

Abstract
During the last 2 years, our laboratory has worked on the elucidation of the molecular basis of capacitative calcium entry (CCE) into cells. Specifically, we tested the hypothesis that CCE channels are formed of subunits encoded in genes related to the Drosophila trp gene. The first step in this pursuit was to search for mammalian trp genes. We found not one but six mammalian genes and cloned several of their cDNAs, some in their full length. As assayed in mammalian cells, overexpression of some mammalian Trps increases CCE, while expression of partial trp cDNAs in antisense orientation can interfere with endogenous CCE. These findings provided a firm connection between CCE and mammalian Trps. This article reviews the known forms of CCE and highlights unanswered questions in our understanding of intracellular Ca2+ homeostasis and the physiological roles of CCE.
Keywords: calcium, cell signaling, hormone action, ion channels
The two primary second messengers mediating rapid responses of cells to hormones, autacoids, and neurotransmitters are cyclic nucleotides and Ca2+. Cyclic nucleotides act, for the most part, by activating protein kinases. The actions of Ca2+ are more complex, in that this cation acts in two ways: directly, by binding to effector proteins, and indirectly, by first binding to regulatory proteins such as calmodulin, troponin C, and recoverin, which in turn associate and modulate effector proteins. Effector proteins regulated in these manners by Ca2+ include not only protein kinases and protein phosphatases but also phospholipases and adenylyl cyclases, which are signaling enzymes in their own right, and an array of proteins involved in cellular responses that range from muscle contraction to glycogenolysis, endo-, exo-, and neurosecretion, cell differentiation, and programmed cell death. A common mechanism used by hormones and growth factors to signal through cytosolic Ca2+ ([Ca2+]i) is activation of a rather complex reaction cascade that begins with stimulation of phosphoinositide-specific phospholipase C (PLC) enzymes, PLCβ and PLCγ, and is followed sequentially by formation of diacylglycerol plus inositol 1,4,5-trisphosphate (IP3), liberation of Ca2+ from intracellular stores, and finally, entry of Ca2+ from the external milieu. Table 1 lists examples of extracellular signals that activate cellular PLCs and use changes in [Ca2+]i as part of their signaling mechanism.
Table 1.
Examples of stimuli that activate PLCs, increase intracellular Ca2+, and activate CCE
| Ligand | Receptor |
|---|---|
| Due to activation of PLCβ enzymes | |
| Mediated by Gi-type G proteins (pertussis toxin-sensitive) | |
| FMLP | FMLP |
| Interleukin 8 | IL-8 |
| Mediated by Gq-type G proteins (pertussis toxin-insensitive) | |
| Epinephrine | α1-Adrenergic |
| Acetylcholine | M1, M3, M5 |
| Serotonin (5-HT) | 5-HT2a, 2c |
| Histamine | H1 |
| ATP | Pu, Py |
| Arg-vasopressin | V1a, V1b |
| Oxytocin | OT |
| Angiotensin II | AT1 |
| Bradykinin | BK2 |
| Thromboxane | TP |
| Prostaglandin F2α | FP |
| Endothelin | ET-1, ET-2 |
| Colecystokinin | CCK-A |
| Gastrin | CCK-B |
| Gastrin-release peptide (bombesin) | GRP |
| Thyrotropin releasing hormone | TRH |
| Gonadotropin releasing hormone | GnRH |
| Mediated by either Gi- or Gq-type G proteins | |
| Thrombin | Thrombin |
| Platelet activating factor | PAF |
| Due to activation of PLCγ via tyrosine phosphorylation | |
| Antigen | T-cell receptor |
| B-cell receptor | |
| Epidermal growth factor | EGF |
The basic mechanisms used to signal through [Ca2+]i are determined by the fact that the resting level of cytosolic Ca2+ is very low, in the neighborhood of 100 nM, while that in intracellular stores and in the surrounding extracellular milieu is in the neighborhood of 2 mM, that is, ≈10,000-fold higher. As a result, [Ca2+]i is set by the balance of two opposing forces. One is passive influx into the cytoplasm. It is driven by the electrochemical gradient and causes cytosolic [Ca2+]i to rise without expenditure of energy. This influx is carefully controlled both at the level of the plasma membrane and at the level of the membranes, which delimit the internal storage compartment. Entry of Ca2+ from the extracellular space occurs through three classes of Ca2+ permeable gates: voltage-dependent Ca2+ channels, ligand-gated Ca2+-permeable cation channels, and the so-called capacitative calcium entry (CCE) channels. While voltage-dependent Ca2+ channels and ligand-gated cation channels are expressed in selected sets of cells (neurons, myocytes, and endocrine cells; for reviews see refs. 1–3), CCE channels are ubiquitous (see ref. 4). Entry of Ca2+ from internal stores into the cytoplasm occurs through two classes of Ca2+-release channels, the IP3 receptors and the ryanodine receptors, which, depending on the tissue, may coexist (reviewed in ref. 5). Functional and molecular diversity and/or distinguishing pharmacological properties characterize all three types of Ca2+ entry pathways. The second force opposes the first and is the active extrusion of Ca2+ from the cytoplasmic compartment against the electrochemical gradient, either into the stores or out of the cell. This process requires expenditure of metabolic energy. Pumping into the internal stores is mediated by sarcoplasmic endoplasmic reticulum Ca-ATPases, while extrusion from the cell is mediated by plasma membrane Ca-ATPases. This article deals with the molecular nature and regulation of CCE from the extracellular space stimulated by signals that activate phospholipases. It is not a comprehensive review; rather, it concentrates on aspects that have caught our attention during the last 2 years and some of the initial results from our investigations into CCE and its role in the regulation of [Ca2+]i.
Studies with the fluorescent Ca2+ indicator dye Fura2 on the effect of stimulating PLC on [Ca2+]i have revealed a now well-established response pattern: [Ca2+]i shows an initial rise to a peak level, which, in the continued presence of receptor ligand and extracellular Ca2+, decays to a plateau level that is maintained for as long as the cell is exposed to the stimulus. Fig. 1 shows typical temporal [Ca2+]i response pattern of cultured human embryonic kidney HEK 293 cells induced by ATP and carbachol (CCh), which bind to receptors that are coupled by the Gq class of G proteins and activate β-type PLCs (7, 8), and by epidermal growth factor, which exerts its actions through a typical tyrosine kinase receptor, which, among other effects, stimulates γ-type PLCs (9, 10). In each case, [Ca2+]i rises to a peak level that immediately begins to drop to a lower level that has been termed sustained phase of elevated [Ca2+]i. The difference between the starting and the sustained levels of [Ca2+]i varies with the stimulus and with the cell type and lasts for as long as agonist is present. In the example of Fig. 1, this difference is best seen after stimulation of the muscarinic receptor, but would be clearly apparent if [Ca2+]i would have been followed for longer times after addition of either epidermal growth factor or ATP. As seen with CCh, the sustained phase of the transient is agonist-dependent, as illustrated by the fall to basal levels upon washout of CCh. The example also illustrates that response times after agonist or growth factor vary. With G protein-coupled responses, it is fast, while with tyrosine kinase-coupled systems, it is considerably slower, presumably because signaling through tyrosine phosphorylation is slower than signaling through G proteins.
Figure 1.
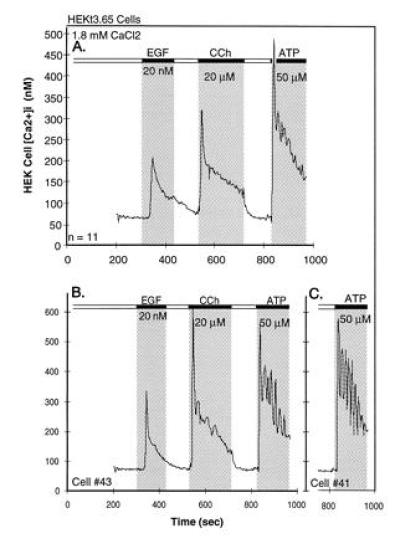
[Ca2+]i transients in HEK-293 cells in response to epidermal growth factor, the muscarinic agonist CCh and the purinergic agonist ATP, as seen in the continuous presence of extracellular Ca2+. (A) Averaged changes in [Ca2+]i, observed in 11 cells. (B) Records of changes in two individual cells of the set. Additions and washouts of the agonists are depicted by the gray areas and the black bars at the top. For details see of [Ca2+]i determination see Zhu et al. (6).
The experiment in Fig. 1 was carried out in the presence of extracellular Ca2+. In the experiment of Fig. 2, the agonist was added in the absence of extracellular Ca2+, and Ca2+ was added subsequently to illustrate activation of the CCE pathway. In the absence of extracellular Ca2+, the initial increase in [Ca2+]i is followed by return to the basal level of [Ca2+]i, the sustained phase of increased [Ca2+]i being absent. Addition of Ca2+ to the extracellular medium after [Ca2+]i has returned to its resting value leads to an increase in [Ca2+]i, due to activation of the CCE pathway. We term the channels that mediate CCE “CCE channels.”
Figure 2.
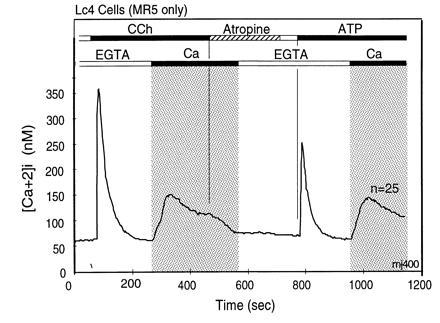
Dissociation of agonist-stimulated [Ca2+]i transients into IP3-induced release and CCE-mediated replenishment components. The first rapid rise does not require extracellular Ca2+ and returns rapidly to the starting [Ca2+]i, due to extrusion of Ca2+ from the cells by plasma membrane Ca-ATPases and reuptake of Ca2+ into stores by sarcoplasmic endoplasmic reticulum Ca-ATPases. The second component, stimulation of CCE, is seen as influx of Ca2+ from the extracellular milieu that occurs upon Ca2+ addition after completion of the first Ca2+ transient. This form of Ca2+ entry is due to stimulation of CCE channels by a signal generated by the IP3-induced store depletion and/or by some other agonist-induced signaling process. [Ca2+]i changes were measured in murine L cells (Lc4 cells) expressing in stable form the M5 muscarinic acetylcholine receptor. Bars on top depict changes in composition of the medium. The lower bars indicate Ca2+ additions; open bar indicates addition of 0.5 mM EGTA to medium without added Ca2+, and the black bar and gray area depicts change to a balanced salt solution containing 1.8 mM CaCl2. The upper bars indicate agonist additions. When added, concentration of CCh was 20 μM and that of ATP was 90 μM. Note that the initial rise of [Ca2+]i after agonist addition in the absence of extracellular Ca2+ returns spontaneously to baseline levels, and also note that addition of extracellular Ca2+ uncovers the presence of an activated influx pathway that causes a robust increase in [Ca2+]i. This influx is referred to as CCE. Note further that in these cells the release of Ca2+ from intracellular stores occurs as a concerted event without hint of oscillations.
A striking aspect of CCE channels is that most share the property of being activated in response to store depletion. Due to this, they have also been called store-operated channels. That store depletion is an activating signal for CCE channels was best inferred from the fact that Ca2+ influx into cells was found to be stimulated when depletion was caused secondary to inhibition of sarcoplasmic endoplasmic reticulum Ca-ATPase pumps, as seen with the tumor promoter thapsigargin or the antioxidant t-butylhydroquinone (refs. 11 and 12; reviewed in ref. 13). Activation by store depletion without involvement of an agonist was shown independently in patch clamp experiments in which the patch pipette was loaded with the Ca2+ chelator bis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetate (BAPTA) acting as a sink for Ca2+ leaking from the stores. In macrophages and lymphocytes, this maneuver leads to the development of a highly selective inward Ca2+ current, ICRAC, which stands for calcium release-activated calcium current (14–16). Attempts to characterize the single channel conductance of the CCE channel responsible for ICRAC have failed, setting the upper limit at <0.2 pS—i.e., at an ≈100-fold lower value than the single channel conductance of a classical voltage-activated calcium channel. Store depletion-activated CCE in COS cells is seen in Fig. 4C.
Figure 4.
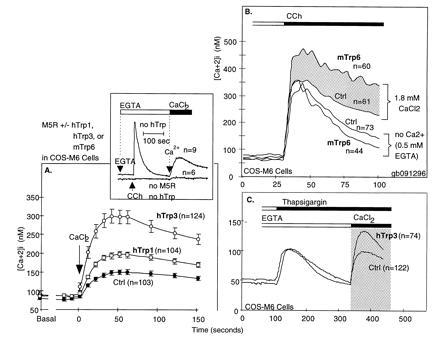
Functional expression of Trp proteins enhances CCE in COS Cells. (A) Enhancement of agonist-stimulated CCE by expression of hTrp3 in COS cells. Note that expression of the Trp protein caused an enhancement of CCE [adapted from Zhu et al. (6)]. (B) Rapid activation of mTrp6-induced Ca2+ influx occurs within the same time frame as IP3-induced Ca2+ release. Note that while, in the absence of external Ca2+, the initial IP3-mediated increase in [Ca2+]i in mTrp6-expressing cells is not significantly different from that seen in cells expressing the M5 receptor alone, the presence of external Ca2+ uncovers rapid stimulation of an additional flux of Ca2+ into the cytoplasm, which we interpret to come from the extracellular space and entering through CCE channels formed of Trp subunits. (C) Trp-based CCE channels are stimulated by store depletion. The figure shows agonist-independent activation of CCE by thapsigargin-induced depletion of Ca2+ stores in COS cells and its enhancement by either wild-type hTrp3.
We define CCE broadly as calcium entry activated by hormones, autacoids, and growth factors, which is apparent upon Ca2+ addition to a cell that has been stimulated in the absence of extracellular Ca2+. Empirically, CCE has been found to be activated secondarily to application of stimuli that cause IP3-induced store depletion, but store depletion is not always a requisite for CCE. By this definition, ICRAC is a form of CCE. Store depletion-independent activation of CCE channels was described by Fasolato et al. (17), also in macrophages. In their experiments, compound 48/80, which stimulates macrophage PLC via a Gi protein (18), stimulated CCE channels under conditions in which store depletion was blocked with heparin. Fluctuation analysis showed this type of CCE channels to have a single channel conductance of ≈50 pS. Since the 50-pS channels did not show up in the fluctuation analysis of store depletion-activated CCE (ICRAC), it must be agonist-activated and store depletion-insensitive. Macrophage agonist-activated CCE differs from macrophage store depletion-activated CCE, not only in the mechanism of activation, but also in that it is a nonselective Ca2+-permeable cation channel instead of a highly Ca2+-selective ion channel. Another type of CCE channel, with properties intermediate between those of the two types of macrophage channels, was described by Lückhoff and Clapham (19) in A431 cells. In these cells, store depletion activates a 16-pS Ca2+-permeable nonselective cation channel. Another form of Ca2+ entry that may or may not be related to classical CCE was described by Ufret-Vicenti et al. (20), who found that pretreatment of cells with thapsigargin may lead to slow development of a caffeine-stimulated Ca2+ influx. The term noncapacitative calcium entry has been used to describe Ca2+ entry that is stimulated by low levels of agonists, induces oscillatory behavior in cells, can be mimicked by arachidonic acid, and does not cause appreciable store depletion (e.g., ref. 21). Numerous studies have now documented the existence of more than one type of Ca2+ entry in response to stimulation of cells by PLC-activating agonists (reviewed in ref. 4; e.g., refs. 22–26). The very different conditions under which Ca2+ entry can be elicited and its different characteristics has led to the conclusion that CCE channels must be complex and heterogeneous in molecular makeup, in mechanism of activation, and in terms of their ion selectivity.
Key questions about CCE channels include: molecular makeup, their mechanism(s) of activation and regulation, and, since under physiologic conditions their activation is preceded by IP3-induced increases in [Ca2+]i, their role in Ca2+ signaling and in modulation of cellular Ca2+ oscillations. Oscillations are a highly variable phenomenon, which has been observed in many nonexcitable cells under conditions where CCE has been activated. In their sustained (continuing) form, they appear to depend on CCE—i.e., on external Ca2+ (27), and the type and abundance of the receptor through which the cells are stimulated (M.J., X.Z., and L.B., unpublished work). In Fig. 1, for example, stimulation of HEK cells by ATP, but not CCh or epidermal growth factor, leads to oscillatory changes in [Ca2+]i. Fundamental to the answers of all these question is the answer to the first: the molecular identity of CCE channels. Experiments from our and other laboratories indicate that CCE channels are made up of subunits encoded in trp genes.
Trps as Elementary CCE Channel Subunits
Role in Invertebrate Photoreceptor Cells.
In contrast to vertebrate visual signal transduction, which leads to activation of a cGMP-specific phosphodiesterase, the target of the G protein activated by light-activated rhodopsin in invertebrates is a β-type PLC, NorpA (28–30). Different also from vertebrate photosignaling is that the electrical response of the invertebrate photoreceptor cells instead of causing membrane hyperpolarization causes membrane depolarization. Accordingly, electroretinograms of compound eyes exposed to a continuous light pulse show a typical light-induced receptor potential with an initial peak followed by a sustained phase that lasts for as long as the eye is subjected to stimulating light. Alterations in electroretinograms of Drosophila eyes were used as a screen to characterize mutants in the photosignaling cascade. Consistent with a mediatory role for PLC, a no receptor potential mutant, NorpA, was found to encode a PLC. NorpA mutants are blind and their eyes lack PLC activity. Minke and colleagues focused on a mutant that has a transient receptor potential, trp (31). These mutants develop a fast depolarizing response but lack the sustained phase of the receptor potential. Hardie and Minke (32) characterized the function of the missing gene as a light-activated Ca2+-permeable channel.
The trp gene was cloned by Montell and Rubin (33) and shown to encode a protein with eight hydrophobic segments that could potentially form transmembrane segments. Independent research in Australia identified and cloned a calmodulin binding protein from Drosophila heads and found it to be a structural homologue of trp, trp-like (trpl; ref. 34). These authors noted that some of the hydrophobic segments of trp and trpl were homologous to transmembrane segments of voltage-dependent Ca2+ and Na+ channels, with the exception that the putative S4 of trp and trpl lacked the positive amino acids responsible for the voltage-sensing function of voltage-dependent ion channels. In 1993, Hardie and Minke (35) formally raised the question of whether the Trp and Trpl proteins might be functional homologues of CCE channels. More recently, Zuker and coworkers (36) showed that a double trp–trpl mutant not only lacks the sustained phase of the electroretinogram, but exhibits no electrical response whatsoever. They concluded that the light-induced depolarization of the invertebrate photoreceptor cell is due to the combined activation of trp- and trpl-encoded channel proteins. Prior studies had shown that the initial depolarizing response is independent of and precedes store depletion (37).
Trp Proteins and CCE in Vertebrate Cells.
Two lines of research have now established that trp genes encode CCE-type channels. The first, developed by Schilling and coworkers (38, 39), showed that when it is expressed in insect Sf9 cells, Drosophila Trp causes appearance of a store depletion-activated Ca2+-selective conductance, while Trpl causes the appearance of a Ca2+-permeable, store depletion-insensitive, rather nonselective cation conductance that tends to activate spontaneously and appears to respond to IP3. Although neither the conductance induced by Trp nor that induced upon expression of Trpl resembled in their ion selectivity the endogenous agonist-activated CCE channel of a nonphotoreceptor insect cell, the results suggested strongly that Trp-related proteins might make up CCE channels.
The second line of research dealt with the search for trp-related genes and investigation of their role in CCE in vertebrates. We, as well as others, indeed found such vertebrate homologues in expressed sequence tag data bases, which led to the molecular cloning of the full-length cDNA encoding human Trp1 (hTrp1; also TRPC1) and a splice variant lacking 34 amino acids in its N-terminal segment (hTrp1Δ34; refs. 40 and 41). Additional searches, both by PCR and of expressed sequence tag data bases, uncovered the existence of six nonallelic trp-related genes in the mouse genome (6), of which four (genes 1, 3, 4, and 6) have also been identified in human mRNA samples. As shown by the phylogenetic analysis of the known Trp proteins, Trp3 and Trp6 and Trp4 and Trp5 are very closely related to each other, reducing vertebrate Trps to four main subfamilies, subfamilies 1–4, where types 3 and 6 belong to the Trp3 class, while types 4 and 5 belong to the Trp4 class of Trps (ref. 6; Fig. 3).
Figure 3.
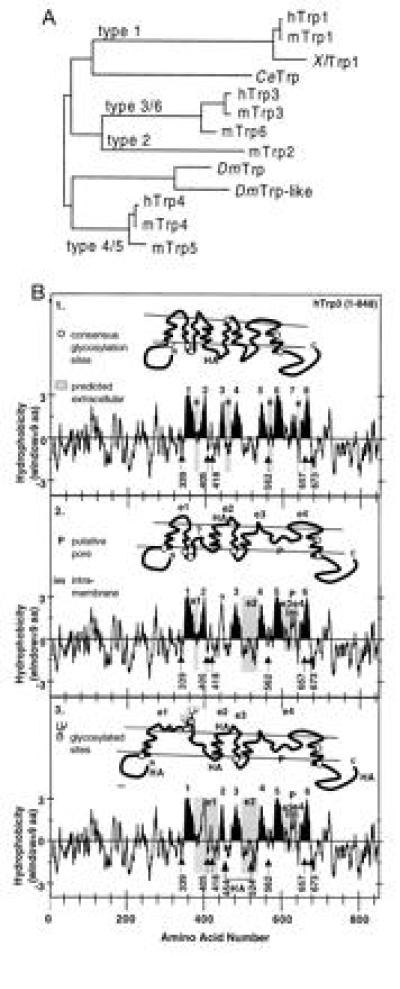
Molecular diversity of 12 Trp sequences as deduced from their corresponding cDNAs and early speculations on secondary structure of CCE channels. (A) Phylogenetic tree obtained by pair-wise comparison of the amino acid sequences that span the putative pore region and the two putative transmembrane segments that flank this region (≈170 amino acids). This assignment assumes a transmembrane topology homologous to that of voltage-gated Ca2+, Na+, and K+ channels (B2 and 3). (B) Kyte–Doolittle plots and models of transmembrane topology of hTrp3 highlighting in black eight or two versions with six hydrophobic regions that may form transmembrane segments based on a possible homologous relation to voltage-gated ion channels. The location of consensus NXS/T glycosylation sites are highlighted (numbers correspond to the asparagines of the NXS/T motifs). In the six-transmembrane models, one of the hydrophobic regions thought not to span the membrane completely, P, is believed to be intramembrane (im) and to form the putative SS1-2 pore region used for the construction of the phylogenetic tree shown in A. e and e1-e4 are sequences predicted by the various models to be exposed to the outside of the cell. Note that, in B2, none of the six consensus glycosylation sites (○) is predicted to be glycosylated, while, in B3, two sites are exposed and presumed to be glycosylated. Locations at which HA epitopes were introduced are also shown.
Two lines of experimental evidence connect mammalian Trp proteins to mammalian CCE channels. The first comes from functional expression in COS, CHO, and HEK cells. In COS cells, hTrp1 and hTrp3 (6) increased CCE (Fig. 4). More recent studies show that murine Trp6 (mTrp6) also increases CCE in COS cells (G.B., X.Z., and L.B., unpublished work). However, these results by themselves did not prove that vertebrate trp genes encode CCE channels. This is because COS cells, like any other cell for that matter, have endogenous CCE channels that respond to both store depletion and agonist stimulation and because detection of functional CCE channels after transfection with a trp involves protocols that activate these endogenous channels.
The second argument for Trp(s) being part of CCE channels is that expression of hTrp1Δ34 (also TRPC1A) in CHO cells (42) and of hTrp3 in HEK 293 cells (Fig. 5) cause the appearance of nonselective Ca2+-permeable cation channels that are not detected in cells transfected with control plasmids. For Trp3, these channels appear to have a single channel conductance of 20 pS, as deduced from noise variance analysis of transmembrane currents recorded under the whole cell configuration (D.P., L.B., and S.E., unpublished work).
Figure 5.
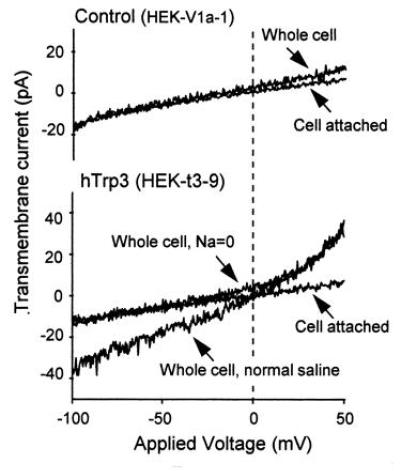
Development of a nonselective cation current in HEK 293 cells expressing human Trp3 after loading with BAPTA. HEK cells were transfected with pcDNA3 carrying either the HA epitope-tagged Trp3 cDNA (hTrp3), or as control, the human type-2 vasopressin receptor cDNA (V2R) using the calcium phosphate/glycerol shock method. G418-resistant colonies were selected and tested for expression of the respective inserts—the HA epitope by immunocytochemistry and the V2R by its ability to stimulate adenylyl cyclase. Clonal cell lines were then isolated by limiting dilution, retested for expression of the proteins, and tested for ionic transmembrane currents using the whole-cell patch clamp technique. The pipette contained 150 mM Cs-aspartate, 4 mM MgCl2, 12 mM Na-BAPTA, and 10 mM Hepes. The extracellular medium contained normal saline: 155 mM NaCl, 2 mM KCl, 2 mM MgCl2, 10 mM glucose, and 10 mM Hepes. Na-free saline contained: 110 mM N-methylglucammonium (NMG)-Mes, 5 mM H-EDTA, 4.3 mM CaCl2, and 10 mM Hepes (free Ca2+, 20 μM). The cells were subjected to a depolarizing ramp from −150 mV to +150 mV. The upper portion shows the records of a control V2R-positive cell; the lower portion shows transmembrane currents of a cell expressing hTrp3. Note presence in the hTrp3-expressing cells of a nonselective conductance that is absent in control HEK cells as well as in the cell attached patch before establishing the whole cell configuration. Exchanging NMG+ for Na+ abolished the inward aspect of the transmembrane current but did not interfere with the outward current carried by Cs+ (data not shown). Other experiments showed that the channel is permeable to Ca2+ as well as Na+.
Unanswered by these results is the question whether Trps are pore-forming subunits of CCE channels or essential regulatory subunits that, when expressed in cells, merely “recruit” the channel proper. Notably in the case of mammalian Trps, the type of CCE channel generated by their expression is a nonselective Ca2+-permeable cation channel, leaving open the possibility that the highly Ca2+-selective channel responsible for ICRAC in macrophages and lymphocytes, may be encoded in an unrelated type of molecule. Against this argument are the results of Sinkins et al. (43) on the ion selectivities of chimeric Trps expressed in Sf9 cells. These authors found that chimera DTrp/DTrpl, composed of the N terminus plus polytransmembrane domain of Drosophila Trp and the C terminus of the Drosophila Trpl, had the ion selectivity of Trp and was insensitive to store depletion, while the reverse chimera (DTrpl/DTrp) had the ion selectivity of Trpl but had acquired Trp’s responsiveness to store depletion. It is difficult to explain how this type of result could be obtained if Trps are not the pore-forming units.
Consistent with the idea that trp genes encode channel subunits and participate as obligatory elements in CCE is the third experimental argument linking vertebrate Trps to CCE: interference with CCE by expression of trp sequences in the antisense direction (ref. 6; Fig. 6). Taken together, the data strongly indicate that CCE channels are composed of Trp subunits.
Figure 6.
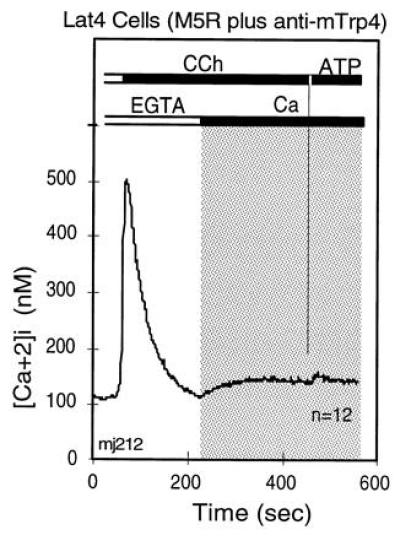
Expression of a 1.2-kb fragment of murine trp4 cDNA in antisense direction inhibits CCE stimulated by cotransfected M5 receptor. Murine L(tk−) cells were transfected with cDNA encoding the M5 receptor alone (Fig. 2) or in combination with a 1.2-kb mTrp4 cDNA fragment transcribed in the antisense direction. Cells were selected for acquisition of G418 resistance encoded in the pcDNA3 vector that carried the M5 receptor cDNA. Lat4 cells were then analyzed for agonist-stimulated Ca2+ transients by video microscopy (6). The figure shows averaged records of changes in [Ca2+]i in Lat4 cells that responded to 20 μM CCh in the absence of external Ca2+ with a typical IP3-induced transient increase in [Ca2+]i, as seen in the absence of extracellular Ca2+. This was followed first by addition of 1.8 mM Ca2+in the presence of CCh, and then of 90 μM ATP in the presence of Ca2+. The addition of external Ca2+ tested for CCh-stimulated CCE and that of ATP for depletion of stores by CCh. Note that CCh indeed depleted the bulk of the intracellular stores, as seen by the failure of the saturating concentration of ATP to elicit a release of Ca2+. Note further that, in spite of full depletion of internal stores, CCE was not activated, indicating interference of the antisense cDNA with expression of one or more components required for CCE.
Although the subunit composition of a CCE channel is not yet known, the limited sequence similarity of some of the hydrophobic regions of Trp to transmembrane segments of voltage-gated Ca2+ channels suggests a tetrameric molecule made up of Trp subunits. Studies on glycosylation of hTrp3 (Fig. 7) and accessibility of the hemagglutinin (HA) epitope, placed in various locations of the protein, to immunostaining in nonpermeabilized cells impose additional constrains and lead us to hypothesize the subunit transmembrane topology depicted in Fig. 3B3. Two tetrameric structures made of Trp subunits are depicted in Fig. 8. The proposed multisubunit structure together with the existence of up to six nonallelic genes in vertebrate genomes offer an easy explanation for functional heterogeneity. Store depletion-activated vs. store depletion-independent CCE channels and highly Ca2+ selective vs. nonselective cation channels are differences that can easily be explained as being due to formation of different homomultimeric or heteromultimeric Trp-based channels.
Figure 7.
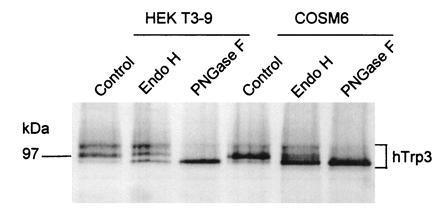
Glycosylation and immunocytochemical analysis of Trp3 suggests the transmembrane topology shown in Fig. 3B3. HEKt3-9 cells expressing HA epitope-tagged hTrp3 in stable form or COS-M6 cells transiently expressing the same Trp, were metabolically labeled with [35S]Met/Cys. Cells were solubilized, and the HA-tagged proteins were immunoprecipitated by incubation with C12A5 monoclonal antibody and protein A-Sepharose, eluted with HA peptide, and either treated or not treated with Endo H or PN-glycosidase F as described in ref. 44. The resulting samples were analyzed by 9% SDS/PAGE, followed by autoradiography. The figure shows a digitally acquired picture of the autoradiogram processed with the aid of photofinish and powerpoint softwares and printed with a Canon CJ10 printer. Note that untreated [35S]hTrp3 runs as a complex set of bands that are converted to a single band of ≈97 kDa by treatment with PN glycosidase F, indicating that protein was glycosylated by the HEK cells.
Figure 8.
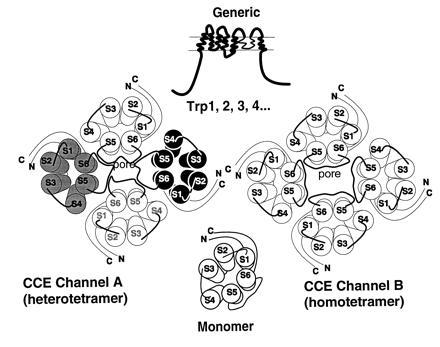
Models of secondary and quaternary structures of CCE channels and their subunits. CCE channels are hypothesized to be made of different Trps and/or different Trp combinations to account for CCE channels with varying ion selectivities and different forms of activation—e.g., store depletion-sensitive vs. store depletion-insensitive. The transmembrane topology chosen is arbitrarily based on that of voltage-gated K+, Na+, and Ca2+ channels and is thus far supported by the finding that hTrp3 is glycosylated and the results on HA localization in expression experiments. However, other transmembrane topologies are possible and the model may need major revisions. Existence of six nonallelic genes encoding six Trp-related proteins, opens the possibility that there are either diverse homomultimeric or diverse heteromultimeric CCE channels that could account for the functional heterogeneity of CCE seen in different tissues and cells.
CCE Channels May Be Activated by Two Distinct Signaling Mechanisms
As is the case for structural organization of CCE channels, there is only scant information available as to how or by what mechanism CCE channels are activated. Among the signaling mechanisms and pathways that have been implicated as mediating activation of CCE are: (i) slowly activating pertussis toxin-sensitive G proteins (45); (ii) small molecular weight GTP-binding proteins (17, 46, 47); (iii) cGMP (48–50); (iv) IP3 (51); (v) tyrosine phosphorylation (52); (vi) activation of cellular phosphatases (53, 54); (vii) arachidonic acid (21, 55); (viii) formation of diffusible messengers of unknown identity (56–58); (ix) direct physical interaction of the CCE channel with an element of the storage compartment, possibly the IP3 receptor itself; and (x) physical translocation of CCE channel containing intracellular vesicles to the plasma membrane. Physical coupling of IP3 receptor to CCE channels is modeled after the mechanism by which the skeletal muscle voltage-sensitive Ca2+ channel activates the sarcoplasmic reticulum Ca2+ release channel, and the translocation model is based on the mechanism by which insulin stimulates glucose uptake in peripheral tissues or that by which antidiuretic hormone via cAMP promotes incorporation of water channels into the apical surface of renal collecting duct cells and thereby increases water reabsorption.
Although it is too early to settle on any one of these mechanisms, the literature clearly suggests that more than one mechanism for regulation of CCE channels must exist, and that in some cases a single CCE channel may be under control of two activating mechanisms. The best evidence for dual regulation comes from studies on visual signal transduction in Drosophila, in which it has now been shown that the initial depolarizing response is due to the combined activation of both Trp and Trpl and that Ca2+ entering through Trp channels is necessary for adaptation to continuous light. Yet, while store depletion activates Trp, as shown upon expression in Sf9 cells (38), the initial depolarizing phase of the receptor potential, which can occur within as little as 10 ms and depends on the presence of Trp and Trpl, arises very rapidly and before any discharge of Ca2+ from stores is detectable. It is thus unlikely to be the result of the generation of a “store depletion” signal (36). Moreover, the time frame is such that it is unlikely that Trp/Trpl activation involves any diffusible messenger or a metabolic reaction such as phosphorylation. Since invertebrates capture light by the same mechanism as do vertebrates (i.e., through photoisomerization of a rhodopsin molecule, which then activates a G protein), it follows that the most likely signal that acts on CCE channels, other than store depletion, diffusible messenger, or phosphorylation, is the activated G protein, either its GTP·α complex or its βγ dimer. The extremely rapid time frame of the activation process also suggests existence of physically precoupled complexes of rhodopsin, G protein, and CCE channels (Fig. 9).
Figure 9.
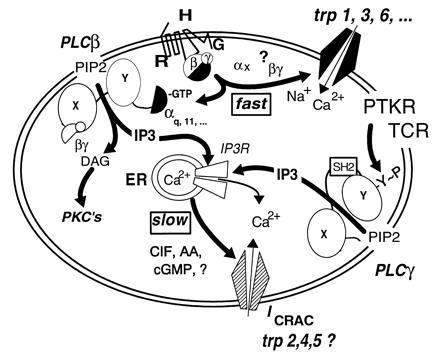
Current hypotheses on the molecular makeup of CCE channels and their regulation. Multiple regulatory pathways can be identified. (i) The activated form of a G protein (Gq or other) directly activates a nonselective CCE channel encoded in Trp proteins, in analogy to activation of Drosophila Trp and Trpl by light. (ii) An activated α subunit of the Gq family or the βγ dimer of any G protein activated by receptor stimulation, stimulate PLCβ to form IP3 and diacylglycerol (DAG). DAG activates cellular PKCs and IP3 promotes Ca2+ release from internal stores through an IP3 receptor, the IP3-activated Ca2+ release channel of the endoplasmic reticulum. Depleted stores then signal to CCE channels of the nonselective and the Ca2+-selective type to allow entry of Ca2+. Whether highly Ca2+-selective CCE channels (ICRAC) are formed of Trps has not yet been determined. Likewise, whether ICRAC channels are regulated both by store depletion and an activated G protein(s) has not been studied. (iii) Receptors coupled by tyrosine kinases either through association with protein tyrosine kinases (PTKs) or by being themselves PTKs, as are the epidermal growth factor and insulin receptors, promote phosphorylation of PLCγ enzymes on tyrosines and thus cause their activation. IP3 formed in this way depletes Ca2+ stores and thus initiates activation of CCE channels. The nature of the store-to-CCE channel signal is not known.
Thus, while it can be taken as an almost foregone conclusion that some CCE channels are regulated directly by a G protein, the mode or mechanism by which CCE channels are activated in a receptor-, G protein-, and IP3-independent form, such as occurs upon store depletion by treatment with thapsigargin or loading of cells with BAPTA, remains obscure, as does which form or forms of CCE are under dual regulation. Some nonselective CCE channels of the type activated by agonist in heparin-loaded macrophages (17) appear to be unresponsive to store depletion, indicating existence of channels responding to G protein only. Whether the opposite situation exists is not known. Drosophila Trp-based channels respond to both the G protein signal and the store depletion signal, as do the recently cloned mammalian Trp-based channels (Figs. 4 and 5).
Conclusion
Even though there are no definitive answers as yet to questions such as what the makeup of a CCE channel actually is or by which mechanism is it activated under which circumstances, it is clear that significant advances are rapidly being made toward achieving this goal. Determining that Trp proteins are part of CCE channels was a critical first step in this process. Knowledge about how CCE channels are regulated will then be the next step for answering the questions such as how a store knows that it is being emptied—i.e., the molecular basis for Ca2+ sensing within the stores. Given that Ca2+ entering cells through CCE is of fundamental physiological importance for processes as disparate as Gs-independent cAMP generation by type III and VIII adenylyl cyclases in the central nervous system (59, 60), the mediation of the mitogenic response of lymphocytes exposed to stimuli that activate the T-cell receptor (61, 62) and sustained secretory responses of endocrine cells, as was shown some years ago by Albert and Tashjian (63), it is to be expected that the unraveling of the mechanisms that regulate CCE will give new insights not only into CCE but also into other Ca2+-dependent processes not yet so well-defined but nevertheless important.
Acknowledgments
This research was supported in part by National Institutes of Health Grants HL45198, DK19318, and AR43411 to L.B., AR38970 to E.S., and DK41244 to M.B., a postdoctoral fellowship from the Canadian Medical Research Council to G.B., and a fellowship from the French Cancer Society to B.V.
Footnotes
Abbreviations: [Ca2+]i, cytosolic Ca2+ concentration; PLC, phospholipase C; IP3, 1,4,5-inositol trisphosphate; CCE, capacitative calcium entry; CCh, carbachol; BAPTA, bis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetate; h, human; m, murine; HA, hemagglutinin.
References
- 1.Birnbaumer L, Campbell K P, Catterall W A, Harpold M M, Hofmann F, Horne W A, Mori Y, Schwartz A, Snutch T P, Tanabe T, Tsien R W. Neuron. 1994;13:505–506. doi: 10.1016/0896-6273(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 2.Perez-Reyes E, Schneider T. Kidney Int. 1995;48:1111–1124. doi: 10.1038/ki.1995.395. [DOI] [PubMed] [Google Scholar]
- 3.Seeburg P H. Trends Neurosci. 1993;16:359–365. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- 4.Fasolato C, Innocenti B, Pozzan T. Trends Pharmacol Sci. 1994;15:77–83. doi: 10.1016/0165-6147(94)90282-8. [DOI] [PubMed] [Google Scholar]
- 5.Striggow F, Ehrlich B E. Curr Opin Cell Biol. 1996;8:490–495. doi: 10.1016/s0955-0674(96)80025-1. [DOI] [PubMed] [Google Scholar]
- 6.Zhu X, Jiang M, Peyton M J, Boulay G, Hurst R, Stefani E, Birnbaumer L. Cell. 1996;85:661–671. doi: 10.1016/s0092-8674(00)81233-7. [DOI] [PubMed] [Google Scholar]
- 7.Smrcka A V, Hepler J R, Brown K O, Sternweis P C. Science. 1991;251:804–807. doi: 10.1126/science.1846707. [DOI] [PubMed] [Google Scholar]
- 8.Taylor S J, Chae H Z, Rhee S G, Exton J H. Nature (London) 1991;350:516–518. doi: 10.1038/350516a0. [DOI] [PubMed] [Google Scholar]
- 9.Margolis B, Rhee S G, Felder S, Mervic M, Lyall R, Levitski A, Ullrich A, Zilberstein A, Schlessinger J. Cell. 1989;57:1101–1111. doi: 10.1016/0092-8674(89)90047-0. [DOI] [PubMed] [Google Scholar]
- 10.Kim H K, Kim J W, Zilberstein A, Margolis B, Kim J G, Schlessinger J, Rhee S G. Cell. 1991;65:435–445. doi: 10.1016/0092-8674(91)90461-7. [DOI] [PubMed] [Google Scholar]
- 11.Thastrup O, Cullen P J, Drobak B K, Hanley M R, Dawson A P. Proc Natl Acad Sci USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwan C Y, Takemura H, Obie J F, Thastrup O, Putney J W., Jr Am J Physiol. 1990;258:C1006–C1015. doi: 10.1152/ajpcell.1990.258.6.C1006. [DOI] [PubMed] [Google Scholar]
- 13.Putney J W, Jr, Bird G S. Endocr Rev. 1993;14:610–631. doi: 10.1210/edrv-14-5-610. [DOI] [PubMed] [Google Scholar]
- 14.Hoth M, Penner R. Nature (London) 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 15.Hoth M, Penner R. J Physiol (London) 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zweifach A, Lewis R S. Proc Natl Acad Sci USA. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fasolato C, Hoth M, Matthews G, Penner R. Proc Natl Acad Sci USA. 1993;90:3068–3072. doi: 10.1073/pnas.90.7.3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura T, Ui M. J Biol Chem. 1985;260:3584–3593. [PubMed] [Google Scholar]
- 19.Lückhoff A, Clapham D E. Biophys J. 1994;67:177–182. doi: 10.1016/S0006-3495(94)80467-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ufret-Vincenty C A, Short A D, Alfonso A, Gill D L. J Biol Chem. 1995;270:26790–26793. doi: 10.1074/jbc.270.45.26790. [DOI] [PubMed] [Google Scholar]
- 21.Shuttleworth T J. J Biol Chem. 1996;271:21720–21725. doi: 10.1074/jbc.271.36.21720. [DOI] [PubMed] [Google Scholar]
- 22.Clementi E, Scheer H, Zacchetti D, Fasolato C, Pozzan T, Meldolesi J. J Biol Chem. 1992;267:2164–2172. [PubMed] [Google Scholar]
- 23.Kass G E N, Chow S C, Gahm A, Webb D-L, Berggren P-O, Llopis J, Orrenius S. Biochim Biophys Acta. 1993;1223:226–233. doi: 10.1016/0167-4889(94)90230-5. [DOI] [PubMed] [Google Scholar]
- 24.Montero M, Garcia-Sancho J, Alvarez J. J Biol Chem. 1994;269:29451–29456. [PubMed] [Google Scholar]
- 25.Byron K, Taylor C W. J Physiol (London) 1995;485:455–468. doi: 10.1113/jphysiol.1995.sp020742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clapham D E. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- 27.Bootman M D, Cheek T R, Moreton R B, Bennet D L, Berridge M J. J Biol Chem. 1994;269:24783–24791. [PubMed] [Google Scholar]
- 28.Devary O, Heichal O, Blumenfeld A, Cassel D, Suss E, Barash S, Rubinstein C T, Minke B, Selinger Z. Proc Natl Acad Sci USA. 1987;84:6939–6943. doi: 10.1073/pnas.84.19.6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Selinger Z, Minke B. Cold Spring Harbor Symp Quant Biol. 1988;53:333–341. doi: 10.1101/sqb.1988.053.01.040. [DOI] [PubMed] [Google Scholar]
- 30.Bloomquist B T, Shortridge R D, Schneuwly S, Perdew M, Montell C, Steller H, Rubin G, Pak W L. Cell. 1988;54:723–733. doi: 10.1016/s0092-8674(88)80017-5. [DOI] [PubMed] [Google Scholar]
- 31.Cosens D J, Manning A. Nature (London) 1969;224:285–287. doi: 10.1038/224285a0. [DOI] [PubMed] [Google Scholar]
- 32.Hardie R C, Minke B. Neuron. 1992;8:643–651. doi: 10.1016/0896-6273(92)90086-s. [DOI] [PubMed] [Google Scholar]
- 33.Montell C, Rubin G M. Neuron. 1989;2:1313–1323. doi: 10.1016/0896-6273(89)90069-x. [DOI] [PubMed] [Google Scholar]
- 34.Phillips A M, Bull A, Kelly L E. Neuron. 1992;8:631–642. doi: 10.1016/0896-6273(92)90085-r. [DOI] [PubMed] [Google Scholar]
- 35.Hardie R C, Minke B. Trends Neurosci. 1993;9:371–376. doi: 10.1016/0166-2236(93)90095-4. [DOI] [PubMed] [Google Scholar]
- 36.Niemeyer B, Suzuki E, Scott K, Jalinik K, Zuker C S. Cell. 1996;83:651–659. doi: 10.1016/s0092-8674(00)81232-5. [DOI] [PubMed] [Google Scholar]
- 37.Ranganthan R, Bakscai B J, Tsien R Y, Zuker C S. Cell. 1994;83:837–848. doi: 10.1016/0896-6273(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 38.Vaca L, Sinkins W G, Hu Y, Kunze D, Schilling W P. Am J Physiol. 1994;267:C1501–C1505. doi: 10.1152/ajpcell.1994.267.5.C1501. [DOI] [PubMed] [Google Scholar]
- 39.Hu Y, Vaca L, Zhu X, Birnbaumer L, Kunze D, Schilling W P. Biochem Biophys Res Commun. 1994;132:346–354. doi: 10.1006/bbrc.1994.1808. [DOI] [PubMed] [Google Scholar]
- 40.Wes P D, Chevesich J, Jeromin A, Rosenberg C, Stetten G, Montell C. Proc Natl Acad Sci USA. 1995;92:9652–9656. doi: 10.1073/pnas.92.21.9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu X, Chu P B, Peyton M, Birnbaumer L. FEBS Lett. 1995;373:193–198. doi: 10.1016/0014-5793(95)01038-g. [DOI] [PubMed] [Google Scholar]
- 42.Zitt C, Zobel A, Obukhov A G, Harteneck C, Kalkbrenner F, Lückhoff A, Schultz G. Neuron. 1996;16:1189–1196. doi: 10.1016/s0896-6273(00)80145-2. [DOI] [PubMed] [Google Scholar]
- 43.Sinkins W G, Vaca L, Hu Y, Kunze D L, Schilling W P. J Biol Chem. 1996;271:2955–2960. doi: 10.1074/jbc.271.6.2955. [DOI] [PubMed] [Google Scholar]
- 44.Innamorati G, Sadeghi H, Birnbaumer M. Mol Pharmacol. 1996;50:467–473. [PubMed] [Google Scholar]
- 45.Berven L A, Hughes B P, Barritt G J. Biochem J. 1994;299:399–407. doi: 10.1042/bj2990399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bird G S J, Putney J W. J Biol Chem. 1993;268:21486–21488. [PubMed] [Google Scholar]
- 47.Jaconi M E, Lew D P, Monod A, Krause K-H. J Biol Chem. 1993;268:26075–26078. [PubMed] [Google Scholar]
- 48.Bahnson T D, Pandol S J, Dionne V E. J Biol Chem. 1993;268:10808–10812. [PubMed] [Google Scholar]
- 49.Gukovskaya A, Pandol S J. Am J Physiol. 1994;266:G350–G356. doi: 10.1152/ajpgi.1994.266.3.G350. [DOI] [PubMed] [Google Scholar]
- 50.Gilon P, Obie J F, Bian X, Bird G S, Putney J W., Jr Biochem J. 1995;311:649–656. doi: 10.1042/bj3110649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong Y, Kunze D, Vaca L, Schilling W P. Am J Physiol. 1995;269:C1332–C1339. doi: 10.1152/ajpcell.1995.269.5.C1332. [DOI] [PubMed] [Google Scholar]
- 52.Sage S O, Sargeant P, Jenner S, Farndale R W. Trends Pharmacol Sci. 1994;15:282. doi: 10.1016/0165-6147(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 53.Sargeant P, Farndale R W, Sage S O. J Biol Chem. 1993;268:18151–18156. [PubMed] [Google Scholar]
- 54.Koike Y, Ozaki Y, Qi R, Satoh K, Kurota K, Yatomi Y, Kume S. Cell Calcium. 1994;15:381–390. doi: 10.1016/0143-4160(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 55.Rzigalinski B A, Blackmore P F, Rosenthal M D. Biochim Biophys Acta. 1996;1299:342–352. doi: 10.1016/0005-2760(95)00224-3. [DOI] [PubMed] [Google Scholar]
- 56.Parekh A B, Terlau H, Stuehmer W. Nature (London) 1993;364:814–815. doi: 10.1038/364814a0. [DOI] [PubMed] [Google Scholar]
- 57.Randriamampita C, Tsien R Y. Nature (London) 1993;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- 58.Randriamampita C, Tsien R Y. J Biol Chem. 1994;269:29–32. doi: 10.1074/jbc.270.1.29. [DOI] [PubMed] [Google Scholar]
- 59.Chiono M, Mahey R, Tate G, Cooper D M F. J Biol Chem. 1995;270:1149–1155. doi: 10.1074/jbc.270.3.1149. [DOI] [PubMed] [Google Scholar]
- 60.Fagan K A, Mahey R, Cooper D M F. J Biol Chem. 1996;271:12438–12444. doi: 10.1074/jbc.271.21.12438. [DOI] [PubMed] [Google Scholar]
- 61.Lewis R S, Cahalan M D. Cell Regul. 1989;1:99–112. doi: 10.1091/mbc.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gardner P, Alcover A, Kuno M, Moigeon P, Weyand C M, Goronzy J, Reinherz E L. J Biol Chem. 1989;269:1058–1076. [PubMed] [Google Scholar]
- 63.Albert P R, Tashjian A H. J Biol Chem. 1984;259:5827–5832. [PubMed] [Google Scholar]