

Abstract
Objective
The chondrocyte response to IGF-1 is reduced with aging and in OA. IGF-1 signals through the PI-3 kinase/Akt pathway. TRB3, a tribbles homolog, has been shown to inhibit IGF-1 activation of Akt in HEK293 cells. In this study, we determined if TRB3 is expressed in chondrocytes and if TRB3 reduces the chondrocyte response to IGF-1.
Methods
Normal human articular cartilage obtained from tissue donors and OA tissue obtained from knee replacement surgery were used to measure TRB3 by RT-PCR, immunoblotting, and immunohistochemistry. Overexpression of TRB3 by transient transfection was used to determine the effects of TRB3 on cell survival and proteoglycan synthesis.
Results
TRB3 RNA was detected in normal human chondrocytes. TRB3 protein levels were low in cells from normal cartilage but significantly increased in OA cells. Incubation with two agents that induce endoplasmic reticulum stress, tunicamycin and thapsigargin, increased TRB3 levels in normal cells. Overexpression of TRB3 inhibited Akt phosphorylation and reduced chondrocyte survival and proteoglycan synthesis.
Conclusion
These results are the first to demonstrate that TRB3 is present in human chondrocytes and its level is increased in OA cartilage and isolated OA chondrocytes. Because it is an inhibitor of Akt activation, increased levels of TRB3 could play a role in the increased cell death as well as the reduced response to IGF-1 seen in OA cartilage.
Aging does not directly cause osteoarthritis (OA); however it is the most important risk factor in the development of the disease (1). A major contributing factor for the development of OA is a loss of anabolic and catabolic homeostasis by chondrocytes leading to a loss of articular cartilage. The balance of anabolic and catabolic processes in cartilage depends on the local activity of regulatory factors such as cytokines and growth factors (2). IGF-1 has the ability to stimulate matrix synthesis (3-5), promote chondrocyte survival (6), and inhibit specific catabolic pathways (7,8). There is evidence that chondrocytes have a decreased response to IGF-1 with age and also in OA (3,9-12), but the mechanisms involved are incompletely understood.
IGF-1 stimulates both the PI-3 kinase/Akt pathway as well as the Ras-Raf-MEK-ERK pathway by acting through the IGF-1 receptor (13). The activation of IGF-1R results in the activation of Shc and members of the insulin receptor substrate (IRS) family. After the phosphorylation of IRS and Shc, both the PI-3 kinase cascade and ERK cascade are activated. The activation of PI-3 kinase leads to the activation of Akt (also called protein kinase B or PKB), a serine-threonine kinase involved in cell-survival (14,15), and p70S6 kinase, a serine-threonine kinase implicated in protein synthesis (16).
IGF-1 activation of PI-3 kinase but not ERK is required for IGF-1 stimulation of chondrogenesis and proteoglycan synthesis by mesenchymal cells (17) and for proteoglycan synthesis by adult human chondrocytes (18). Although IGF-1 acts as an autocrine survival factor for chondrocytes cultured at low density (6,16), the requirement for PI-3 kinase and/or ERK activity for IGF-1 mediated cell survival is not clear and may depend on the experimental conditions. Inhibition of the ERK pathway has been reported to reduce survival of chondrocytes plated on collagen (19) but not fibronectin (20). Inhibition of PI-3 kinase reduced survival of chondrocytes treated with sodium nitroprusside (17) but did not reduce survival of chondrocytes in high density monolayer or alginate cultures (18).
It has been shown that the tribbles homolog TRB3 (also known as SKIP3, NIPK and SINK), inhibits IGF-1 as well as insulin activation of Akt/PKB in the liver (21). TRB3 has been classified as a pseudokinase because it lacks the Asp-Phe-Gly motif in subdomain VII of the kinase domain (22). This truncated kinase domain, which lacks the adenosine 5’-triphosphate binding site, allows TRB3 to act as a negative modulator of Akt through dose-dependent inhibition of Akt/PKB phosphorylation at Serine 473 (Ser473) and Threonine 308 (Thr308) in HEK293 cells (21). TRB3 has also been shown to inhibit the phosphorylation of Akt at Thr308 in FGC-4 cells (23) and to block the insulin activation of p70S6Kinase, Akt, mTOR, p70 S6 ribosomal protein and 4EBP1 in mouse hepatocytes (24). However, a contradicting report could not find evidence that TRB3 inhibited the insulin signaling pathway in rat hepatocytes (25). This discrepancy suggests that the function of TRB3 may be dependent on the cell type and/or experimental conditions.
Regulation of TRB3 expression has focused on the role of endoplasmic reticulum (ER) stress. Agents which induce ER stress have been found to increase TRB3 RNA levels in HepG2 cells (26) as well as p53 null osteosarcoma cells, MCF-7 breast cancer cells, H1299 lung cancer cells and DU145 prostate cancer cells (27). TRB3 protein levels have also been shown to increase with an increase in ER stress in HepG2, HEK 293 and A375 cells (28). Whether or not chondrocytes express TRB3 under normal conditions or conditions of ER stress has not been examined.
Because of the potential of TRB3 to serve as an inhibitor of IGF-1 signaling, we examined whether chondrocytes express TRB3 and determined if this expression changes with age or in the presence of OA. We also examined if an increase in ER stress would increase chondrocyte expression of TRB3 and whether an increased expression of TRB3 might contribute to a decrease in proteoglycan synthesis and cell survival. The results suggest that increased TRB3 in OA cartilage may play an important role in inhibiting chondrocyte IGF-1 signaling.
MATERIALS AND METHODS
Reagents
DMEM (Dulbecco's modified Eagle's medium), Ham's F-12, phosphate buffered saline (PBS) and antibiotics were purchased from Gibco BRL (Gaithersburg, MD, U.S.A.) while fetal bovine serum (FBS) was purchased from Hyclone (Logan, UT, U.S.A.). Pronase was purchased from Calbiochem (San Diego, CA, U.S.A.), Collagenase-P from Boehringer Mannheim (Germany), Keltone LVCR sodium alginate from Kelco (Chicago, IL, U.S.A.) and [35S]sulphate from Amersham Biosciences (San Francisco, CA, U.S.A.). Phosphospecific and non-phosphospecific antibodies directed to Akt and ERK were purchased from Cell Signaling Technology (Beverly, MA, U.S.A.). The antibody directed to TRB3 was custom made against a peptide (sequence SRKKRLELDDNLDTERPVC) by Quality Controlled Biochemicals (Hopkinton, MA, U.S.A.). IGF-1 was purchased from Austral Biologicals (San Ramon, CA, U.S.A.). The TRB3 and HA tagged Akt plasmids were provided by Dr. Montminy from the Salk Institute (La Jolla, CA, U.S.A). The PI-3 kinase dominant negative (PI3KDN) plasmid was provided by Dr. Shu Chien at the University of California, San Diego (U.S.A). The pcDNA 3.0 Empty Vector was purchased from Invitrogen (Carlsbad, CA, U.S.A.) and the RFP plasmid was purchased from Clontech (Mountain View, CA, U.S.A.)
Tissue acquisition
Human ankle (talar) articular cartilage was obtained from tissue donors within 48 hours of death through the National Disease Research Interchange (Philadelphia, PA) or the Gift of Hope Organ and Tissue Donor Network (Elmhurst, IL). Human osteoarthritic knee articular cartilage was obtained from tissue removed during knee replacement surgery in accordance with institutional guidelines and review board approval. Each donor specimen was graded for degenerative changes based on the five-point Collins scale, as modified by Muehleman et al.(29). Unless stated, cells from tissue graded 0 or 1 were used in experiments.
Protein extraction and TRB3 immunoblotting
Full thickness cartilage was removed from all surfaces of the tali or from the medial and lateral condyle surfaces of the femur. The cartilage slices were rinsed with cold PBS and then frozen dry at −80°C. The frozen slices were ground using a mortar and pestle while in liquid nitrogen. The ground cartilage powder was placed in cell lysis buffer containing 20 mM Tris/HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM PMSF and 1 μg/ml each of aprotinin, leupeptin and pepstatin. This mixture was allowed to rotate end-over-end for 4 hours at 4°C at which time the mixture was centrifuged for 10 min at 1400 rpm and the supernatant was kept. The supernatant was centrifuged again twice more to remove any particulates. The supernatants (containing soluble proteins from cartilage extracts) were then measured for total protein (bicinchoninic acid kit; Pierce, Rockford, IL, U.S.A.). Samples with equal amounts of total protein were separated by SDS-PAGE and transferred to nitrocellulose for immunoblotting with anti-TRB3 antibody or with an antibody to β-actin as a control. Immunoreactivity was detected with enhanced chemiluminescence (ECL®; Amersham Biosciences). Densitometry was performed on immunoblots after scanning and then using Kodak 1D 3.6 software in order to analyze the regions of interest (ROI).
Chondrocyte isolation and culture
Full-thickness cartilage was removed from all surfaces of the tali or from the medial and lateral condyle surfaces of the femora. Full-thickness bovine tissue was collected from all surfaces of the metacarpophalangeal joint. Cartilage slices were digested in a sequential manner with 0.2% pronase for 1 h and then overnight with 0.025% collagenase as described previously (30). For experiments using monolayer cultures, short-term high-density monolayers were used where six-well plates were plated with 2mls/well of cells at 1×106 cells/ml in DMEM with 10% FBS. The cells were incubated at 37°C and 5% CO2 until confluent (usually 5−7 days).
Cell lysates were prepared using the cell lysis buffer described above. The cells were scraped, transferred in to microcentrifuge tubes, agitated end-over-end for 30 min at 4 °C, centrifuged at 14000g for 10 min and the supernatants were removed and measured for total protein for immunoblotting as described above.
Immunohistochemistry
10% formalin fixed paraffin embedded sections of cartilage from humans knee joints were deparaffinized and rehydrated. Protein Block (Serum-Free Protein Block; Dako, Carpinteria, CA) was applied for 10 minutes prior to incubation with primary antibody. Anti-TRB3 antibodies were diluted 1:500 and antibody-treated sections were incubated at 4°C overnight. Antibody binding was detected using biotinylated anti-rabbit link (Biogenex, San Ramon, CA) at 1:20 for 30 minutes at 33°C. Alkaline Phosphatase label (Biogenex) was used at 1:20 for 30 minutes at 3°C. Vector Red (Vector Laboratories, Burlingame, CA) was used for visualization, and sections were counterstained with Mayer's hematoxylin (Dako). Negative Control Supersensitive Rabbit Serum for SuperSensitive Antibodies (BioGenex) was substituted for the primary antibody for the negative control sections.
Chondrocyte stimulation and analysis of ER stress
Confluent monolayer cultures were placed in DMEM/Ham's F-12 medium supplemented with 10% FBS and then stimulated with 2μg/mL tunicamycin or 2μM thapsigargin for 6 or 24 hours. After stimulation, media was removed and the cells were washed once with ice-cold PBS containing 0.1 mM Na3VO4. Cell lysates were immediately prepared by solubilization in cell lysis buffer as described above.
Transfection
Transfection was by nucleofection performed according to instructions of the human chondrocyte nucleofector kit (Amaxa Biosystems, Gaithersburg, MD, U.S.A.) and as previously described (31) with some modification. Briefly, after plating in monolayers, normal human chondrocytes were allowed to recover for 72hrs. The cells were then treated with media containing 0.2% pronase/0.025% collagenase and 10% FBS serum for 3 hours to detach the cells. The media was centrifuged and the cell pellet was washed twice with 10% serum and once with PBS. Following the wash with PBS, the cells were resuspended in the nucleofector solution and plasmid DNA was added. Chondrocytes were routinely transfected with 5μg of total plasmid DNA in the Amaxa Nucleofector machine using program U-24. After transfection, the cells from each cuvette were immediately placed in 900uL of culture medium containing 20% FBS. This solution was mixed and then placed into 1ml culture medium containing 20% FBS per well of a twelve well plate which was then incubated at 37°C in 5% CO2.
The cells were allowed to recover from the nucleofection process for 24 hours in 20% FBS, then changed to media containing 10% FBS for another 24 hours, and then finally serum starved overnight. The cells were treated with 100ng/mL IGF-1 for 30 minutes and then whole cell lysates were made as described above. For the immunoprecipitation experiments, the lysates were agitated end over end for 30 minutes at 4°C and then centrifuged at 14000g for 10 min and the supernatants were removed and incubated with 40μL 1:1 ImmunoPure© Immobilized Protein A (Pierce, Rockford, IL, U.S.A.) for 1 hour at room temperature with end over end agitation. The slurry was centrifuged at 5000rpm for 5 minutes, the supernatant collected, and 5μL of anti-HA antibody was added and the supernatant/antibody mixture was incubated with gentle agitation overnight at 4°C. Next 40μL 1:1 ImmunoPure© Immobilized Protein A beads were added to the supernatant/antibody mixture and gently agitated overnight at 4°C. The slurry was centrifuged at 5000rpm for 5 minutes and the supernatant discarded, the beads washed 6 times with RIPA buffer and then 40μL of Laemmli buffer was added to the beads and the Laemmli/bead mixture boiled for 5 minutes, centrifuged. The solubilized proteins were separated by SDS-PAGE and transferred to nitrocellulose for immunoblot analysis using an antibody directed at phosphorylated Akt Thr308.
Cell survival experiments
Normal human articular chondrocytes were transfected with expression plasmids that expressed TRB3, red fluorescent protein (RFP) as a control, a dominant negative form of PI3K (PI3K DN) and a Hemagglutinin (HA) tagged Akt. 24 hours after the nucleofection process, the media was changed to DMEM/F-12 with 10% FBS. After 24 hours in media with 10% serum, the cells were lifted from their respective wells with trypsin treatment, washed, and transferred to serum-free suspension culture, a condition where IGF-1 mediates cell survival, by placing them into alginate beads at a density of 2×105 cells/bead. Cells were cultured in alginate as previously described (6) with some modifications to make the low density suspension culture. Briefly, freshly isolated cells were suspended in 0.9% NaCl containing low viscosity alginate (1.2%) at a density of 2 × 106 cells/ml and added in a drop wise fashion through a 22−1/2-gauge needle into a 102 mM CaCl2 solution. Beads were allowed to gel in the CaCl2 for 10 minutes and washed 3 times with DMEM/Ham's F12 medium. The beads were placed in serum-free DMEM/F-12 media and cell death was determined after 72 hours using the LIVE/DEAD cell assay (Molecular Probes, Eugene, OR, U.S.A.) as described previously(20). The percentage of cells that remained alive after treatment was measured in triplicate with at least 100 cells counted for each data point.
Proteoglycan synthesis experiments
[35S]Sulfate incorportation was used to measure PG synthesis in monolayer cultures of normal human chondrocytes which were isolated and cultured at high density as described above. The cells were treated with IGF-1 overnight and then pulsed with [35S]Sulfate for 4 hours. [35S]Sulfate incorporation was measured in the media as well as in the cell pellet after Alcian Blue precipitation as previously described (18;32).
Statistical analysis
Results comparing the means between multiple groups were analysed using a one-way ANOVA with a post hoc Bonferroni correction using the Windows-based StatView software (SAS Institute, Cary, NC, U.S.A.). Samples for sulphate incorporation were performed in triplicate, and P<0.05 was considered to be statistically significant for all statistical analyses.
RESULTS
Chondrocytes express TRB3
TRB3 expression by human articular chondrocytes was initially assessed using RT-PCR. TRB3 mRNA was detected in normal articular chondrocytes, with the lowest levels found in tissue from an 8 year-old donor (Fig.1A). Immunoblot analysis was used to determine if chondrocytes synthesize the TRB3 protein. TRB3 was detected in chondrocytes isolated from either the knee or ankle joint with higher levels noted in cells from Grade 4 knee tissue from an 83 year-old donor as compared to Grade 0 from the ankle of the same donor and Grade 0 knee and ankle tissue from a 28 year-old donor (Fig.1B).
Figure 1. Chondrocytes express TRB3.
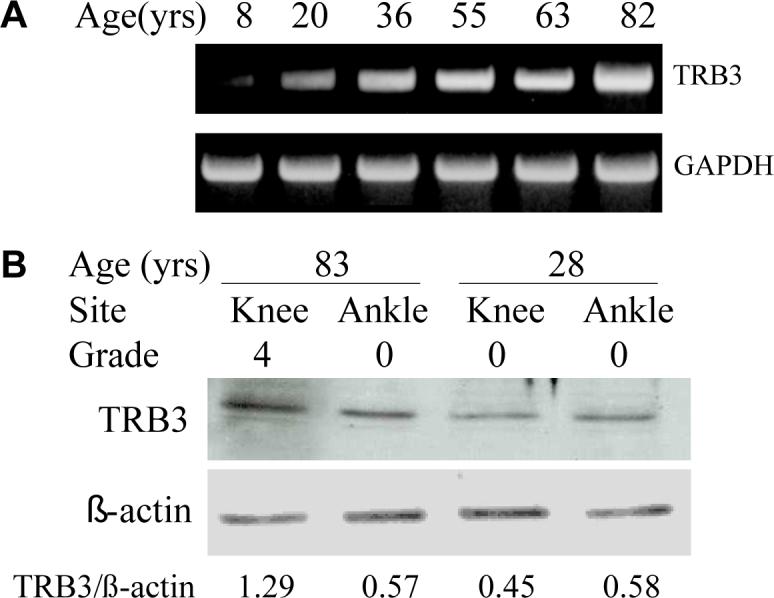
(A) Human chondrocytes isolated from donors of different ages were plated in monolayer culture. At confluency, RNA was isolated from these cells, RT-PCR was performed, and the cDNA products were stained with ethidium after separation on an agarose gel. GAPDH expression was used as a control. (B) Chondrocytes were isolated from matched pairs of knee and ankle cartilage from an old (83 years) and young (28 years) donor. Cell lysates were prepared after overnight incubation in serum-free media and were used for immunoblot analysis with anti-TRB3 antibody. Immunoblotting for actin was used as a protein loading control. Densitometry was used to quantitate the band intensity which is reported beneath the blots as a ratio of TRB3 to actin.
In order to determine if the protein levels of TRB3 increase with age and/or degeneration, TRB3 was measured in protein extracts made from cartilage having a range of Collin's grades that had been removed from human tissue donors of different ages. In these extracts (made directly from cartilage rather than isolated cells) the TRB3 bands were fainter (Figs.2A and 2B) than those seen in cell lysates (Figs.1 and 2C). Somewhat higher TRB3 levels, quantitated by densitometry and normalized for β-actin, were noted in extracts from normal ankle cartilage of 4 older adults (0.63±0.1, mean ±SEM) when compared to 4 younger adults (0.37±0.6) on the same blot (Fig.2A) but this difference did not reach statistical significance (p=0.7 by t-test). A difference by Collin's grade in TRB3 levels among older adult tissues with grades ranging from 0−3 was not seen (Fig.2B).
Figure 2. The effect of donor age, tissue grade and OA on TRB3 protein levels.
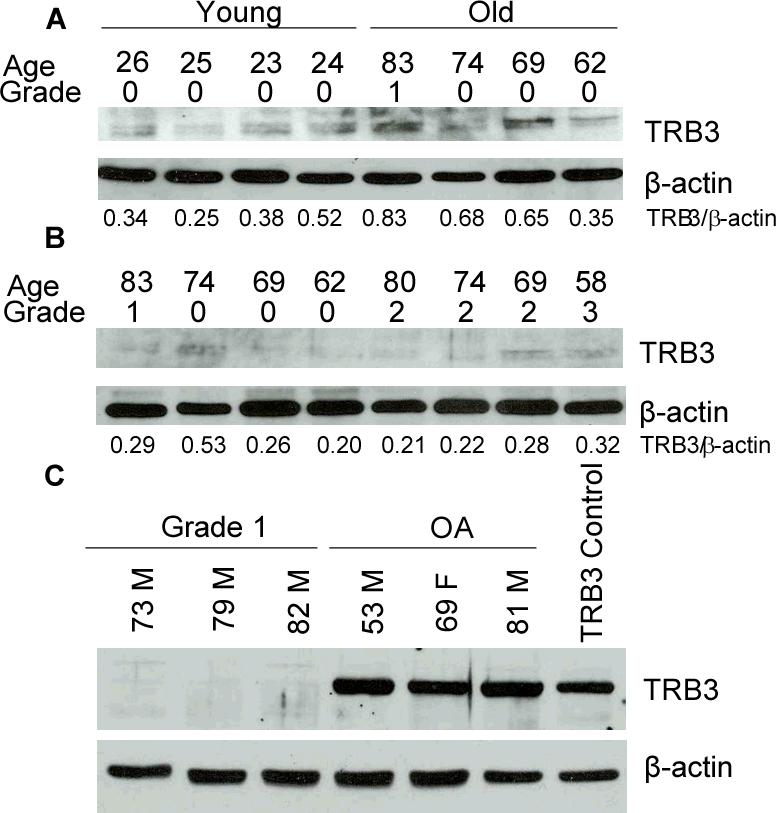
(A) Protein extracts were made directly from ankle tissue graded 0−1 and obtained from young and old tissue donors without a known history of arthritis. Immunoblot analysis was performed using anti-TRB3 antibody and with anti-β-actin antibody as a loading control. Densitometry results are shown under the blots. (B) Protein extracts prepared and analyzed as in panel A using samples obtained from older adult ankle cartilage with grades from 0−3. (C) Cell lysates were prepared from cultured, age-matched, untreated human chondrocytes from normal tissue donors and tissue harvested at time of knee joint replacement for OA. The lysates were used for immunoblot analysis with an anti-TRB3 antibody along with anti-β-actin antibody as a loading control. An additional cell lysate made from human chondrocytes transfected with a TRB3 expression vector was used as a positive control.
These results suggested that measuring TRB3 by immunoblotting protein extracted from cartilage was not sensitive enough to detect significant differences related to age or grade. We then examined TRB3 in cell lysates prepared from cells isolated from age-matched normal and OA cartilage and noted a striking increase in TRB3 in the OA cells (Fig.2C). In this experiment, a cell lysate made from normal chondrocytes transfected with a TRB3 expression construct was included as a positive control.
To confirm these results and to examine the location of the TRB3 in human cartilage, immunohistochemistry was performed on intact cartilage slices using the anti-TRB3 antibody. Age-matched normal tissue contained very low numbers of TRB3 immunopositive chondrocytes, whereas tissue with OA changes contained many TRB3 immonopositive cells (Fig. 3). The majority of the TRB3 immunostaining was noted in the cells in the more superficial regions in which chondrocyte clusters were prominent and the matrix was fibrillated.
Figure 3. Immunohistochemical staining for TRB3 in normal and OA knee tissue.
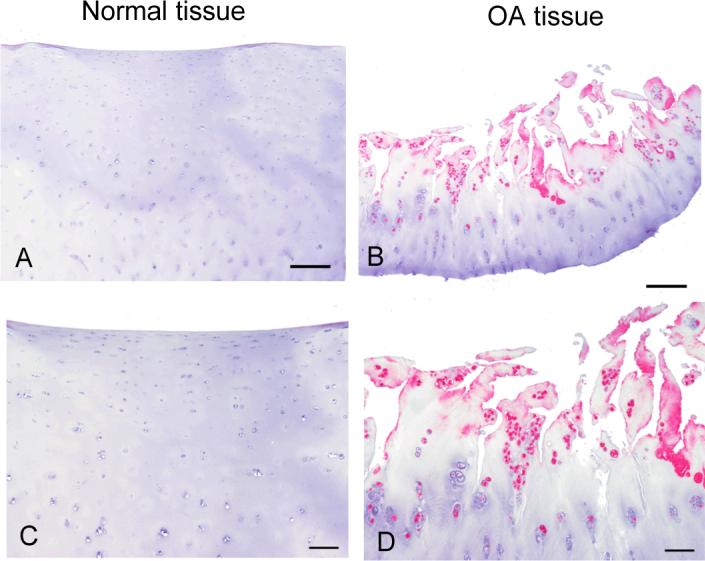
Immunohistochemistry was done on tissue from a normal knee joint and from one obtained at joint replacement for OA. The sections were immunostained with anti-TRB3 at a 1:500 dilution. An alkaline phosphatase detection system was used and the product was visualized with Vector Red. The negative control images (not shown) were similar to the images from normal tissue which did not detect TRB3. Bar = 250μm in the upper images and 100μm in the lower images.
ER stress promotes TRB3 expression
ER stress has been shown to stimulate TRB3 expression in a number of cell types (26-28). ER stress has also been shown to inhibit chondrocyte growth, down-regulate expression of the cartilage matrix proteins collagen type II and aggrecan, and induce chondrocyte apoptosis (33) suggesting that it might play a role in the development of OA. We induced ER stress in normal primary chondrocytes using tunicamycin, which blocks protein glycosylation, and thapsigargin, which depletes Ca2+ stores. GADD-153 was used as a marker for ER stress. Both treatments increased protein levels of TRB3 (Fig. 4).
Figure 4. ER stress induces TRB3 expression.
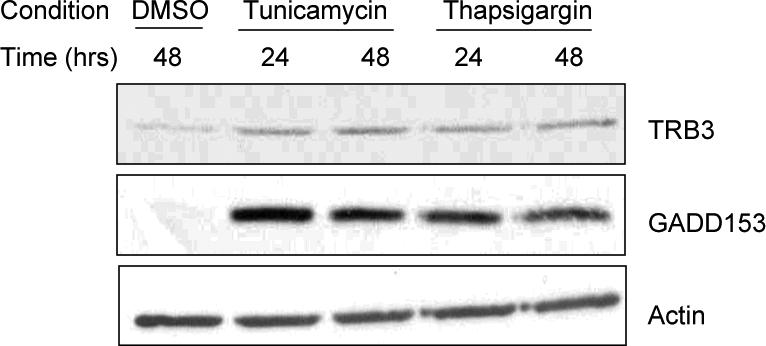
Normal human chondrocytes cultured in monolayer were treated with ER stress inducing agents tunicamycin (2μg/mL) or thapsigargin (2μM) or 0.001% DMSO for the indicated times and cell lysates were used for immunoblot analysis with anti-TRB3 antibody, anti-GADD153 antibody (ER stress marker) and anti-β-actin antibody as a loading control.
Overexpression of TRB3 decreases chondrocyte survival and inhibits IGF-1 stimulation of Akt phosphorylation
Since the inhibition of Akt has been shown to be a causal mediator of cell death (34), the blocking of Akt activation by TRB3 might induce cell death under conditions where IGF-1 stimulation of Akt is necessary for chondrocyte survival. We had previously found that chondrocytes cultured in suspension in serum-free media at low density require autocrine signaling by IGF-1 to survive (6); therefore, similar culture conditions were used to test the effects of TRB3. Overexpression of TRB3 reduced the survival of normal human chondrocytes by almost 40% compared to cells transfected with a control plasmid. Furthermore, addition of IGF-1 to cells overexpressing TRB3 was not able to improve survival (Fig. 5A). However, the cell death induced by TRB3 overexpression could be prevented by co-transfection of a construct designed to overexpress Akt, consistent with the notion that TRB3 overexpression caused cell death through inhibition of endogenous Akt activation. PI-3 kinase activates Akt in response to IGF-1. Therefore, as an additional control to support the role of IGF-1 mediated Akt activation in promoting cell survival under these conditions, we transfected chondrocytes with a dominant negative PI-3 kinase construct and observed cell death similar to the overexpression of TRB3.
Figure 5. Overexpression of TRB3 reduces chondrocyte survival and IGF-1 stimulated Akt phosphorylation.
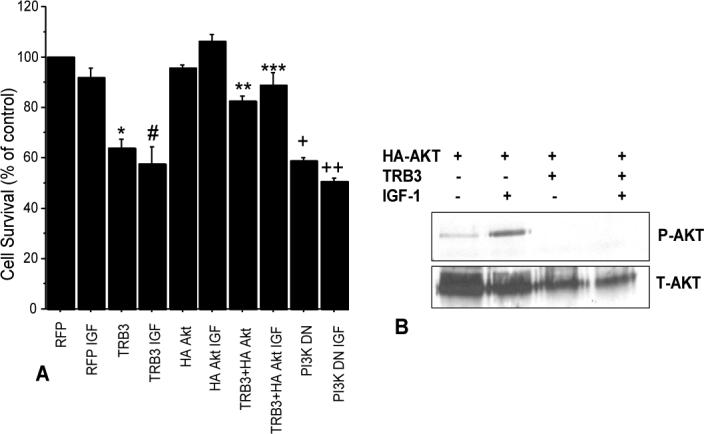
(A) Human chondrocytes isolated from articular cartilage of normal donor ankles were nucleofected with plasmid constructs to express TRB3, HA-Akt, red fluorescent protein (RFP control), or a dominant-negative PI-3 kinase. After 48 hours cells were harvested and placed into alginate beads at 20,000 cells/bead and cultured in serum-free media for 72 hours with either 50ng/ml IGF-1 or vehicle as a control. Live/Dead survival assay was then performed (n=3 experiments with cells from different donors). p <0.0001: RFP control vs TRB3 (*), RFP IGF vs TRB3 IGF (#), RFP vs PI3KDN (+), and RFP IGF to PI3KDN IGF(++). p=0.001: TRB3+HA Akt vs TRB3(**). p=0.03: TRB3+HA Akt IGF vs TRB3 IGF(***). (B) Normal human chondrocytes were transfected with an HA tagged Akt expression construct or cotransfected with HA tagged Akt and TRB3 expression constructs. After 48hours cultures were made serum free overnight. Indicated wells were treated with 100ng/mL IGF-1 for 30 minutes and then all wells were lysed. Immunoprecipitation was performed using anti-HA antibody and then the lysates were immunoblotted using anti-phosphorylated Akt antibody (P-AKT). The membrane was stripped and reprobed using anti-Akt antibody (T-AKT). This is a representative blot from an n of 4 experiments.
In order to determine if overexpression of TRB3 inhibited Akt phosphorylation, human chondrocytes were transfected with a plasmid expressing HA tagged Akt or co-transfected with HA-Akt and TRB3 constructs and then stimulated with IGF-1. The HA-Akt construct was phosphorylated at Thr308 in the presence of IGF-1 and co-transfection with TRB3 was inhibitory (Fig.5B).
TRB3 inhibits IGF-1 induced proteoglycan synthesis
Previous work has shown that stimulation of the PI-3 kinase/Akt pathway is required for the chondrocyte production of PG in response to IGF-1 (18). To examine if TRB3 overexpression affected PG synthesis, normal human chondrocytes (from grade 0−1 tissue) were transfected with the TRB3 construct or pcDNA 3.0 empty vector as a control. Because overexpression of TRB3 in low density serum-free alginate cultures resulted in cell death, the PG synthesis studies were performed with cells in high density monolayer and with 5% FBS added to the media, which promoted survival under these conditions. PG synthesis was measured by [35S]sulfate incorporation and the results were normalized to DNA content in order to control for any loss of cells. In this culture system, overexpression of TRB3, but not the control vector, reduced basal PG synthesis and completely blocked IGF-1 stimulation of PG synthesis (Fig. 6.).
Figure 6. Overexpression of TRB3 decreases IGF-1 stimulation of PG synthesis.
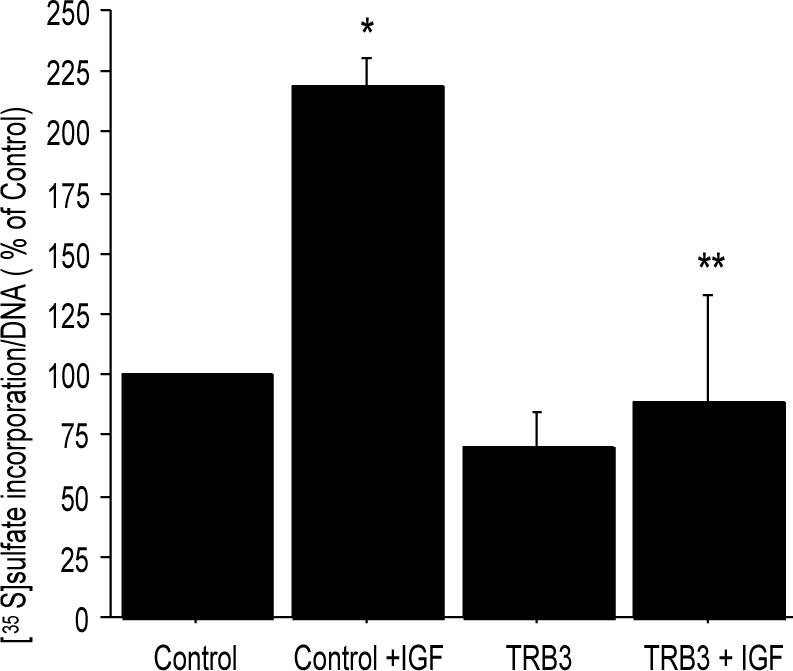
Normal human chondrocytes were transfected with 5μg pcDNA 3.0 empty vector or 5μg TRB3 overexpression plasmids. The cells were allowed to recover from the transfection in monolayer culture and then placed in media containing 5% serum. 100ng/mL IGF-1 was added to indicated wells for an overnight treatment and then [35S]sulfate incorporation was performed. The counts per minute were normalized to DNA concentration of the samples and the results (mean±SEM) expressed as a % of the unstimulated control (n=3 experiments with cells from different donors). *p=0.02 control vs control+IGF-1; **p=0.02 control+IGF-1 vs TRB3+IGF-1.
DISCUSSION
This study is the first to show that chondrocytes express TRB3, a protein previously identified in other cell types (21,23,24) as an inhibitor of insulin- and IGF-induced activation of Akt/PKB. PI-3 kinase mediated activation of Akt is required for IGF-1 stimulation of chondrocyte PG synthesis (18) as well survival under conditions dependent on autocrine IGF-1 signaling (6). The PI-3 kinase/Akt pathway has also been shown to be required for TGF-β induction of TIMP-3 expression (46). Significantly greater amounts of TRB3 were detected in OA chondrocytes relative to age-matched cells from normal appearing cartilage. Over expression of TRB3 in normal chondrocytes reduced survival and blocked IGF-1 stimulation of PG synthesis. Together, these findings suggest that an increased level of TRB3 in OA chondrocytes could contribute to the previously observed decreased response of OA chondrocytes to IGF-1 (3,9) and might contribute to the cell death and matrix loss seen during the development of OA.
A potential mechanism for increased expression of TRB3 is ER stress (26-28). Chondrocytes exhibit a variety of stresses, including osmotic stress, nutrient deprivation, oxidative stress, and mechanical stress (35-37) which can potentially impair ER function resulting in ER stress (38). Recently it has been shown that ER stress can be induced in chondrocytes under certain conditions (33,39,40). We found that an increase in ER stress induced by either tunicamycin or thapsigargin caused an increase in chondrocyte TRB3. ER stress in chondrocytes has been shown to decrease expression of cartilage matrix genes (39) and prolonged ER stress leads to apoptosis (33,40). The present findings demonstrating that overexpression of TRB3 can inhibit IGF-1 mediated PG synthesis and reduce chondrocyte survival suggest a potential mechanism for TRB3 as a mediator of the effects of ER stress in chondrocytes.
TRB3 is a pseudokinase that binds Akt and prevents phosphorylation of Akt which is required for Akt to be active (21). We confirmed in human chondrocytes, that, similar to other cell types (21,23,24), TRB3 blocks the IGF-1 stimulated phosphorylation of Akt. Inhibition of Akt activation by TRB3 was associated with reduced chondrocyte survival in low-density serum free alginate cultures, a condition in which we previously demonstrated that IGF-1 autocrine signaling promoted survival (6). This is consistent with other reports showing that TRB3 caused cell death in several different cell types (28,41,42). The IGF-1-PI-3-kinase-Akt pathway is a well characterized survival pathway in many cell types (14,15,43,44). The PI3-kinase inhibitor wortmannin was previously shown to decrease the survival of rabbit chondrocytes (45). In the present study we found that over expression of a PI-3 kinase dominant negative construct decreased survival of human chondrocytes. These finding suggest that increased levels of TRB3 cause cell death through inhibition of the IGF-1-PI3-kinase-Akt survival pathway.
When TRB3 was overexpressed in high density monolayer cultures in media supplemented with 5% FBS, the negative effects of TRB3 on survival could be overcome. Using these conditions we determined that increased TRB3 could inhibit PG synthesis. This is consistent with previous studies that showed the PI-3-kinase-Akt pathway was required for IGF-1 stimulation of PG synthesis by chondrocytes (18). The finding of increased TRB3 in OA chondrocytes suggests that it could play a role in the reduced response to IGF-1 previously noted in OA (10). It was less clear if TRB3 levels were increased with aging in cartilage without OA-changes; therefore, we do not know at this time if TRB3 also contributes to the age-related decline in IGF-1 response.
In conclusion, we have identified TRB3 production by human chondrocytes and shown that TRB3 levels are dramatically increased in OA cells. TRB3 blocks the IGF-1 stimulated phosphorylation of Akt in chondrocytes. Overexpression of TRB3 causes chondrocyte cell death by inhibiting the PI-3 kinase/Akt pathway in culture conditions where this pathway is important for chondrocyte survival. Overexpressing TRB3 also inhibits the IGF-1 stimulated PG synthesis by chondrocytes. It was also seen that an increase in ER stress can upregulate TRB3 in chondrocytes. Together, these results suggest that TRB3 may play a role in the reduced response to IGF-1 in OA chondrocytes and therefore contribute to the imbalance in anabolic and catabolic activity that is characteristic of OA. Further studies will be necessary to determine if inhibition of TRB3 during the development of OA can slow the progression of cartilage loss.
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health Grant AG-16697. We would like to thank Dr. Mark Montminy for supplying the TRB3 and HA Akt plasmids, Dr. Shu Chien for providing the PI-3 kinase DN plasmid and Carol Pacione and Anne Undersander for technical assistance. We gratefully acknowledge the Gift of Hope Organ and Tissue Donor Network, NDRI and the donor families for providing donor tissue and Dr. David Martin for assistance in collecting OA tissue.
Supported by NIH grant AG-16697 (RL)
REFERENCES
- 1.Aigner T, Haag J, Martin J, Buckwalter J. Osteoarthritis: aging of matrix and cells - going for a remedy. Current Drug Targets. 2007;8:325–331. doi: 10.2174/138945007779940070. [DOI] [PubMed] [Google Scholar]
- 2.Goldring MB. The role of the chondrocyte in osteoarthritis. Arthritis Rheum. 2000;43(9):1916–26. doi: 10.1002/1529-0131(200009)43:9<1916::AID-ANR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 3.Loeser RF, Shanker G, Carlson CS, Gardin JF, Shelton BJ, Sonntag WE. Reduction in the chondrocyte response to insulin-like growth factor 1 in aging and osteoarthritis: studies in a non-human primate model of naturally occurring disease. Arthritis Rheum. 2000;43(9):2110–20. doi: 10.1002/1529-0131(200009)43:9<2110::AID-ANR23>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 4.van Osch GJ, van den Berg WB, Hunziker EB, Hauselmann HJ. Differential effects of IGF-1 and TGF beta-2 on the assembly of proteoglycans in pericellular and territorial matrix by cultured bovine articular chondrocytes. Osteoarthritis Cartilage. 1998;6(3):187–95. doi: 10.1053/joca.1998.0111. [DOI] [PubMed] [Google Scholar]
- 5.Guenther HL, Guenther HE, Froesch ER, Fleisch H. Effect of insulin-like growth factor on collagen and glycosaminoglycan synthesis by rabbit articular chondrocytes in culture. Experientia. 1982;38(8):979–81. doi: 10.1007/BF01953688. [DOI] [PubMed] [Google Scholar]
- 6.Loeser RF, Shanker G. Autocrine stimulation by insulin-like growth factor 1 and insulin-like growth factor 2 mediates chondrocyte survival in vitro. Arthritis Rheum. 2000;43(7):1552–9. doi: 10.1002/1529-0131(200007)43:7<1552::AID-ANR20>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 7.Hui W, Rowan AD, Cawston T. Insulin-like growth factor 1 blocks collagen release and down regulates matrix metalloproteinase-1, -3, -8, and -13 mRNA expression in bovine nasal cartilage stimulated with oncostatin M in combination with interleukin 1alpha. Ann Rheum Dis. 2001;60(3):254–61. doi: 10.1136/ard.60.3.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Im HJ, Pacione C, Chubinskaya S, Van Wijnen AJ, Sun Y, Loeser RF. Inhibitory effects of insulin-like growth factor-1 and osteogenic protein-1 on fibronectin fragment- and interleukin-1beta-stimulated matrix metalloproteinase-13 expression in human chondrocytes. J Biol Chem. 2003;278(28):25386–94. doi: 10.1074/jbc.M302048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin JA, Ellerbroek SM, Buckwalter JA. Age-related decline in chondrocyte response to insulin-like growth factor-I: the role of growth factor binding proteins. J Orthop Res. 1997;15(4):491–8. doi: 10.1002/jor.1100150403. [DOI] [PubMed] [Google Scholar]
- 10.Dore S, Pelletier JP, DiBattista JA, Tardif G, Brazeau P, Martel-Pelletier J. Human osteoarthritic chondrocytes possess an increased number of insulin-like growth factor 1 binding sites but are unresponsive to its stimulation. Possible role of IGF-1-binding proteins. Arthritis Rheum. 1994;37(2):253–63. doi: 10.1002/art.1780370215. [DOI] [PubMed] [Google Scholar]
- 11.Barone-Varelas J, Schnitzer TJ, Meng Q, Otten L, Thonar EJ. Age-related differences in the metabolism of proteoglycans in bovine articular cartilage explants maintained in the presence of insulin-like growth factor I. Connect Tissue Res. 1991;26(1−2):101–20. doi: 10.3109/03008209109152167. [DOI] [PubMed] [Google Scholar]
- 12.Messai H, Duchossoy Y, Khatib A, Panasyuk A, Mitrovic DR. Articular chondrocytes from aging rats respond poorly to insulin-like growth factor-1: an altered signaling pathway. Mech Ageing Dev. 2000;115(1−2):21–37. doi: 10.1016/s0047-6374(00)00107-x. [DOI] [PubMed] [Google Scholar]
- 13.Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16(1):3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 14.Kulik G, Weber MJ. Akt-dependent and -independent survival signaling pathways utilized by insulin-like growth factor I. Mol Cell Biol. 1998;18(11):6711–8. doi: 10.1128/mcb.18.11.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parrizas M, Saltiel AR, LeRoith D. Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3'-kinase and mitogen-activated protein kinase pathways. J Biol Chem. 1997;272(1):154–61. doi: 10.1074/jbc.272.1.154. [DOI] [PubMed] [Google Scholar]
- 16.Lehman JA, Calvo V, Gomez-Cambronero J. Mechanism of ribosomal p70S6 kinase activation by granulocyte macrophage colony-stimulating factor in neutrophils: cooperation of a MEK-related, Thr421/Ser424 kinase and a rapamycin-sensitive, mTOR-related Thr389 kinase. J Biol Chem. 2003;278(30):28130–28138. doi: 10.1074/jbc.M300376200. [DOI] [PubMed] [Google Scholar]
- 17.Oh CD, Chun JS. Signaling mechanisms leading to the regulation of differentiation and apoptosis of articular chondrocytes by insulin-like growth factor-1. J Biol Chem. 2003;278(38):36563–71. doi: 10.1074/jbc.M304857200. [DOI] [PubMed] [Google Scholar]
- 18.Starkman BG, Cravero JD, Delcarlo M, Loeser RF. IGF-I stimulation of proteoglycan synthesis by chondrocytes requires activation of the PI 3-kinase pathway but not ERK MAPK. Biochem J. 2005;389(Pt 3):723–9. doi: 10.1042/BJ20041636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shakibaei M, Schulze-Tanzil G, De Souza P, John T, Rahmanzadeh M, Rahmanzadeh R, et al. Inhibition of mitogen-activated protein kinase kinase induces apoptosis of human chondrocytes. J Biol Chem. 2001;16:16. doi: 10.1074/jbc.M010859200. [DOI] [PubMed] [Google Scholar]
- 20.Del Carlo M, Jr., Loeser RF. Nitric oxide--mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum. 2002;46(2):394–403. doi: 10.1002/art.10056. [DOI] [PubMed] [Google Scholar]
- 21.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300(5625):1574–7. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- 22.Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends Cell Biol. 2006;16(9):443–452. doi: 10.1016/j.tcb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 23.He L, Simmen FA, Mehendale HM, Ronis MJ, Badger TM. Chronic ethanol intake impairs insulin signaling in rats by disrupting Akt association with the cell membrane. Role of TRB3 in inhibition of Akt/protein kinase B activation. J Biol Chem. 2006;281(16):11126–11134. doi: 10.1074/jbc.M510724200. [DOI] [PubMed] [Google Scholar]
- 24.Matsushima R, Harada N, Webster NJ, Tsutsumi YM, Nakaya Y. Effect of TRB3 on insulin and nutrient-stimulated hepatic p70 S6 kinase activity. J Biol Chem. 2006;281(40):29719–29729. doi: 10.1074/jbc.M511636200. [DOI] [PubMed] [Google Scholar]
- 25.Iynedjian PB. Lack of evidence for a role of TRB3/NIPK as an inhibitor of PKB-mediated insulin signalling in primary hepatocytes. Biochem J. 2005;386(Pt 1):113–118. doi: 10.1042/BJ20041425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ord D, Ord T. Characterization of human NIPK (TRB3, SKIP3) gene activation in stressful conditions. Biochem Biophys Res Commun. 2005;330(1):210–218. doi: 10.1016/j.bbrc.2005.02.149. [DOI] [PubMed] [Google Scholar]
- 27.Corcoran CA, Luo X, He Q, Jiang C, Huang Y, Sheikh MS. Genotoxic and endoplasmic reticulum stresses differentially regulate TRB3 expression. Cancer Biol Ther. 2005;4(10):1063–1067. doi: 10.4161/cbt.4.10.2205. [DOI] [PubMed] [Google Scholar]
- 28.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24(6):1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muehleman C, Bareither D, Huch K, Cole AA, Kuettner KE. Prevalence of degenerative morphological changes in the joints of the lower extremity. Osteoarthritis Cartilage. 1997;5(1):23–37. doi: 10.1016/s1063-4584(97)80029-5. [DOI] [PubMed] [Google Scholar]
- 30.Loeser RF, Todd MD, Seely BL. Prolonged treatment of human osteoarthritic chondrocytes with insulin-like growth factor-I stimulates proteoglycan synthesis but not proteoglycan matrix accumulation in alginate cultures. J Rheumatol. 2003;30(7):1565–70. [PubMed] [Google Scholar]
- 31.Pulai JI, Del Carlo M, Jr., Loeser RF. The alpha5beta1 integrin provides matrix survival signals for normal and osteoarthritic human articular chondrocytes in vitro. Arthritis Rheum. 2002;46(6):1528–1535. doi: 10.1002/art.10334. [DOI] [PubMed] [Google Scholar]
- 32.Masuda K, Shirota H, Thonar EJ. Quantification of 35S-labeled proteoglycans complexed to alcian blue by rapid filtration in multiwell plates. Anal Biochem. 1994;217(2):167–75. doi: 10.1006/abio.1994.1105. [DOI] [PubMed] [Google Scholar]
- 33.Yang L, Carlson SG, McBurney D, Horton WE. Multiple signals induce endoplasmic reticulum stress in both primary and immortalized chondrocytes resulting in loss of differentiation, impaired cell growth, and apoptosis. J Biol Chem. 2005;280(35):31156–31165. doi: 10.1074/jbc.M501069200. [DOI] [PubMed] [Google Scholar]
- 34.Luo HR, Hattori H, Hossain MA, Hester L, Huang YF, Lee-Kwon W, et al. Akt as a mediator of cell death. Proc Nat Acad Sci USA. 2003;100(20):11712–11717. doi: 10.1073/pnas.1634990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erickson GR, Northrup DL, Guilak F. Hypo-osmotic stress induces calcium-dependent actin reorganization in articular chondrocytes. Osteoarthritis and Cartilage. 2003;11(3):187–197. doi: 10.1053/s1063-4584(02)00347-3. [DOI] [PubMed] [Google Scholar]
- 36.Del Carlo M, Jr., Loeser RF. Increased oxidative stress with aging reduces chondrocyte survival: Correlation with intracellular glutathione levels. Arthritis Rheum. 2003;48(12):3419–30. doi: 10.1002/art.11338. [DOI] [PubMed] [Google Scholar]
- 37.Lee RB, ilkins RJ, azaq S, Urban JPG. The effect of mechanical stress on cartilage energy metabolism. Biorheology. 2002;39(1):133–143. [PubMed] [Google Scholar]
- 38.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13(10):1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 39.Yang L, McBurney D, Tang SC, Carlson SG, Horton WE., Jr. A novel role for Bcl-2 associated-athanogene-1 (Bag-1) in regulation of the endoplasmic reticulum stress response in mammalian chondrocytes. J Cell Biochem. 2007;102(3):786–800. doi: 10.1002/jcb.21328. [DOI] [PubMed] [Google Scholar]
- 40.Oliver BL, Cronin CG, Zhang-Benoit Y, Goldring MB, Tanzer ML. Divergent stress responses to IL-1beta, nitric oxide, and tunicamycin by chondrocytes. J Cell Physiol. 2005;204(1):45–50. doi: 10.1002/jcp.20261. [DOI] [PubMed] [Google Scholar]
- 41.Carracedo A, Lorente M, Egia A, Blazquez C, Garcia S, Giroux V, et al. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell. 2006;9(4):301–312. doi: 10.1016/j.ccr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 42.Carracedo A, Gironella M, Lorente M, Garcia S, Guzman M, Velasco G, et al. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006;66(13):6748–6755. doi: 10.1158/0008-5472.CAN-06-0169. [DOI] [PubMed] [Google Scholar]
- 43.Butler AA, Yakar S, Gewolb IH, Karas M, Okubo Y, LeRoith D. Insulin-like growth factor-I receptor signal transduction: at the interface between physiology and cell biology. Comp Biochem Physiol B Biochem Mol Biol. 1998;121(1):19–26. doi: 10.1016/s0305-0491(98)10106-2. [DOI] [PubMed] [Google Scholar]
- 44.Clemmons DR, Maile LA. Minireview: Integral Membrane Proteins that Function Coordinately with the Insulin-Like Growth Factor I Receptor to Regulate Intracellular Signaling. Endocrinology. 2003;144(5):1664–1670. doi: 10.1210/en.2002-221102. [DOI] [PubMed] [Google Scholar]
- 45.Todd Allen R, Robertson CM, Harwood FL, Sasho T, Williams SK, Pomerleau AC, et al. Characterization of mature vs aged rabbit articular cartilage: analysis of cell density, apoptosis-related gene expression and mechanisms controlling chondrocyte apoptosis. Osteoarthritis and Cartilage. 2004;12(11):917–923. doi: 10.1016/j.joca.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 46.Qureshi HY, Ahmad R, Sylvester J, Zafarullah M. Requirement of phosphatidylinositol 3-kinase/Akt signaling pathway for regulation of tissue inhibitor of metalloproteinases-3 gene expression by TGF-beta in human chondrocytes. Cell Signal. 2007;19(8):1643–51. doi: 10.1016/j.cellsig.2007.02.007. [DOI] [PubMed] [Google Scholar]