstrated on the separate red and green channels. Unlike the other patient, the GCL on both sides of the WM is full of JCV-infected cells, which express only T Ag (T Ag+/VP1−, red). (Asterisk, T Ag−/VP1+ cell; Arrow, T Ag+/VP1+ cell; Arrowhead, T Ag+/VP1− cell; ML, molecular layer.)
We investigated this surprising pattern of distribution of both protein in the GCL, first with a quantitative analysis described in the section “Quantification of VP1 and T Ag-Expressing Cells in the GCL” above. The counting on sections stained for NeuN and PAB597 showed that the number of cells expressing VP1/mm2 were 0.08, 0.35, 2.38 and 0.87 in patients with type I to IV cerebellar demyelination, respectively. The counting, done on contiguous sections (double IF staining assays with either mouse NeuN or MAP-2 and rabbit T Ag) resulted in a much higher number of cells that expressed the T Ag/mm2, that is, 2.5, 13.4, 1467.0 and 110.0 in patients with type I-IV cerebellar demyelination, respectively. This analysis, in which cells expressing T Ag and/or VP1 were counted in the whole GCL area, confirmed the predominance of the T Ag-expressing cells in the GCL (including glial cells and GCN), which were 1 to 3 log higher than the VP1-expressing cells.
The predominance of T Ag-expressing cells in the GCL was confirmed by a second quantitative analysis (see “Ratio of T Ag+ cells/ VP1+ Cells in the GCL and Adjacent WM” above) in which T Ag and VP1 counts were done on the same section. The ratio of JCV-infected cells expressing T Ag/VP1 ranged from 0.3 to 430. Conversely, the counts done in the WM showed that the ratio of JCV-infected glial cells that expressed T Ag/VP1 ranged from 0.3 to 2.6, indicating that the numbers of T Ag- and VP1-expressing cells in the WM were in the same order of magnitude as had been observed in the qualitative observations (Fig. 3B, D).
We then performed a semiquantitative estimation of the relative numbers of T Ag and VP1 in the WM and GCL in 24 cases of PML (3 cases were excluded for technical reasons) and in the 15 cases with JCV-positive GCNs. As shown in Figure 4A, 79 % (19/24) of PML cases tested had T Ag > VP1 expression in GCL and 82% (9/11) in GCNs compared to only 41% (9/22) in the glial cells of the WM. These results suggest that the expression of VP1 was reduced in GCNs compared to glial cells.
Figure 4.

Comparison of the frequency of T Ag and VP1-expressing cells in the cerebellum. (A) Percentage of patients with number of cells expressing T Ag greater than VP1 (T Ag > VP1) or T Ag equal to VP1 (T Ag = VP1) in the white matter (WM) (glial cells), granule cell layer (GCL) (glial cells and granule cell neurons [GCN]), and GCN. (B) Percentage of patients with a ratio of T Ag/VP1 expression superior than 10 (T Ag/VP1 ≥ 10) or comprised between 1 and 10 (T Ag/VP1 = 1–10) for different types of demyelination in cerebellum.
To determine the magnitude of the difference between T Ag and VP1 expression in JCV-infected cells in the GCL, we plotted the percentage of cases having T Ag+/VP1+ ratio >10, or between 1 and 10 within the four types of demyelination. As shown in Figure 4B, the 5 analyzed patients with nascent and isolated JCV-infection of GCL (glial cells and/or GCNs) in Type I had a T Ag/VP1 ratio >10, while this was the case of 60% (3/5) of patients in Type II who had isolated lesions at the GCL/WM junction and 67% (4/6) patients in Type III who had JCV infection in GCL confluent to PML lesions in cerebellar white matter. Interestingly, all 6 analyzed patients with cavitated PML lesions in cerebellar WM (Type IV) had a T Ag/VP1 ratio >10 in adjacent GCL.
Discovery of an Overlooked Case of JCV GCN in an HIV-positive Control Subject
None of the 37 HIV-negative control subjects had JCV-positive cells in the cerebellum. However, 1/35 (3%) HIV-positive controls without clinical or histological evidence of PML anywhere in the CNS had JCV infection in the GCL. As shown in Figure 5, we detected 2 foci of JCV infection that were mainly restricted to the GCL surrounding preserved white matter tracts in this case. In these foci, numerous GCNs expressed exclusively JCV T Ag, whereas only few glial cells in WM or GCL expressed the capsid protein VP1 or VP2/VP3. These results indicated that this was a previously overlooked case of JCV GCN (9).
Figure 5.
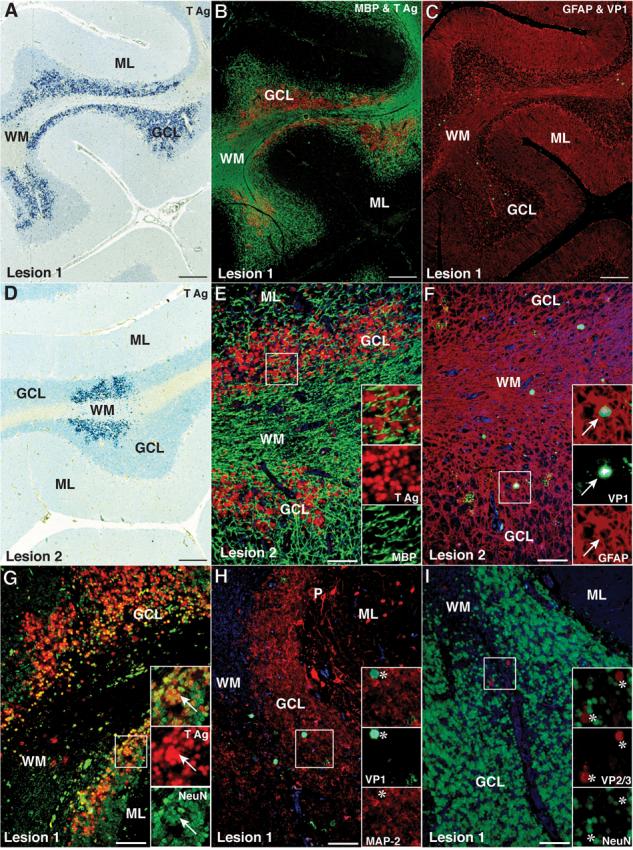
Numerous JCV-infected GCNs expressing T Ag are present in the cerebellum of an HIV-positive control patient without PML. (A, D) Immunohistochemistry (IHC) staining for T Ag (v-300, blue) with hematoxylin counterstaining showing 2 areas of the cerebellum containing numerous T Ag- expressing cells in the granule cell layer (GCL) and a few in the white matter (WM) (bar = 250 μm). (B, E) Double IFA staining for oligodendrocytes and myelin (MBP, SMI-94, Alexa Fluor 488, green) and T Ag (v-300, Alexa 568, red) reveals preservation of the myelin in the WM (B, bar = 250 μm). A higher magnification view (E, bar = 50 μm) reveals no T Ag+/MBP+ oligodendrocytes in the GCL (inset, and separate red and green channels). (C, F) Double IFA staining for astrocytes (rabbit polyclonal anti-GFAP, Z0334, Alexa Fluor 568, red) and VP1 capsid protein (PAB597, Alexa Fluor 488, green) shows that the rare cells expressing VP1 (C, bar = 250 μm) are GFAP+ astrocytes (F, bar = 50 μm) located in WM or at the WM-GCL junction (inset, arrow, and separate green and red channels). (G) Double IFA with the neuronal marker mouse monoclonal anti-NeuN (Alexa Fluor 488, green) and the T Ag (v-300, Alexa Fluor 568, red) shows that most of the numerous JCV-infected cells in the GCL are T Ag+/NeuN+ GCNs (inset, yellow, arrows) (bar = 50 μm). (H) Double IFA for the neuronal marker mouse monoclonal anti-MAP-2 (HM-2, Alexa Fluor 488, green) and the rabbit polyclonal anti-VP1 capsid protein (ab53977, Alexa Fluor 568, red) show only MAP-2−/VP1+ glial cells in the GCL (asterisk, insets) and no VP1-expressing GCNs (bar = 50 μm). (I) Double IFA with the neuronal marker mouse monoclonal anti-NeuN (A60, Alexa Fluor 488, green) and the rabbit polyclonal anti-VP2 and VP3 (VP2-3) capsid proteins (ab53983, Alexa Fluor 568, red) reveals only NeuN−/VP2-3+ glial cells in the GCL (asterisk, insets) and no GCNs expressing VP2 or VP3 (bar = 50 μm). (ML, molecular layer).
DISCUSSION
The present results considerably expand previous case reports of GCN infection by JCV in HIV−positive (7-10) or HIV-negative immunosuppressed individuals (11), and indicate that this infection is a major, and previously entirely unappreciated, aspect of JCV pathogenesis. Indeed, 65% of PML patients had JCV infection in the GCL, and among these, 79% of patients tested had JCV-infected GCNs. Our inability to perform double staining for JCV and neuronal markers in 9/28 cases that had JCV-positive cells in GCL, due to loss of antigenicity for neuronal markers, precluded an exact evaluation of the number of cases with JCV-positive GCNs in this study. This may be due to technical differences in the fixation and embedding procedures of these archival samples, some of which had been collected up to 19 years ago. Since a variety of physiological changes have been shown to influence NeuN expression (18), loss of NeuN antigenicity could also possibly be a direct consequence of JCV infection. Therefore, if we assume that the same proportion (79%) applies to all the 28 samples from the PML patients who had JCV-positive cells in the GCL, it is possible that up to 51% (22/43) of all PML patients in this series may indeed have JCV-infected GCNs. Since the majority of PML patients in our series were HIV-infected, our results are more representative of GCN infection in the context of AIDS, including patients with and without HAART. Moreover, we also demonstrated JCV infection in GCNs of an HIV-negative PML case.
Since the discovery of JCV in 1971 (19), several groups had observed either focal areas of cell loss and in GCL of PML patients (20-22), or unexplained cerebellar atrophy with degeneration of the GCL of HIV-positive patients without PML (23); some of these had JCV DNA detected by PCR in the cerebellar biopsy tissue. These studies did not, however, characterize the phenotype of infected cells. In retrospect, it is likely that many of these cases actually had JCV-infected GCNs. The high frequency of GCN infection detected in the present study has important clinical implications. Indeed, granule cell neurons are a major class of cerebellar interneurons, and their destruction may in and of itself cause cerebellar atrophy and cerebellar dysfunction, irrespective of the concomitant presence of PML lesions in cerebellar WM. In addition, GCN infection was also present in 7% of PML patients who had lesions in the hemispheric, but not cerebellar WM. Therefore, JCV should be considered as a potential cause of cerebellar dysfunction in PML patients even in the absence of PML lesions in the cerebellum.
Our study also reveals another novel aspect of JCV biology: unlike JCV infection of oligodendrocytes, which is always productive and lytic, the majority of infected GCNs expressed JCV T Ag more frequently than JCV VP1. JCV T Ag is a regulatory protein expressed early in the viral cycle and is not part of the mature virion, whereas the VP1 protein is expressed late in viral cycle and is the major component of the viral capsid, which is constituted of 72 VP1 pentamers (24). Therefore, it is possible that, in most cases, JCV causes a semi-permissive, abortive infection of GCNs. Indeed, in many areas where numerous JCV-infected GCNs mainly expressed T Ag, no focal cell loss could be seen. This was especially evident in patients with Type I and IV cerebellar demyelination, where infected GCNs were either distant from active PML lesions in WM or adjacent to cavitated PML lesions. These findings suggest that GCNs may be a target of early and possibly slowly developing infection by JCV before any demyelinating lesions appear in the WM, and remain a site of JCV latency in patients with burnt-out lesions of PML where all oligodendrocytes have been destroyed. The discovery of foci of GCN infection in an HIV-positive control subject who did not have any clinical or histological evidence of PML clearly underscores that GCNs may be the initial target of JCV infection in the CNS. Conversely, productive JCV infection is more likely to occur in GCNs in patients with lesions at the GCL/WM junction (Type II) or those with lesions of GCL extending into PML lesions in the cerebellar WM (Type III)
We previously demonstrated that the granule cell neuron-tropic JCVGCN1 had a 10 nt deletion in the C terminus of the VP1 gene that was not present in the strain JCVHWM isolated from demyelinating lesions in the hemispheric white matter of the same individual (25). JCV DNA containing this mutation was also present in cerebrospinal fluid and blood of other PML patients, together with the undeleted form of JCV. Whether patients with JCV-infected GCNs in our current study also carry this mutation in the VP1 gene deserves further studies that are now in progress in our laboratory.
Regardless of the mechanisms leading to entry and replication of JCV in GCNs, the fact that JCV may reside in these cells sheds a new light on a common inflammatory reaction occurring in HIV-positive PML patients treated with HAART. These patients present with contrast enhancement in PML lesions, often associated with swelling and mass effect as well as worsening of neurological deficits, and may require treatment with corticosteroids. This is a manifestation of the immune reconstitution inflammatory syndrome (IRIS) (26-28), which is concomitant with an increase in CD4+ T cell counts in peripheral blood, and appears to be immune-mediated. It is possible that JCV-infected GCNs may constitute one of the targets of a recovering immune system in PML IRIS.
Finally, this study indicates that GCNs may be the initial site of JCV infection in the CNS of HIV-positive patients. Such infection can cause cerebellar dysfunction, but may remain undiagnosed in the absence of demyelinating lesions seen on MRI or in histological samples. Therefore, to establish the diagnosis of JCVGCN, clinicians should request JCV PCR in the cerebrospinal fluid (11), and histological confirmation should include immunostaining for JCV in cerebellar tissue of immunosuppressed individuals presenting with an unexplained cerebellar syndrome or cerebellar atrophy.
ACKNOWLEDGMENTS
We are grateful to Dr. Susan Morgello, Benjamin B. Gelman, H. Aaron Aronow, Elyse Singer and Deborah Commins for providing PML samples through National NeuroAIDS Tissue Consortium (NNTC). The NNTC is supported by grants R24MH59724, R24NS38841, R24MH59745, and R24MH59656 from the NIH. Samples were also received from the UK Medical Research Council HIV brain and tissue bank in Edinburgh. We also thank Dr. W. Atwood for the PAB597 hybridoma.
This study was supported in part by NIH grant R21 NS 051124 to IJK.
REFERENCES
- 1.Weber F, Goldmann C, Kramer M, Kaup FJ, Pickhardt M, Young P, et al. Cellular and humoral immune response in progressive multifocal leukoencephalopathy. Ann Neurol. 2001;49:636–42. [PubMed] [Google Scholar]
- 2.Koralnik IJ, Boden D, Mai VX, Lord CI, Letvin NL. JC virus DNA load in patients with and without progressive multifocal leukoencephalopathy. Neurology. 1999;52:253–60. doi: 10.1212/wnl.52.2.253. [DOI] [PubMed] [Google Scholar]
- 3.Monaco MC, Jensen PN, Hou J, Durham LC, Major EO. Detection of JC virus DNA in human tonsil tissue: Evidence for site of initial viral infection. J Virol. 1998;72:9918–23. doi: 10.1128/jvi.72.12.9918-9923.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koralnik IJ. Progressive multifocal leukoencephalopathy revisited: Has the disease outgrown its name? Ann Neurol. 2006;60:162–73. doi: 10.1002/ana.20933. [DOI] [PubMed] [Google Scholar]
- 5.Richardson EP, Jr., Webster HD. Progressive multifocal leukoencephalopathy: Its pathological features. Prog Clin Biol Res. 1983;105:191–203. [PubMed] [Google Scholar]
- 6.Post MJ, Yiannoutsos C, Simpson D, Booss J, Clifford DB, Cohen B, et al. Progressive multifocal leukoencephalopathy in AIDS: Are there any MR findings useful to patient management and predictive of patient survival? AIDS Clinical Trials Group, 243 Team. AJNR Am J Neuroradiol. 1999;20:1896–1906. [PMC free article] [PubMed] [Google Scholar]
- 7.Du Pasquier RA, Corey S, Margolin DH, Williams K, Pfister LA, De Girolami U, et al. Productive infection of cerebellar granule cell neurons by JC virus in an HIV+ individual. Neurology. 2003;61:775–82. doi: 10.1212/01.wnl.0000081306.86961.33. [DOI] [PubMed] [Google Scholar]
- 8.Tyler KL. The uninvited guest: JC virus infection of neurons in PML. Neurology. 2003;61:734–35. doi: 10.1212/wnl.61.6.734. [DOI] [PubMed] [Google Scholar]
- 9.Koralnik IJ, Wuthrich C, Dang X, Rottnek M, Gurtman A, Simpson D, et al. JC virus granule cell neuronopathy: A novel clinical syndrome distinct from progressive multifocal leukoencephalopathy. Ann Neurol. 2005;57:576–80. doi: 10.1002/ana.20431. [DOI] [PubMed] [Google Scholar]
- 10.Otis CN, Moral LA. Images in pathology: Granule cell loss in AIDS-associated progressive multifocal leukoencephalopathy. Int J Surg Pathol. 2005;13:360. doi: 10.1177/106689690501300409. [DOI] [PubMed] [Google Scholar]
- 11.Hecht JH, Glenn OA, Wara DW, Wu YW. JC virus granule cell neuronopathy in a child with CD40 ligand deficiency. Pediatr Neurol. 2007;36:186–89. doi: 10.1016/j.pediatrneurol.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Morgello S, Gelman BB, Kozlowski PB, Vinters HV, Masliah E, Cornford M, et al. The National NeuroAIDS Tissue Consortium: A new paradigm in brain banking with an emphasis on infectious disease. Neuropathol Appl Neurobiol. 2001;27:326–35. doi: 10.1046/j.0305-1846.2001.00334.x. [DOI] [PubMed] [Google Scholar]
- 13.Ashok A, Atwood WJ. Contrasting roles of endosomal pH and the cytoskeleton in infection of human glial cells by JC virus and simian virus 40. J Virol. 2003;77:1347–56. doi: 10.1128/JVI.77.2.1347-1356.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wuthrich C, Kesari S, Kim WK, Williams K, Gelman R, Elmeric D, et al. Characterization of lymphocytic infiltrates in progressive multifocal leukoencephalopathy: Co-localization of CD8(+) T cells with JCV-infected glial cells. J Neurovirol. 2006;12:116–28. doi: 10.1080/13550280600716604. [DOI] [PubMed] [Google Scholar]
- 15.Kim WK, Corey S, Chesney G, Knight H, Klumpp S, Wuthrich C, et al. Identification of T lymphocytes in simian immunodeficiency virus encephalitis: Distribution of CD8+ T cells in association with central nervous system vessels and virus. J Neurovirol. 2004;10:315–25. doi: 10.1080/13550280490505382. [DOI] [PubMed] [Google Scholar]
- 16.Williams KC, Corey S, Westmoreland SV, Pauley D, Knight H, deBakker C, et al. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: Implications for the neuropathogenesis of AIDS. J Exp Med. 2001;193:905–15. doi: 10.1084/jem.193.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ordonez NG. The diagnostic utility of immunohistochemistry in distinguishing between epithelioid mesotheliomas and squamous carcinomas of the lung: A comparative study. Mod Pathol. 2006;19:417–28. doi: 10.1038/modpathol.3800544. [DOI] [PubMed] [Google Scholar]
- 18.Lind D, Franken S, Kappler J, Jankowski J, Schilling K. Characterization of the neuronal marker NeuN as a multiply phosphorylated antigen with discrete subcellular localization. J Neurosci Res. 2005;79:295–302. doi: 10.1002/jnr.20354. [DOI] [PubMed] [Google Scholar]
- 19.Padgett BL, Walker DL, ZuRhein GM, Eckroade RJ, Dessel BH. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet. 1971;1:1257–60. doi: 10.1016/s0140-6736(71)91777-6. [DOI] [PubMed] [Google Scholar]
- 20.Kuchelmeister K, Bergmann M, Gullotta F. Cellular changes in the cerebellar granular layer in AIDS-associated PML. Neuropathol Appl Neurobiol. 1993;19:398–401. doi: 10.1111/j.1365-2990.1993.tb00460.x. [DOI] [PubMed] [Google Scholar]
- 21.Weidenheim KM, Nelson SJ, Kure K, Harris C, Biempica L, Dickson DW. Unusual patterns of Histoplasma capsulatum meningitis and progressive multifocal leukoencephalopathy in a patient with the acquired immunodeficiency virus. Hum Pathol. 1992;23:581–86. doi: 10.1016/0046-8177(92)90137-r. [DOI] [PubMed] [Google Scholar]
- 22.Sweeney BJ, Manji H, Miller RF, Harrison MJ, Gray F, Scaravilli F. Cortical and subcortical JC virus infection: two unusual cases of AIDS associated progressive multifocal leukoencephalopathy. J Neurol Neurosurg Psychiatry. 1994;57:994–97. doi: 10.1136/jnnp.57.8.994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tagliati M, Simpson D, Morgello S, Clifford D, Schwartz RL, Berger JR. Cerebellar degeneration associated with human immunodeficiency virus infection. Neurology. 1998;50:244–51. doi: 10.1212/wnl.50.1.244. [DOI] [PubMed] [Google Scholar]
- 24.Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev. 1992;5:49–73. doi: 10.1128/cmr.5.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dang X, Koralnik IJ. A granule cell neuron-associated JC virus variant has a unique deletion in the VP1 gene. J Gen Virol. 2006;87:2533–37. doi: 10.1099/vir.0.81945-0. [DOI] [PubMed] [Google Scholar]
- 26.Du Pasquier RA, Koralnik IJ. Inflammatory reaction in progressive multifocal leukoencephalopathy: Harmful or beneficial? J Neurovirol. 2003;9(Suppl 1):25–31. doi: 10.1080/13550280390195315. [DOI] [PubMed] [Google Scholar]
- 27.Cinque P, Bossolasco S, Brambilla AM, Boschini A, Mussini C, Pierotti C, et al. The effect of highly active antiretroviral therapy-induced immune reconstitution on development and outcome of progressive multifocal leukoencephalopathy: Study of 43 cases with review of the literature. J Neurovirol. 2003;9(Suppl 1):73–80. doi: 10.1080/13550280390195351. [DOI] [PubMed] [Google Scholar]
- 28.Gray F, Bazille C, Adle-Biassette H, Mikol J, Moulignier A, Scaravilli F. Central nervous system immune reconstitution disease in acquired immunodeficiency syndrome patients receiving highly active antiretroviral treatment. J Neurovirol. 2005;11(Suppl 3):16–22. doi: 10.1080/13550280500511741. [DOI] [PubMed] [Google Scholar]