

Abstract
Putative functions of the cellular prion protein, PrPC, include resistance to oxidative stress, copper uptake, cell adhesion, and cell signaling. Here, we report NADPH oxidase-dependent reactive oxygen species (ROS) production and extracellular regulated kinases 1/2 (ERK1/2) phosphorylation on PrPC stimulation in the 1C11 neuroectodermal precursor, in its neuronal differentiated progenies, and in GT1-7 neurohypothalamic and BW5147 lymphoid cells. In neuroprogenitor, hypothalamic, and lymphoid cells, ERK1/2 activation is fully controlled by the NADPH oxidase-dependent ROS production. In 1C11-derived bioaminergic cells, ROS signaling and ERK1/2 phosphorylation are both controlled by Fyn kinase activation, introducing some specificity in PrPC transduction associated with this neuronal context. These data argue for an ubiquitous function of PrPC in cell-redox homeostasis through ROS production.
Although the central role of the cellular prion protein (PrPC) in transmissible spongiform encephalopathies (TSEs) has been recognized for many years, the identification of PrPC normal function(s) still constitutes a great challenge in the field of prion biology. PrPC is a ubiquitous glycoprotein anchored to the cell surface by a glycosyl phosphatidyl inositol (GPI) moiety. It is majorly expressed in neuronal cells where TSE-associated damages occur. Knockout experiments (1) have not unraveled any physiological role of PrPC. However, ex vivo studies using cells from PrP null mice or prion-infected cells revealed alterations in copper metabolism and enhanced susceptibility to oxidative stress (2, 3). Biochemical and structural studies (4) of the copper-binding ability of PrPC bring support to the view of an involvement of PrPC in copper homeostasis. Altogether, these data have led to the assumption that PrPC protects neurons from oxidative damage (5). However, the exact relation between PrPC and oxidative stress remains elusive.
Another set of investigations focuses on the localization of PrPC on the cell membrane, consistent with a possible role of this protein in cell adhesion or cell signaling. Indeed, PrPC binds N-CAM (6), laminin, and the laminin receptor (7–9). Such interactions were assumed to reflect some role of PrPC in neurite extension and maintenance (10) or even in cell survival. The observation that PrP knock-out mice expressing N-terminally truncated PrP suffer from neurodegeneration soon after birth (11) also fits in with the hypothesis that PrPC may control neuronal functions.
In a recent report (12), we showed a caveolin-dependent coupling of PrPC to the tyrosine kinase Fyn. This coupling was evidenced by using the murine 1C11 neuronal cell line. The 1C11 clone behaves as a committed neuroepithelial progenitor that lacks neuron-associated functions (13). On induction, nearly all 1C11 cells develop bipolar extensions, express neuronal markers, such as neurofilaments, N-CAM, γγ-enolase, or synaptophysin, and convert into either 1C115-HT serotonergic or 1C11NE nor-adrenergic cells. In the 1C11 progenitor and throughout differentiation, PrPC, caveolin, and Fyn are endogenously expressed (12, 14). However, the PrPC-dependent Fyn activation is restricted to 1C115-HT and 1C11NE progenies, which have implemented a complete neuronal phenotype including serotonin or noradrenaline synthesis, storage, and transport (12).
In the present study we searched for intracellular targets recruited by PrPC stimulation. Because of the potential link between the prion protein and the cellular redox state, we investigated whether PrPC might be coupled with a reactive-oxygen-species (ROS) production pathway in the 1C11 neuronal differentiation model (13) and in GT1-7 hypothalamic (15) and BW5147 lymphoid (16) cells. Indeed, beyond their role in cell toxicity and oxidative stress, ROS may also behave as chemical mediators in signal transduction processes involved in cell proliferation and/or differentiation (17). We identified NADPH oxidase, a major ROS generator in cells, and extracellular regulated kinases 1/2 (ERK1/2), two mitogen-activated protein kinases (MAPKs), as targets of the PrPC-mediated signaling. Similar responses were observed in neuronal and nonneuronal cells. Moreover, in 1C11 fully differentiated neuronal cells only, an involvement of the Fyn kinase in the control of the PrPC-induced transduction cascade was established, in agreement with the idea of some specificity of PrPC signaling related to the expression of a complete serotonergic or noradrenergic phenotype.
Materials and Methods
Antibodies. Monoclonal PrP-targeted antibodies, SAF61, SAF32, and Bar221, were kindly provided by the Service de Pharmacologie et d'Immunologie (Commissariat à l'Energie Atomique, Saclay, France). The polyclonal 1A8 antibody was from C. Farquhar. SAF61, Bar221, and 1A8 target C-ter epitopes, and SAF32 encompasses the 79–92 PrP-epitope. C-20, a monoclonal antibody against the p47PHOX NADPH oxidase subunit, was purchased from Santa Cruz Biotechnology. A polyclonal antibody recognizing the dually phosphorylated region of the active form of ERK1/2 [pTpY185/187] was from BioSource International (Camarillo, CA).
Cell Culture and Enzyme Inhibition. 1C11 cells were cultured and differentiated along the serotonergic (1C115-HT) or the catecholaminergic (1C11NE) pathway as described (13). GT1-7 cells (15) were grown in DMEM/10% FCS. T lymphocytes, BW5147, and the BW5147-derived Thy1-e mutant deficient for GPI synthesis (16), were grown in RPMI/7% FCS.
Specific enzyme inhibition was performed by pretreating cells at 37°C for 45 min in their culture medium with the inhibitor. The NADPH oxidase activity was inhibited with diphenyleneiodonium (DPI, Sigma). PP2 and PD98059 (Calbiochem) were used to inhibit the Fyn kinase and the MAPK kinase (MEK), respectively.
Reactive Oxygen Species (ROS) Detection by Fluorescence. Extracellular release of ROS from 1C11 precursor cells, their neuronal progenies (1C115-HT and 1C11NE), GT1-7 cells, or T lymphocytes was followed by using the fluorogenic reagent OxyBurst Green H2HFF BSA (Molecular Probes). In a typical assay, cells were washed twice with PBS, supplemented with 1 mM Ca2+ and Mg2+, and preincubated for 2 min at 37°C in the presence of the fluorogenic reagent (10 μg·ml-1). Along cell stimulation with PrP antibodies, the fluorescence was continuously recorded at λ = 528 nm (slit width = 10 nm) with excitation at λ = 488 nm (slit width = 10 nm) by using a Cary Eclipse fluorometer (Varian). Fluorescence measurements with 1C11 or GT1-7 adherent cells and with lymphoid cells were performed in a 96-well microculture plate and in a 1-ml, stirred, thermostated four-optical fluorescence cuvette, respectively.
Preparation of Cytosolic Extracts. 1C11 precursor, 1C115-HT, 1C11NE, GT1-7, and lymphoid cells were washed in PBS with 1 mM Ca2+ and Mg2+ and incubated for 30 min at 4°C in NET lysis buffer [50 mM Tris·HCl (pH 7.4)/150 mM NaCl/5 mM EDTA/1% Triton X-100/1 mM Na3VO4 and a mixture of protease inhibitors; Roche]. Extracts were centrifuged at 14,000 × g for 15 min. Protein concentrations in the supernatant were measured by using the bicinchoninic acid method (Pierce). Cytosolic extracts were stored at -80°C before analysis.
Assessment of ERK1/2 Phosphorylation by Western Blotting. Twenty micrograms of cytosolic proteins were resolved by SDS/10% PAGE and transferred to Immobilon membranes (Millipore). Membranes were blocked in 5% nonfat dried milk and Tris-buffered saline with 0.1% Tween 20 for 1 h at room temperature and then incubated overnight at 4°C with 0.5 μg·ml-1 ERK1/2 [pTpY185/187] primary antibody. Bound antibody was revealed by enhanced chemiluminescence detection (Amersham Pharmacia).
Immunoprecipitation of 32P-Labeled p47PHOX NADPH Oxidase Subunit. 1C11 precursor, 1C115-HT, or 1C11NE cells were washed in PBS and incubated in phosphate-free DMEM containing 50 μCi of 32P per ml per 106 cells for 1 h at 37°C. After cell stimulation with PrP antibodies, 32P-labeled cells were scrapped off into ice-cold NET lysis buffer and centrifuged at 14,000 × g for 15 min at 4°C. The p47PHOX NADPH oxidase subunit was immunoprecipitated overnight at 4°C under gentle mixing by using Sepharose G beads (Amersham Pharmacia) coupled to C-20 antibody. Immunocomplexes were resolved by SDS/10% PAGE and transferred to Immobilon membranes. 32P-labeled p47PHOX was detected by using a PhosphorImager (Molecular Dynamics). For each immunoprecipitate, equal amounts of p47PHOX subunit were loaded on the gel, as estimated by Western blotting by using the C-20 antibody.
Results
PrPC-Dependent NADPH Oxidase Activation Causes ROS Production in Neuronal, Hypothalamic, and Lymphoid Cells. Antibody-mediated ligation was used to mimic an extracellular signal acting on PrPC. On binding of distinct antibodies against PrP, ROS markedly accumulated in 1C11 precursor cells, as revealed by monitoring the fluorescence of OxyBurst Green H2HFF BSA (Molecular Probes) (Fig. 1a). The PrPC-mediated ROS release became detectable as soon as 5 min after antibody addition and reached a plateau after 40 min. This response resembles that observed on exposure of 1C11 cells to the phorbol ester PMA, a potent activator of NADPH oxidase through PKCs (ref. 18 and data not shown). ROS production was also observed with 1C115-HT and 1C11NE differentiated cells (Fig. 1a). PrPC ligation also elicited a ROS signal in GT1-7 hypothalamic cells (Fig. 1a). The intensity of the ROS response was in the same order of magnitude as that monitored in the 1C11 cell line.
Fig. 1.

PrPC-induced ROS production through NADPH oxidase activity. Extracellular release of ROS was detected by using the fluorogenic reagent OxyBurst Green H2HFF BSA. (a Inset) The PrPC-mediated ROS production was followed during 60 min with 1C11 precursor cells stimulated by 10 μg·ml-1 of PrP-targeted antibodies 1A8 (•) or Bar221 (▴). Background ROS production was also recorded with unstimulated cells (▪). (a) Antibodies SAF61, Bar221, 1A8, and SAF32 were used at 10 μg·ml-1 to elicit ROS production with 1C11 precursor cells, either 1C115-HT serotonergic or 1C11NE noradrenergic cells, and with GT1-7 hypothalamic cells. ROS release was completed within 45 min of stimulation. ROS levels at 45 min are expressed in fluorescence intensity per mg of protein. The basal level of ROS production was substracted to the shown signals of PrPC-induced ROS release. (b) ROS release induced by Bar221-mediated PrPC ligation was also monitored with the BW5147 lymphoid cell line. As a control, ROS production was followed in the BW5147-derived Thy1-e mutant, deficient for GPI synthesis. (c) PrPC stimulation induces NADPH oxidase p47PHOX phosphorylation. After metabolic labeling with 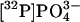 , 1C11 precursor, 1C115-HT and 1C11NE cells were stimulated with 1A8 antibody for 1 h. 32P-labeled p47PHOX (47 kDa) was immunoprecipitated from cytosolic extracts and detected as described in Materials and Methods. Control experiments (Ctrl) without antibody stimulation are also shown. (d) Involvement of NADPH oxidase in the PrPC-induced ROS production was confirmed by using DPI, a specific inhibitor of NADPH oxidase activity. 1C11 precursor cells were preincubated with DPI concentrations ranging from 1 to 100μM and exposed to PrP antibodies in the presence of DPI. ROS release was followed over a period of 45 min. Shown are the mean ROS release velocities deduced from these experiments. Values of fluorescence intensity per mg of protein per time unit are normalized with respect to the value in the absence of DPI (100%). Data shown are representative of a set of three independent experiments.
, 1C11 precursor, 1C115-HT and 1C11NE cells were stimulated with 1A8 antibody for 1 h. 32P-labeled p47PHOX (47 kDa) was immunoprecipitated from cytosolic extracts and detected as described in Materials and Methods. Control experiments (Ctrl) without antibody stimulation are also shown. (d) Involvement of NADPH oxidase in the PrPC-induced ROS production was confirmed by using DPI, a specific inhibitor of NADPH oxidase activity. 1C11 precursor cells were preincubated with DPI concentrations ranging from 1 to 100μM and exposed to PrP antibodies in the presence of DPI. ROS release was followed over a period of 45 min. Shown are the mean ROS release velocities deduced from these experiments. Values of fluorescence intensity per mg of protein per time unit are normalized with respect to the value in the absence of DPI (100%). Data shown are representative of a set of three independent experiments.
To possibly enlarge this observation to cell systems of nonneuronal origin, experiments were performed with BW5147, a T lymphoid cell line, which expresses PrPC at its surface (data not shown), and with the Thy1-e cell line, a glycosylation mutant derived from the BW5147 cell line and deficient for GPI synthesis (16). PrPC activation on BW5147 cells triggered a burst of ROS (Fig. 1b). No significant response was obtained with the Thy1-e cell line lacking PrPC at its cell surface (Fig. 1b). The PrPC-dependent coupling therefore appears not to be restricted to neuronal or neuroendocrine cells but to also participate to the signaling networks of lymphoid cells.
Because NADPH oxidase is known to be a powerful ROS generator, we investigated whether this enzyme was involved in the PrPC-mediated ROS response. NADPH oxidase is a multi-component protein, whose recruitment involves phosphorylation steps and participation of members of the Ras superfamily of small G proteins (19). We observed that the binding of specific antibodies to PrPC on 1C11 cells markedly raised the level of phosphorylation of p47PHOX, a NADPH oxidase subunit described as a substrate of PKCs (20). Similar effects were obtained whatever the differentiation state of the 1C11 cells (Fig. 1c). Exposure to diphenyleneiodonium (DPI), a selective inhibitor of NADPH oxidase, cancelled the PrPC-induced ROS production in 1C11 precursor cells (Fig. 1d). Comparable results were obtained with 1C11-derived bioaminergic neurons and with GT1-7 and BW5147 cells (data not shown). Altogether, these data identify a NADPH oxidase-dependent ROS production pathway as a feature of PrPC signaling activity.
PrPC Stimulation Promotes Phosphorylation of ERK1/2 in Neuronal and Lymphoid Cells. A large number of signaling pathways appears to be regulated by ROS. The broad range of ROS action includes kinase activation, down-regulation of phosphatase activity and direct regulation of transcription factors (17). We focused our search of downstream targets of the PrPC-NADPH cascade on members of the MAPK family of serine/threonine kinases. Indeed, the redox-mediated activation of MAPK has been established in a number of studies (17). Three distinct groups of kinases belong to the MAPK family: ERKs or MAPK which are involved in cell proliferation and differentiation, c-Jun NH2-terminal kinase and p38MAPK. The two latter kinases are implicated in inflammatory responses, cell cycle arrest, DNA repair, and cell death.
In 1C11 cells, the antibody-mediated ligation of PrPC did not elicit any modification in the phosphorylation level of c-Jun NH2-terminal kinase or of p38MAPK, at least within 30 min (data not shown). By contrast, by 5 min after PrPC ligation by various antibodies, phosphorylation of both p44ERK1 and p42ERK2 was promoted (Fig. 2a). This ERK stimulation was transient. ERK phosphorylation returned to its basal level by 30 min after antibody ligation (data not shown). Similar effects on phosphorylation of p44ERK1 and p42ERK2 were observed in 1C115-HT and 1C11NE neuronal cells (Fig. 2b). PrPC-induced ERK activation also occurred in GT1-7 hypothalamic cells and in the BW5147 lymphoid cell line (Fig. 2b). As expected, PrP-antibodies had no effect on the Thy1-e cell line (Fig. 2b).
Fig. 2.

PrPC stimulation induces MEK-ERK1/2 phosphorylation. 1C11 precursor, 1C115-HT serotonergic and 1C11NE noradrenergic cells, GT1-7 hypothalamic cells, and BW5147 and Thy1-e lymphoid cell lines were stimulated for 5 min by using various PrP-targeted antibodies (10 μg·ml-1) or PMA (1 μM), a potent activator of PKCs. (a) Stimulated 1C11 progenitor cells lysates were immunoblotted by using ERK1/2 [pTpY185/187] antibody. Control experiments (Ctrl) with unstimulated cells were also performed. (b) ERK1/2 phosphorylation profiles of 1C115-HT, 1C11NE, and GT1-7 cells and of BW5147 and Thy1-e lymphoid cell lines are compared. All cells were submitted to stimulation by the SAF61 antibody. Also shown are the control experiments without antibody addition. (c) Selective inhibition of MEK1/2 abrogates PrPC-induced ERK activation. Before PrPC–antibody stimulation, cells were preincubated for 45 min in the presence of PD98059, a selective inhibitor of MEK (5 μM with 1C11 precursor, 1C115-HT and 1C11NE cells and with BW5147 T lymphocytes and 20 μM with GT1-7 cells). Cytosolic extracts were immunoblotted by using the ERK1/2 [pTpY185/187] antibody. In all sets of experiments, it was checked that the PD98059 concentration used did not impair cell viability according to trypan blue staining. Data shown are representative of a set of three independent experiments.
To examine a possible role of MEKs in the PrPC-induced ERK1/2 signaling cascade, we used PD98059, a specific inhibitor of MEK1/2. With 1C11 precursor cells and their bioaminergic progenies, as well as with GT1-7 hypothalamic and BW5147 lymphoid cells, the addition of this inhibitor completely cancelled ERK1/2 phosphorylation (Fig. 2c). We therefore concluded that MEK1/2 activation mediates the PrPC signal targeting ERK1/2.
In 1C11 Precursor, GT1-7, and BW5147 Cells, PrPC-Mediated ERK1/2 Phosphorylation Is Strictly Controlled by NADPH Oxidase Activity. In all cell types examined here, NADPH oxidase and the MEK/ERK1/2 module are downstream intracellular targets of the PrPC-induced signaling. We wondered whether ROS could take part in ERK activation. The idea of an involvement of ROS in the PrPC-dependent ERK activation pathway was indeed supported by our observation that PMA increases ERK phosphorylation in the 1C11 and GT1-7 cell lines as well as in T lymphocytes. Moreover, PMA action on ERK phosphorylation was switched off with DPI, an inhibitor of NADPH oxidase (Fig. 3a).
Fig. 3.

ROS involvement in the PrPC-induced ERK1/2 phosphorylation. (a) NADPH oxidase implication in the PrPC-ERK1/2 activation pathway was assessed in 1C11 precursor, 1C115-HT and 1C11NE cells, in GT1-7 hypothalamic cells and in BW5147 and Thy1-e lymphoid cell lines. Cells were incubated for 45 min in the presence of DPI (100 μM with 1C11 cells and its differentiated progenies, 25 μM with GT1-7 hypothalamic cells, 5 μM with BW5147, and 1 μM with Thy1-e) and then stimulated for 5 min by addition of SAF61 or PMA in the presence of DPI. Cytosolic extracts were immunoblotted by using the ERK1/2 [pTpY185/187] antibody. (b) Fyn tyrosine kinase implication in the PrPC-ERK1/2 signal transduction pathway was assessed in 1C11 precursor and in either 1C115-HT or 1C11NE cells. Cells were preincubated for 45 min with 50 pM PP2 before SAF61-mediated stimulation. ERK1/2 phosphorylation was detected as above. (c) Fyn tyrosine kinase involvement in the PrPC-mediated NADPH oxidase recruitment was assessed in 1C11 precursor and in 1C115-HT cells. Before SAF61 stimulation, cells were treated with PP2 concentrations ranging from 5 pM to 0.5 nM. PrPC-induced ROS production was monitored during 45 min as described in Fig. 1d. In all sets of experiments, it was checked that the DPI or PP2 concentrations used did not impair cell viability according to trypan blue staining. Data shown are representative of a set of three independent experiments.
In 1C11 precursor, GT1-7 hypothalamic and BW5147 T cells, the PrPC-dependent ERK phosphorylation was also quenched on addition of DPI (Fig. 3a). However, as shown in Fig. 3a, in 1C115-HT cells, nearly 40% of the PrPC-induced ERK1/2 phosphorylation persisted despite DPI addition. In 1C11NE cells, >90% of the PrPC-dependent ERK1/2 phosphorylation escaped the PrPC-NADPH oxidase cascade. To account for these observations, one must admit that, in 1C11 cells harboring a complete serotonergic or noradrenergic phenotype, alternative signaling pathway(s), independent of NADPH oxidase, also contribute(s) to the PrPC-induced ERK1/2 phosphorylation.
In 1C11-Derived Serotonergic or Noradrenergic Neurons, the PrPC-Fyn Coupling Exerts an Upstream Control on ROS Production and ERK1/2 Activation. The identification of a PrPC-Fyn coupling in the 1C11 cell line prompted us to examine whether Fyn was involved in the PrPC-ERK signaling pathways. In either 1C115-HT or 1C11NE cells, inhibition of the Fyn kinase with PP2 (50 pM) fully cancelled the PrPC-related ERK signal (Fig. 3b). PP2 addition also quenched the PrPC-mediated ROS production (Fig. 3c). In contrast, in 1C11 precursor cells, PP2 treatment (50 pM) neither reduced the amplitude of ERK1/2 phosphorylation (Fig. 3b) nor interfered with the PrPC-dependent NADPH oxidase activation (Fig. 3c). These results are in good agreement with our previous observation that the PrPC-Fyn coupling is restricted to 1C11 cells which have acquired neuronal and neurotransmitter-associated functions (12). In both serotonergic and noradrenergic 1C11-derived cells, recruitment of the tyrosine kinase Fyn hence appears to be a prerequisite to both NADPH oxidase activation and ERK1/2 phosphorylation.
Discussion
The biological function(s) of prion protein is fervently debated and still enigmatic. The prime evidence of a PrPC-Fyn coupling mediated by caveolin in an in vitro inducible neuronal cell line (1C11) lent support to the hypothesis that PrPC acts as a cell surface signaling molecule. The aim of the present study was to identify ubiquitous intracellular responses mediated by PrPC stimulation. Because the physiological signals acting on PrPC remain unidentified, the experimental approach was to mimic an extracellular ligand with PrP-targeted antibodies. We show that antibody-mediated PrP ligation promotes NADPH oxidase activation and ERK1/2 phosphorylation in the 1C11 neuroectodermal progenitor and its neuronal derivatives, in GT1-7 hypothalamic and BW5147 lymphoid cells. These results indicate for the first time that PrPC signaling activity is not restricted to neuronal cells. Moreover, common intracellular targets are recruited by the PrPC-mediated signals whatever the cell type studied here. Finally, the nature of the signaling intermediates involved in the PrPC transduction cascade varies depending on the cell context. In both differentiated 1C115-HT and 1C11NE cells, which have acquired the overall neurotransmitter-associated functions of serotonergic or noradrenergic neurons, Fyn activation controls the downstream targets, thereby constituting a level of specificity in PrPC signaling.
One main observation is the ROS generation through NADPH oxidase activation on PrPC ligation. ROS act as “second message” signals and control the activation of the MEK-ERK1/2 module (Fig. 4). In 1C11 precursor, GT1-7 and BW5147 cells, the NADPH oxidase-dependent ROS signaling fully governs the PrPC-induced ERK pathway. By analogy with the mode of action of PMA on ROS production, PKCs are likely to participate to the PrPC-dependent NADPH oxidase recruitment at the plasma membrane through direct phosphorylation of the NADPH oxidase p47PHOX cytosolic subunit (18). Because PrPC is anchored to the outer membrane, the PrPC-dependent signaling mechanisms engaging the linear PKC-NADPH oxidase-MEK-ERK1/2 cascade are likely to also involve yet-to-be identified cellular partners (Fig. 4). In 1C11-derived bioaminergic neurons, we find that Fyn governs all of the PrPC-induced pathways that converge to the MEK-ERK1/2 module. A first pathway clearly involves the ROS signaling. The second one is NADPH oxidase-independent and possibly involves the Shc-Grb2/SOS-Ras-MEK cascade (21–23) (Fig. 4). In line with this idea, in integrin signaling, a Ras-ERK pathway has been shown to depend on the recruitment of the caveolin-Fyn complex (24). Moreover, Grb2 has been identified as a PrP-interacting protein (25).
Fig. 4.

Model of PrPC-dependent signaling pathways in 1C11 precursor cells, in 1C11-derived serotonergic (1C115-HT) or noradrenergic (1C11NE) cells, in GT1-7 hypothalamic cells and in BW5147 T lymphocytes. See text for further details.
In fully differentiated 1C11 progenies, caveolin appears to be one of the protagonists involved in PrPC-coupling to the tyrosine kinase Fyn (12). Some cell specificity of PrPC signaling may hence be related to the onset of a PrPC-caveolin-Fyn complex. Neither GT1-7 hypothalamic (26) nor BW5147 lymphoid cells (27) appear to express caveolin. Therefore, in these cells, the direct control of NADPH oxidase on PrPC-induced ERK activation, within a single transduction pathway, may be accounted for by the lack of a functional PrPC-caveolin-Fyn ternary complex. In 1C11 precursor cells, despite the presence of caveolin, the phosphorylation level of Fyn is not sensitive to PrPC stimulation. Actually, in these cells, PrPC was shown not to physically and functionally interact with caveolin. In contrast, in fully differentiated serotonergic or noradrenergic progenies, which express the overall neurotransmitter-associated functions of bioaminergic neurons, PrPC-caveolin interaction and Fyn activation were shown to occur preferentially at neurites (12, 28). Therefore, in these bioaminergic neuronal cells, one can imagine a localization of PrPC, caveolin and Fyn within specialized microdomains known to concentrate other signaling effectors (29, 30). Potential partners would include the adaptator molecule Shc and the signaling proteins Ras, Raf, MEK and ERK (22, 23). The targeting of PrPC in such caveolin-rich microdomains would thus expand the repertoire of PrPC downstream pathways. At this stage, one difficulty is to explain why the PrPC-dependent signaling is fully governed by Fyn in 1C11-derived neuronal cells. One would expect that the PrPC molecules on the cell bodies elicit a Fyn–independent response, similar to that observed in the precursor cells. Possibly, in a neuronal fully differentiated context, prion proteins on the cell stroma are switched off from any signaling function or involved in transduction pathway(s) distinct from that described here. Another idea is that, among the PrPC isoforms, those competent for signaling are selectively targeted to neuritic varicosities known as major sites of signal transduction.
To conclude, by linking ERKs phosphorylation to PrPC, the present study strengthens the view that PrPC activation may participate to the survival of neurons and also plays a role in lymphoid cell homeostasis. Indeed, ERKs have emerged as key mediators of cell survival and as proliferation signals (31). Our results also shed some new light on the relationship between PrPC and ROS. In this work, emphasis has been mainly placed on PrPC-dependent production of ROS acting as signaling molecules. The demonstration of a link between PrPC and cellular ROS enables to establish a bridge between PrP and the redox state of the cell. Actually, enhanced susceptibility to oxidants of prion-infected or PrP-deficient cells has been observed (3, 32). Clearly, in such cases, an alteration of PrPC function is relatable to some loss in the control of the cell redox equilibrium (5, 33). Moreover, induced-oxidative stress has been reported to trigger a sustained activation of ERK, itself promoting neuronal cell death (34, 35). Hence, the occurrence of a PrPC-mediated NADPH oxidase coupling provides foundation for uncovering whether prion infection contributes to neurode-generation through modifications of ROS production and ERK stimulation. Hopefully, the 1C11 and the GT1-7 cell systems, which combine PrPC signaling activity and susceptibility to prions (unpublished data, 36), will help to challenge these ideas.
Acknowledgments
We thank Professor J. M. Launay for constant support and fruitful discussions, Professor S. Blanquet for critical reading of the manuscript, and Dr. André Sobel (Institut National de la Santé et de la Recherche Médicale, Paris) for the kind gift of GT1-7 cells. S.M.-R. is a member of the Direction Générale de l'Alimentation, Ministère de l'Agriculture. This work was supported by grants from the Groupement d'intérêt scientifique “Infections à prions.”
Abbreviations: ROS, reactive oxygen species; ERK1/2, extracellular regulated kinases 1/2; PrPC, cellular prion protein; DPI, diphenyleneiodonium chloride; PMA, phorbol 12-myristate 13-acetate; 1C115-HT, 1C11 serotonergic cells; 1C11NE, 1C11 noradrenergic cells; MAPK, mitogen-activated protein kinase; MEK, MAPK kinase.
References
- 1.Raeber, A. J., Brandner, S., Klein, M. A., Benninger, Y., Musahl, C., Frigg, R., Roeckl, C., Fischer, M. B., Weissmann, C. & Aguzzi, A. (1998) Brain Pathol. 8, 715-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown, D. R., Qin, K., Herms, J. W., Madlung, A., Manson, J., Strome, R., Fraser, P. E., Kruck, T., von Bohlen, A., Schulz-Schaeffer, W., et al. (1997) Nature 390, 684-687. [DOI] [PubMed] [Google Scholar]
- 3.Brown, D. R., Schulz-Schaeffer, W. J., Schmidt, B. & Kretzschmar, H. A. (1997) Exp. Neurol. 146, 104-112. [DOI] [PubMed] [Google Scholar]
- 4.Kretzschmar, H. A., Tings, T., Madlung, A., Giese, A. & Herms, J. (2000) Arch. Virol. Suppl. 239-249. [DOI] [PubMed]
- 5.Milhavet, O. & Lehmann, S. (2002) Brain Res. Brain Res. Rev. 38, 328-339. [DOI] [PubMed] [Google Scholar]
- 6.Schmitt-Ulms, G., Legname, G., Baldwin, M. A., Ball, H. L., Bradon, N., Bosque, P. J., Crossin, K. L., Edelman, G. M., DeArmond, S. J., Cohen, F. E. & Prusiner, S. B. (2001) J. Mol. Biol. 314, 1209-1225. [DOI] [PubMed] [Google Scholar]
- 7.Rieger, R., Edenhofer, F., Lasmezas, C. I. & Weiss, S. (1997) Nat. Med. 3, 1383-1388. [DOI] [PubMed] [Google Scholar]
- 8.Graner, E., Mercadante, A. F., Zanata, S. M., Forlenza, O. V., Cabral, A. L., Veiga, S. S., Juliano, M. A., Roesler, R., Walz, R., Minetti, A., et al. (2000) Brain Res. Mol. Brain Res. 76, 85-92. [DOI] [PubMed] [Google Scholar]
- 9.Gauczynski, S., Peyrin, J. M., Haik, S., Leucht, C., Hundt, C., Rieger, R., Krasemann, S., Deslys, J. P., Dormont, D., Lasmezas, C. I. & Weiss, S. (2001) EMBO J. 20, 5863-5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martins, V. R., Mercadante, A. F., Cabral, A. L., Freitas, A. R. & Castro, R. M. (2001) Braz. J. Med. Biol. Res. 34, 585-595. [DOI] [PubMed] [Google Scholar]
- 11.Shmerling, D., Hegyi, I., Fischer, M., Blattler, T., Brandner, S., Gotz, J., Rulicke, T., Flechsig, E., Cozzio, A., von Mering, C., et al. (1998) Cell 93, 203-214. [DOI] [PubMed] [Google Scholar]
- 12.Mouillet-Richard, S., Ermonval, M., Chebassier, C., Laplanche, J. L., Lehmann, S., Launay, J. M. & Kellermann, O. (2000) Science 289, 1925-1928. [DOI] [PubMed] [Google Scholar]
- 13.Mouillet-Richard, S., Mutel, V., Loric, S., Tournois, C., Launay, J. M. & Kellermann, O. (2000) J. Biol. Chem. 275, 9186-9192. [DOI] [PubMed] [Google Scholar]
- 14.Mouillet-Richard, S., Laurendeau, I., Vidaud, M., Kellermann, O. & Laplanche, J. L. (1999) Microbes Infect. 1, 969-976. [DOI] [PubMed] [Google Scholar]
- 15.Radovick, S., Wray, S., Lee, E., Nicols, D. K., Nakayama, Y., Weintraub, B. D., Westphal, H., Cutler, G. B., Jr., & Wondisford, F. E. (1991) Proc. Natl. Acad. Sci. USA 88, 3402-3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chapman, A., Fujimoto, K. & Kornfeld, S. (1980) J. Biol. Chem. 255, 4441-4446. [PubMed] [Google Scholar]
- 17.Sauer, H., Wartenberg, M. & Hescheler, J. (2001) Cell. Physiol. Biochem. 11, 173-186. [DOI] [PubMed] [Google Scholar]
- 18.Kawakami, N., Kita, K., Hayakawa, T., Yamaguchi, T. & Fujimoto, S. (2000) Biol. Pharm. Bull. 23, 1100-1104. [DOI] [PubMed] [Google Scholar]
- 19.Hancock, J. T., Desikan, R. & Neill, S. J. (2001) Biochem. Soc. Trans. 29, 345-350. [DOI] [PubMed] [Google Scholar]
- 20.Fontayne, A., Dang, P. M., Gougerot-Pocidalo, M. A. & El-Benna, J. (2002) Biochemistry 41, 7743-7750. [DOI] [PubMed] [Google Scholar]
- 21.Abe, J. & Berk, B. C. (1999) J. Biol. Chem. 274, 21003-21010. [DOI] [PubMed] [Google Scholar]
- 22.Sternberg, P. W. & Schmid, S. L. (1999) Nat. Cell Biol. 1, E35-E37. [DOI] [PubMed] [Google Scholar]
- 23.Roy, S., Luetterforst, R., Harding, A., Apolloni, A., Etheridge, M., Stang, E., Rolls, B., Hancock, J. F. & Parton, R. G. (1999) Nat. Cell Biol. 1, 98-105. [DOI] [PubMed] [Google Scholar]
- 24.Wary, K. K., Mariotti, A., Zurzolo, C. & Giancotti, F. G. (1998) Cell 94, 625-634. [DOI] [PubMed] [Google Scholar]
- 25.Spielhaupter, C. & Schatzl, H. M. (2001) J. Biol. Chem. 276, 44604-44612. [DOI] [PubMed] [Google Scholar]
- 26.Prioni, S., Loberto, N., Prinetti, A., Chigorno, V., Guzzi, F., Maggi, R., Parenti, M. & Sonnino, S. (2002) Neurochem. Res. 27, 831-840. [DOI] [PubMed] [Google Scholar]
- 27.Alonso, M. A. & Millan, J. (2001) J. Cell Sci. 114, 3957-3965. [DOI] [PubMed] [Google Scholar]
- 28.Kellermann, O., Lafay-Chebassier, C., Ermonval, M., Lehmann, S. & Mouillet-Richard, S. (2002) C. R. Acad. Sci. Ser. III 325, 9-15. [DOI] [PubMed] [Google Scholar]
- 29.Anderson, R. G. & Jacobson, K. (2002) Science 296, 1821-1825. [DOI] [PubMed] [Google Scholar]
- 30.Gorodinsky, A. & Harris, D. A. (1995) J. Cell Biol. 129, 619-627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grewal, S. S., York, R. D. & Stork, P. J. (1999) Curr. Opin. Neurobiol. 9, 544-553. [DOI] [PubMed] [Google Scholar]
- 32.White, A. R., Collins, S. J., Maher, F., Jobling, M. F., Stewart, L. R., Thyer, J. M., Beyreuther, K., Masters, C. L. & Cappai, R. (1999) Am. J. Pathol. 155, 1723-1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee, D. W., Sohn, H. O., Lim, H. B., Lee, Y. G., Kim, Y. S., Carp, R. I. & Wisniewski, H. M. (1999) Free Radical Res. 30, 499-507. [DOI] [PubMed] [Google Scholar]
- 34.Stanciu, M. & DeFranco, D. B. (2002) J. Biol. Chem. 277, 4010-4017. [DOI] [PubMed] [Google Scholar]
- 35.Kulich, S. M. & Chu, C. T. (2001) J. Neurochem. 77, 1058-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schatzl, H. M., Laszlo, L., Holtzman, D. M., Tatzelt, J., DeArmond, S. J., Weiner, R. I., Mobley, W. C. & Prusiner, S. B. (1997) J. Virol. 71, 8821-8831. [DOI] [PMC free article] [PubMed] [Google Scholar]