

Abstract
We examined the anion binding behavior of the Mg(Mn) site in cytochrome c oxidase to test a possible role of this center in proton pumping. Rhodobacter sphaeroides grown in a Mn(II)-rich medium replaces the intrinsic Mg(II) ion with an EPR-detectable Mn(II) ion without change in activity. Due to its close proximity and a shared ligand, oxidized CuA is spin-coupled to the Mn(II) ion, affecting the EPR spectrum. An examination of both bovine and R.s. oxidase crystal structures reveals a hydrogen-bonding pattern in the vicinity of the Mg(II) site that is consistent with three water ligands of the Mg(Mn) center when CuA is oxidized. In the reduced structure, one water molecule in the vicinity of the CuA ligand, E198, moves closer, appearing to be converted into an ionically bonded hydronium ion, while a second water molecule bonded to Mg(Mn) shows evidence of conversion to a hydroxide. The implied proton movement is proposed to be part of a redox-linked export of a pumped proton from the binuclear center into the exit pathway. To test the model, cyanide and azide were added to the oxidized and reduced forms of the enzyme, and Mn(II) CW-EPR and ESEEM spectra were recorded. Addition of azide broadened the CW-EPR spectra for both oxidized and reduced enzyme. Cyanide addition affected the Mn(II) CW-EPR spectrum of reduced cytochrome c oxidase by increasing Mn(II) zero field splitting and broadening the spectral line shapes but had no effect on oxidized enzyme. ESEEM measurements support a differential ability of Mn(II) to bind cyanide in the reduced state of cytochrome c oxidase. This new observation of anion binding at the Mg/Mn site is of interest in terms of accessibility of the buried site and its potential role in redox-dependent proton pumping.
Cytochrome c oxidase is a proton pump, and this pumping activity is a property of other members of the heme–copper oxidase superfamily (1, 2). Despite an extensive body of work using site-directed mutagenesis to examine enzymatic function in detail and the availability of a number of crystal structures, no model has been able to explain how all the members of this structurally diverse family of oxidases are able to pump protons (3-7). A recent examination of high-resolution structures has led us to a focus on the role of water in the facilitation of proton movement (8). Herein, we test a model of how the Mg/Mn site in all eukaryotic and some bacterial heme–copper oxidases may augment the proton pumping mechanism (9). Oxidases that contain a CuA–Mg site are part of the superfamily, and the inclusion of Mg(II) appears to aid their efficiency; i.e., the proton pumping stoichiometry of CuA–Mg-type oxidases appears to be closer to one than in other types (10-12). A role for the CuA–Mg site in pumping would suggest that the CuA redox potential should have a pH dependence, but the relationship between the redox potential of CuA and the bulk pH is quite complex. The redox potential of CuA appears to be insensitive to bulk pH, but oxidase releases stoichiometric amounts of protons upon CuA oxidation (13, 14). Studies of cytochrome c oxidase vesicles using flash injection of electrons show that the changes in membrane potential during the transfer of electrons from CuA to heme a have two components (15, 16). The major component is due to vectorial electron transfer, and the second component is due to intraprotein proton relocation step(s) (inside to outside) that adds to the electrogenicity of the electron transfer step, as indicated by electrometric techniques (15-21).
An examination of Rhodobacter sphaeroides (R.s.) and bovine oxidase crystal structures (Figure 1) has led us to propose that changes in the positions and protonation state of two water molecules, W1Mg and W1E198II,1 are linked to the redox status of CuA and thereby facilitate a gated exit pump (9). In Figure 1 we show the bovine structures before and after CuA undergoes reduction (oxidized 2DYR and reduced 2EIJ). W1Mg is in the sixth coordination position on the Mg(II) ion and is part of a water chain connecting the binuclear center to the external bulk solvent. W1E198II is close to the enzyme surface. In the oxidized enzyme both W1Mg and W1E198II are best modeled as neutral water molecules (Figure 1A). Upon CuA reduction (Figure 1B) a proton appears to be gained by W1E198II as evidenced by a change in the O–O distance between W1E198II and the E198II carboxyl from 3.7 Å (oxidized) to 2.6 Å (reduced). The reduced structure is also consistent with loss of a proton from W1Mg leaving a (1−)OH ligated to the Mg(II) ion, as indicated by an increase in the ionic character of the waters around this ligand (Figure 1 inserts). It should be noted that very similar distance and structure changes are observed in the reduced (Qin et al., unpublished) and oxidized (22) structures of R.s. cytochrome c oxidase (9).
Figure 1.
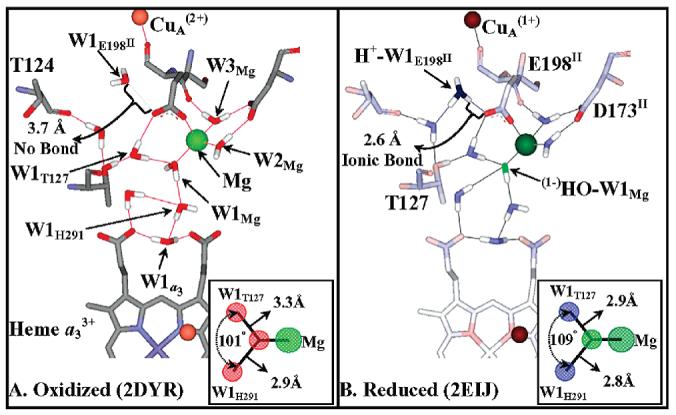
Formation of a hydroxide/hydronium ion pair upon CuA reduction, modeled on bovine oxidase oxidized and reduced structures. A comparison of the oxidized (A) and reduced (B) structures of bovine oxidase indicates a change in the protonation status and bonding of two water molecules. (i) W1E198II. W1E198II is HOH2289 in the bovine 2DYR (Ox) and HOH2291 in the bovine 2EIJ (Red) structures. The distances indicate that W1E198II switches between a neutral water molecule and a hydronium ion upon reduction. This switch in protonation status is accompanied by its movement of 1.1 Å toward the free carboxylate oxygen of E198II (OE2 in both structures). Upon CuA reduction H+−W1E198II becomes ionically bonded to the free carboxylate oxygen of E198II, the bond distance shortening from 3.7 to 2.6 Å. (ii) W1Mg. There are three waters bonded to the Mg ion, but only one can undergo deprotonation, W1Mg (HOH2032). W2Mg and W3Mg (HOH2031, HOH2033) are pinned by the Mg(II) ion and the carboxylate D173II (in the neutral state). A change in bond length between (1−)HO−W1Mg/W1Mg and the Mg(II) cation is not resolved in the bovine crystal structures; i.e., it is 2.3 Å in both structures. However, the bond lengths of W1Mg to the two nearest water molecules, W1H291 and W1T127 (HOH2020 and HOH2054), do show a considerable shrinkage upon reduction, 2.9 to 2.8 Å and 3.3 to 2.9 Å, respectively, due to increased ionic bonding character as W1Mg is converted to (1−)HO−W1Mg. The bond angles and bond lengths of W1Mg to W1H291 and W1T127 are shown in the insert of panels A and B.
From these observations, we postulate the formation of two salt pairs, H+−W1E198II/(1−)E198II and (1−)HO−W1Mg/Mg(II), upon the reduction of CuA. We suggest that this disproportionation occurs because most of the anionic charge on the carboxylate group of E198II shifts from the Mg(II) ion toward the hydronium. When CuA is oxidized, E198II is an anionic ligand toward the Mg(II) ion, but upon the reduction of CuA and the ensuing formation of the E198II/(1+)H3O salt, E198II becomes neutral toward the Mn(II) ion (9). That being the case, it should be possible to replace the hydroxide ligand of the Mg(II) ion with a weak acid, such as cyanide (eq 1).
| (1) |
However, although weak acids like cyanide are generally good at replacing anionic ligands like hydroxide (eq 1), they are generally poor at replacing water ligands (eq 2). A
| (2) |
somewhat analogous situation occurs with the differential binding of cyanide to ferri/ferromyoglobin, where cyanide can replace hydroxide anions but is a very poor at replacing neutral water molecules (23-29).
To test this hypothesis, that W1Mg switches between a neutral water ligand and an anionic hydroxide ion in response to the reduction of CuA, we prepared R.s. oxidase that was purified from R.s. grown in the presence of Mn(II) so that we could monitor an EPR-detectable Mn(II) atom in the Mg(II) site. We then probed the aqueous ligands of Mn(II) with deuterium, cyanide, and azide under reduced and oxidized conditions.
The results of our studies support the hypothesis, providing evidence that cyanide binds to the Mn(II) ion of the CuA–Mg(Mn) site when CuA is reduced but not when CuA is oxidized.
MATERIALS AND METHODS
Modeling the Hydrogen-Bonding Structure in Cytochrome Oxidase
The arrangement of hydrogen bonding of the water molecules from bovine and R.s. crystal structures was determined manually using the package Accelrys ViewerPro.
Spectroscopy
All samples consisted of 90 μM R.s. oxidase, 100 mM K+-HEPES, and 4 mM KCl, pH 7.0. Sodium azide and cyanide were added to a final concentration of 5 mM, and the samples were reduced using 10 mM dithionite after the sample had been deaerated using three vacuum/argon cycles.
CW-EPR
CW-EPR spectra were recorded on a Bruker ESP300E X-band EPR spectrometer equipped with a TE102 cavity and an Oxford model 900 liquid helium cryostat. Sample conditions were as folllows: sample temperature, 4.5 K; microwave frequency, 9.46 GHz; microwave power, 50 μW; 100 kHz magnetic field modulation frequency; 13 G field modulation amplitude; 327 ms conversion time. CW-EPR simulations were done using the XSOPHE program available from Bruker.
ESEEM
Pulsed EPR measurements were made on a Bruker E-680X spectrometer operating at X band and equipped with a model ER 4118X-MD-X5-W1 probe that employs a 5 mm dielectric resonator. The temperature was maintained at 4.5 K using an Oxford Instruments liquid helium flow system equipped with a CF-935 cryostat and an ITC-503 temperature controller. ESEEM data were collected using a three-pulse (stimulated echo) sequence, 90°−τ−90°−T−90°, with 90° microwave pulse widths of 16 ns (fwhm). An integration window of 24 ns was used to acquire spin–echo amplitude, and data set lengths were 512 points. The conditions were chosen so as to suppress the contribution of matrix (bulk water) protons to the signal; the success of the suppression is evident in the absence of a broad feature centered around 14–15 MHz in the ESEEM spectra (30, 31). Deuterium contributions to ESEEM spectra were often elucidated using the ratio method introduced by Mims (30, 31) together with processing tools provided by Bruker. Three-pulse ESEEM data were normalized by dividing each data set by its maximum amplitude. The normalized 2H ESEEM data were then divided by the corresponding, normalized 1H ESEEM data, resulting in ESEEM data dominated by the 2H. The time domain data which resulted from the ratio method were tapered with a Hamming window function and Fourier transformed. ESEEM spectra were obtained by taking the absolute value of the real part of the transforms. Simulations of ESEEM spectra were accomplished with MATLAB scripts written in-house.
ESEEM Simulation
We combined the simulation strategy of Ashtashkin and Raitsimring (32) for a S = 5/ 2 spin system with the density matrix formalism developed by Mims (30, 31) to compute time domain ESEEM functions for deuterium. The expressions developed for the average electron spin operators in the presence of a weak perturbation due to the zero-field splitting interaction (eqs 17 and 18 of ref 32) were used in the nuclear spin Hamiltonian to model the electron–nuclear hyperfine coupling. After combining nuclear Zeeman and quadrupole terms, the Hamiltonian matrices for electron spin manifolds being considered were diagonalized, and the resulting eigenfrequencies and eigenvector coefficients were used to compute the ESEEM function, for the specific EPR transition being considered, according to the recipe provided by Mims (30, 31). Subspectra were combined using the weightings given in the caption for Figure 4C. The deuterium hyperfine interaction was considered axial with principal values of Axx = Ayy = −0.34 MHz and Azz = 1.16 MHz that were gleaned from the results of ENDOR studies of Mn(H2O)6(2+) (33). The deuterium nuclear quadrupole interaction was taken as axial and a value of e2qQ/h = 0.2 MHz was used. Simulations for multiple coupled nuclei were generated using the spherical model approximation to the product rule (34).
Figure 4.
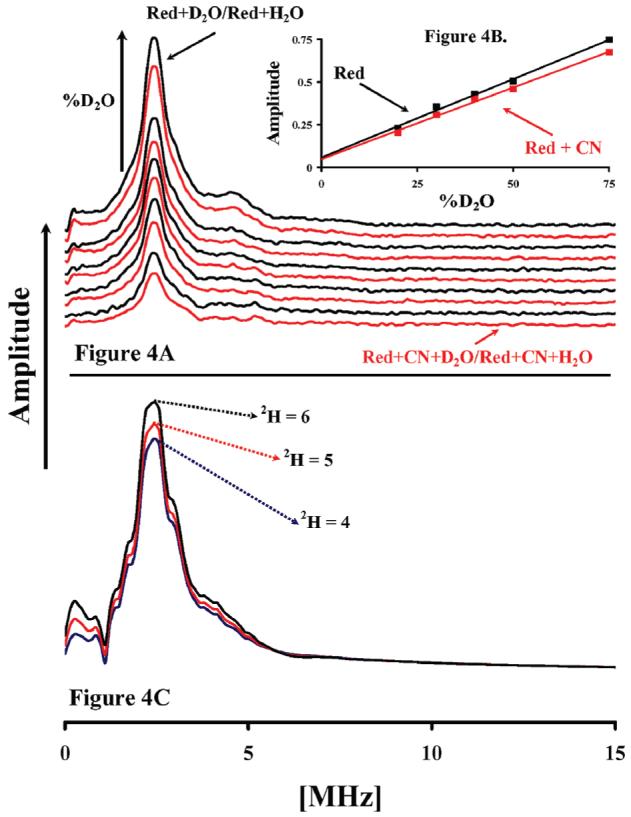
ESEEM is used to measure the displacement of D2O from the Mn site by CN. Panel A shows the Fourier transform of the normalized three-pulse ESEEM of reduced enzyme (black) in 20%, 30%, 40%, 50%, and 75% (v/v) D2O buffer ratioed against reduced enzyme in H2O and reduced enzyme with cyanide (Red) in D2O buffer ratioed against reduced enzyme with cyanide in H2O. The spectra were integrated from 0.25 to 5 MHz and plotted vs % D2O of the buffer. The points were fit with a linear function, the slope of which corresponds to the amplitude of interacting 2H (panel B, insert). Experimental conditions were the same as in Figure 3. Panel C shows a simulation of the ESEEM spectra of the Mn center where it is coupled to six, five, and four deuterons, respectively. The simulation program is described in the text. The simulation parameters are as follows: central field, 3700 G; microwave frequency, 9.72 GHz; starting T, 40 ns; τ, 204 ns; T increment, 16 ns; Axx, Ayy for 2H, −0.34 MHz, and Azz, 1.16 MHz. Relative weights of these transitions: 0.36 for |−1/2> ↔ |+1/2>, 0.20 for |−3/2> ↔ |−1/2>, 0.20 for |+1/2> ↔ |+3/2>, 0.12 for |−5/2> ↔ |−3/2>, and 0.12 for |+3/2> ↔ |+5/2>.
RESULTS
CW-EPR spectra of Mn(II)-enriched R.s. oxidase, in the fully oxidized and reduced states, were essentially identical to those previously obtained by our group (35) (Figure 2). The biggest difference between the CW-EPR spectrum of the Mn(II) center in R.s. oxidase studies and those using the Paracoccus denitrificans (P.d.) oxidase is that the R.s. enzyme has a much greater degree of Mn(II) incorporation (>70%) than the P.d. enzyme (<10%).
Figure 2.
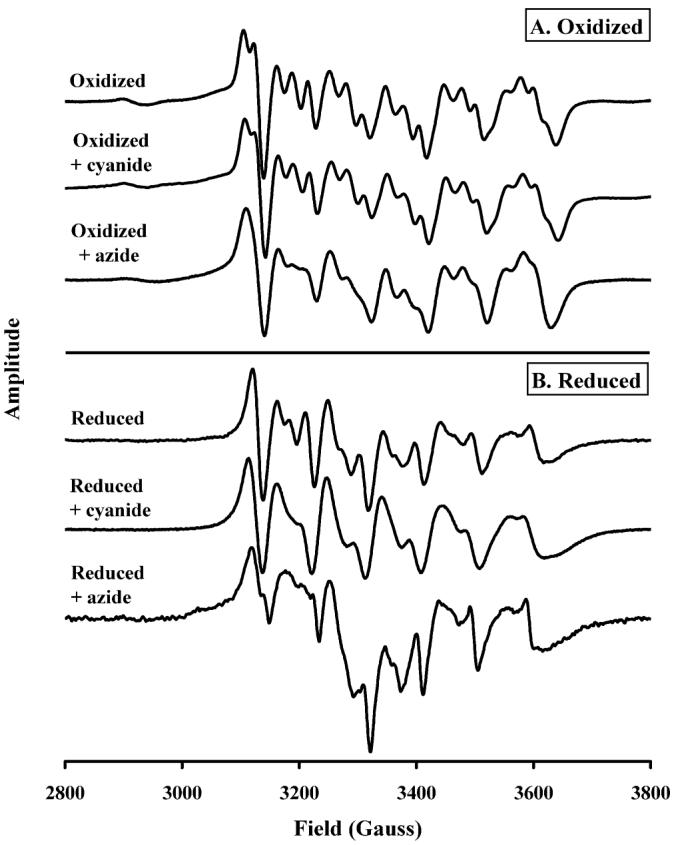
The CuA redox-dependent binding of cyanide and azide to the Mn center of R.s. oxidase. Spectra taken at 9.46 GHz, 50 μW power, 100 kHz modulation frequency, 13 G modulation amplitude, and 327 ms conversion time. Conditions: 90 μM R.s. oxidase, 100 mM K+-HEPES, and 4 mM KCl, pH 7.0; cyanide/azide, 5 mM; dithionite, 10 mM.
In the CW-EPR spectrum of the oxidized enzyme (Figure 2A, top spectrum), the Mn(II) center is spin coupled to CuA, resulting in an enrichment of the fine structure of the Mn(II) signal. Reduction of CuA (Figure 2B, top spectrum) eliminates this dipolar coupling and leads to a simpler line structure, more typical of Mn(II) model compounds (35, 36).
Figure 2A also shows a comparison of the CW-EPR spectra of oxidized R.s. oxidase in the presence of the externally added ligands, cyanide and azide. Addition of cyanide to the oxidized enzyme had no effect on the line shape of the Mn(II) CW-EPR signal, but it did give the typical heme-a3/CN CW-EPR spectrum (37) (data not shown). Interestingly, azide broadened the Mn(II) CW-EPR line shape, indicating that it might be coordinated to the Mn(II) ion itself.
Figure 2B shows the effect of dithionite reduction on the Mn(II) CW-EPR spectra of oxidase pretreated with either cyanide or azide.2 In the reduced enzyme, upper trace, the Mn(II) spectrum is typical of that seen previously (35). However, when cyanide is present and the CuA center is reduced, the CW-EPR spectrum is altered. To understand these changes, we undertook spectral simulations of the Mn(II) CW-EPR spectra using the XSOPHE program distributed by Bruker. Using a spin Hamiltonian that consisted of isotropic electronic Zeeman, zero-field splitting, and isotropic 55Mn hyperfine terms (eq 3)
| (3) |
we were able to simulate the spectrum of reduced cytochrome c oxidase (Figure 2B, top) using the following parameters: |D|, 140 G; E/D, 0.20; g-value, 2.00; aiso, 96.5 G. A Gaussian line shape model that included D- and E/D-strain was employed in our simulations. Line shape parameters that provided satisfactory simulations of the spectra were as follows: intrinsic line width, 8.0 G; D-strain, 20 G; E/D-strain, 0.03. Simulations are shown along with the experimental spectrum in the Supporting Information, Figure S1. The spin Hamiltonian parameters used for these simulations compare well with those found for reduced cytochrome c oxidase from P.d., where D = 120 G and E/D = 0.18 provided satisfactory predictions (35).
The addition of cyanide to reduced oxidase leads to an increase in |D| from 140 G to 170 G. This accounts for the skewing of the individual line shape features of the central fine structure transition toward lower field. Our simulations also showed that E/D = 0.20, unchanged for that determined above for reduced, untreated cytochrome c oxidase, and that line shape parameters showed a modest increase in D-strain, 30 G, and E/D strain, 0.05 (see Figure S1).
In the presence of azide (Figure 2B, bottom) there appears to be both a broadening of the fine structure and the superimposition of another broad line shape. The new broad feature appears in a region where oxidized CuA is found, and so it is conceivable that the change is partially due to the oxidation of CuA. But this can be ruled out because of the following: (1) We have used a large excess of dithionite (10 mM), and addition of further dithionite has no effect on the line shape. (2) The reduced plus azide spectrum differs greatly from that of the oxidized plus azide (Figure 2). At Mn enrichment levels used (>70%), the signal of the oxidized CuA is much smaller than that seen in the reduced plus azide sample. (3) It is very unlikely that azide can act as an oxidant, from a chemistry standpoint.
The broadening of the intrinsic line width upon azide addition is in line with what we reported above for cyanide addition. However, the skewing of the central fine structure transition line shapes that was observed for cyanide addition was not observed for azide-treated cytochrome c oxidase. We conclude that the Mn(II) zero field splitting parameters, D and E, are unchanged by azide addition to reduced cytochrome c oxidase.
The data presented in Figure 2 show that both cyanide and azide are capable of perturbing the Mn(II) CW-EPR spectrum, and this interaction is influenced by the redox status of the oxidase. It does not however show that these extrinsic anions are binding to the Mn(II) ion. There are six ligands to the Mn(II) ion, H368, D369, E198II, and three water molecules, W1Mn, W2Mn, and W3Mn, and it is possible that any of these ligands could be displaced by either cyanide or azide without altering the X-band CW-EPR spectrum because ligand hyperfine couplings for Mn(II) are typically small when compared to the inhomogeneous line widths of the central fine structure transition. Electron spin–echo envelope modulation (ESEEM) spectroscopy was used in an attempt to measure weak ligand hyperfine interactions and, thus, report on coordination chemistry at the Mn(II) ion that results from cyanide addition.
The ESEEM spectra shown in Figure 3 were recorded at 3700 G to avoid potential contributions from CuA or heme a. In addition, the sample concentration, <100 μM enzyme, and the pulse sequence repetition rate used for these measurements would severely limit the amplitudes of any of the CW-EPR signals except those from Mn(II). The conditions used also suppress ESEEM signals from weakly coupled protons. Each ESEEM spectrum was obtained by ratioing the time domain data sets against parallel data collected for a control sample in an attempt to remove the ESEEM of the coordinating histidine. This division procedure is essentially the same as performing a difference spectrum in optical spectroscopy; however, it should be noted that because our ESEEM spectra are absolute value spectra, all contributions are rendered positive in amplitude, so both the disappearance and appearance of a spin–spin interaction generate spectral peaks, there are no troughs.
Figure 3.
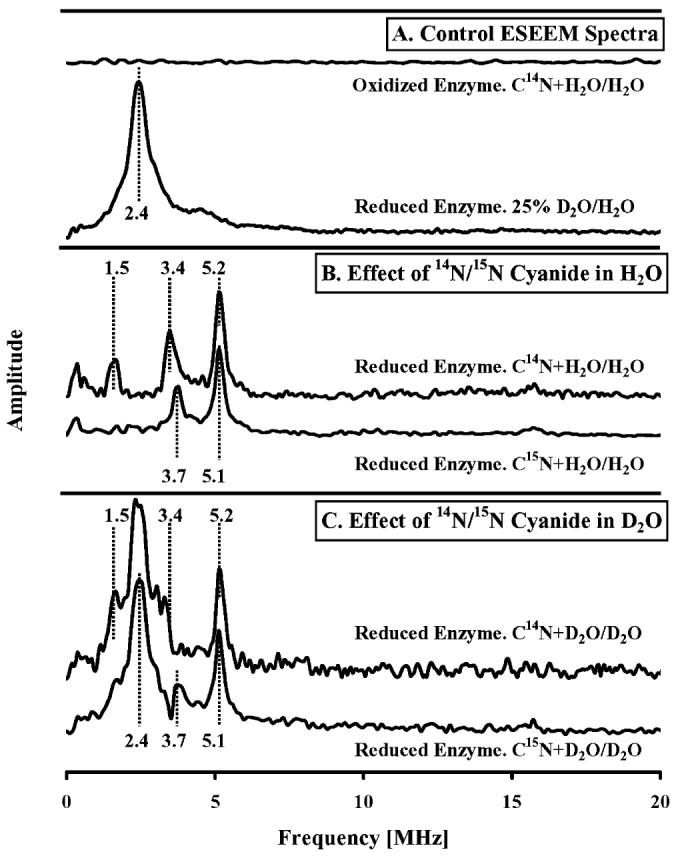
Effect of 14N and 15N cyanide on the Mn ESEEM spectra of R.s. oxidase in H2O and D2O. Ratioed ESEEM spectra obtained for (A) oxidized oxidase + C14N/oxidized oxidase in aqueous buffer (top trace); reduced oxidase in 25% D2O buffer/reduced oxidase in H2O buffer; (B) reduced oxidase + C14N/reduced oxidase in H2O buffer (top trace); reduced oxidase + C15N/reduced oxidase in H2O buffer (bottom trace); and (C) reduced oxidase + C14N/reduced oxidase in 75% D2O buffer (top trace); reduced oxidase + C15N/reduced oxidase in 75% D2O buffer (bottom trace). Conditions common to these measurements were as follows: sample temperature, 4.5 K; microwave frequency, 9.68 GHz; magnetic field strength, 3700 G; pulse sequence repetition rate, 1 kHz. ESEEM data were collected using a three-pulse (stimulated echo) sequence of 90°–τ–90°–T–90° with 90° pulse widths of 16 ns (fwhm); tau value, 128 ns; integration window, 24 ns. Common sample conditions were 90 μM R.s. oxidase, 100 mM K+-HEPES, and 4 mM KCl, pH 7.0; cyanide, 5 mM; dithionite, 10 mM.
The upper trace of Figure 3A shows the ESEEM difference spectrum of the Mn(II) center that results from the addition of cyanide to the oxidized enzyme in H2O. The ESEEM data from the oxidized enzyme in the presence cyanide were ratioed against the oxidized enzyme without cyanide, and the result was Fourier transformed to obtain the difference spectrum. No signal above noise level is detected, consistent with the CW-EPR data shown in Figure 2A, which indicated that cyanide does not perturb the Mn(II) center in the fully oxidized enzyme. The lack of ESEEM signals indicates that the conditions chosen, together with the ratioing procedure, have fully suppressed matrix 1H interactions (expected at 15.7 MHz) and 14N modulations from coordinating His-411 (32). The lower control trace in Figure 3A shows the ESEEM difference spectrum that is obtained when a reduced sample in 25% D2O is divided by the data from a reduced sample in 100% H2O. The resulting spectrum shows the interaction of 2H nuclei with the Mn(II) center and is directly comparable with similar experiments done previously (38). The interaction of D2O with Mn(II) generates a broad peak centered 2.4 MHz and a smaller peak at 4.8 MHz.
In Figure 3B we show the interaction of cyanide with the Mn(II) center in reduced oxidase in H2O buffer. These difference spectra were obtained by ratioing of reduced enzyme without cyanide against reduced enzyme that was incubated with either 14N cyanide (top trace) or 15N cyanide (bottom trace). Peaks were resolved using 14N cyanide, at 1.5, 3.4, and 5.2 MHz, and using 15N cyanide, at 3.7 and 5.1 MHz. We cannot assign these peaks to the direct interactions of 14N cyanide or 15N cyanide with Mn(II), with complete confidence, as Mn(II) is also coupled to a nitrogen atom of its histidine ligand. In their examination of the Mn(II) center of reduced R.s. cytochrome c oxidase, Espe and coworkers observed in the ESEEM low-frequency peaks centered around 1.7, 3.6, and 5.0 MHz (35). They concluded that their peaks were a result of the interaction of a nitrogen atom of the sole histidine ligand, H411 (H368 in bovine), coordinating the Mn(II) center. Because the addition of cyanide to reduced cytochrome c oxidase leads to a 20% increase in |D|, we know that the electronic structure of the Mn(II) center is perturbed. This could lead to small changes in ligand hyperfine coupling for His-411, and it will lead to a slight change in the EPR resonance conditions upon which the ESEEM spectrum is measured. Recent theoretical work by Ashtashkin and Raitsimring shows that the Mn(II) zero field splitting parameters affect the amplitudes of the observed ESEEM frequencies (38). Because the ratioed ESEEM spectra we obtained with both 14N and 15N cyanide incubations show peaks that coincide with those assigned to His-411, we infer that a portion of these signals stems from modest changes in 14N-H411 ESEEM amplitudes and/or frequencies that occur upon cyanide binding to Mn(II), rather than just from the direct interaction of 14N and 15N cyanide with Mn(II).
The differences in the ratio spectra of Figure 3B,C, which stem from the use of C14N vs C15N, are best explained by contributions of cyanide-derived 14N at 1.5, 3.4, and 5.2 MHz. The positions of the peaks at 3.4 and 5.2 MHz may be distorted by the presence of overlapping contributions from H411 that are observed at 3.7 and 5.1 MHz for the 15N cyanide-treated samples (Figure 3B,C, bottom traces). In this interpretation, the ESEEM from cyanide-derived 15N is too weak to be resolved in the ratioed ESEEM spectra (see Discussion).
As an additional test of whether cyanide binds and displaces water from the Mn(II) ion, we exchanged oxidase samples into D2O and studied the effect of cyanide addition on the ESEEM spectrum of coupled deuterons. If cyanide binds directly to the Mn(II) center in the reduced enzyme and displaces a hydroxide, it should lessen the peak at 2.4 MHz that is mainly derived from the replacement by deuterium of the hydrogen atoms of the three inner sphere aqueous ligands bound to Mn(II). To observe changes that result from an altered interaction of Mn(II) with its aqueous ligands upon the binding of cyanide, the experiments shown in Figure 3B were repeated in the presence of 75% D2O (Figure 3C). All changes in ESEEM ratioed spectra are positive; therefore, if cyanide were to displace an aqueous ligand of Mn(II), the resulting signal would be expected to be a combination of the cyanide/H411 spectrum shown in Figure 3B and the D2O control signal shown in Figure 3A.
Figure 3C shows the ratioed ESEEM spectra obtained when 14N or 15N cyanide is added to reduced oxidase in 75% D2O buffer against reduced enzyme in 75% D2O without cyanide. The spectra show the same large deuterium signal at 2.4 MHz, seen in the 25% D2O/H2O control trace (Figure 3A, bottom trace), and the same underlying peaks that are present in the cyanide spectra shown in Figure 3B. If the ESEEM spectra of Figure 3B,C arose solely from exchangeable deuterons and the 14N of H411, the data for C14N- and C15N-treated samples would be identical. The differences are most likely due to the added contributions of cyanide-derived 14N for the top traces of Figure 3B,C.
An additional way to study the proposed cyanide chemistry at the Mn site is to use ESEEM spectroscopy on samples exchanged against 2H2O to measure the displacement of a putative H2O or (1−)OH ligand. The amount of exchangeable 1H/2H proximal to the Mn(II) center was probed using ESEEM in reduced oxidase in buffer solutions made in various ratios of H2O/D2O, in the absence and presence of cyanide. Figure 4A shows the change in the 2H/Mn ESEEM signal at various H2O/D2O ratios, in the absence (black traces) and presence (red traces) of cyanide. Figure 4B (insert) shows the slope of the signal amplitude, as integrated from 0.2 to 5 MHz, as a function of increasing deuterium enrichment. The difference in the slopes of the two lines in Figure 4B is approximately 9%.
The intensity of the ESEEM signal of the 2H–Mn(II) hyperfine interaction is the product of all the individual 2H–Mn(II) interactions. The observed loss of 2H-ESEEM amplitude that resulted from cyanide addition was modeled using simulation of ESEEM modulation depths calculated from first principles, as described in Materials and Methods.
We found that a change in one of the inner sphere 2H from 5 to 4 resulted in a 9.5% decrease in intensity, whereas 6 to 4 resulted in a change of 15.5% (Figure 4C). The drop in ESSEM signal intensity, shown in Figure 4A,B, for reduced oxidase in the presence of cyanide is approximately 9%, and this supports the model presented above, in which cyanide replaces an inner sphere hydroxide ligand to Mn(II) in the reduced enzyme.
DISCUSSION
In this paper we report experimental observations that indicate that cyanide can bind to the Mg/Mn site in cytochrome oxidase when the enzyme is in the reduced state. This finding is consistent with the model that the CuA–Mg(Mn) site is part of the oxidase proton pumping cycle, participating in the movement of a proton from the vicinity of Mg(II) toward the outer bulk phase (9). The model was derived from the careful examination of high-resolution crystal structures of mammalian and bacterial oxidases in which changes in the bonding distances were observed for waters in the vicinity of the CuA–Mg site in the reduced and oxidized forms as depicted in the bovine oxidase in Figure 1 and reported for the R.s. oxidase previously (9).
We have shown for the first time that the Mn(II) center of oxidase is capable of binding nonaqueous extrinsic ligands using a combination of CW-EPR and ESEEM. CW-EPR spectra show that cyanide addition to reduced oxidase causes at 20% increase in the zero field splitting constant, |D|, with little or no change in the symmetry of the interaction. An increase in D- and E/D-strain also accompanies cyanide addition to reduced enzyme and that leads to a broadening of the central fine structure transition features.
Direct evidence that cyanide affects these changes by coordination to Mn(II) comes from ESEEM studies where the results were complicated by the above-mentioned increase in |D| and a change in ligand hyperfine coupling to H411. Despite this complication the reproducible difference between 14N and 15N cyanide, explained by the weaker C15N interactions, shows that cyanide is indeed a Mn(II) ligand. This weaker interaction of 15N is a fairly common feature of nitrogen isotopic substitution experiments in ESEEM spectroscopy because the higher spin and nuclear quadrupole moment of 14N make it more amenable to the interference effects that lead to modulations (31). Because the nuclear quadrupole coupling constant, e2qQ, for cyanide 14N is in the vicinity of 4 MHz, the hyperfine coupling is most likely in the neighborhood of 0.5 MHz for the bound anion. We are currently using the two-dimensional ESEEM method of HYSCORE to separate contributions from cyanide- and histidyl-14N so an analysis of the ligand hyperfine coupling can be completed.
Our use of 2H-ESEEM to quantify the change in (1−)OH/H2O binding to Mn(II) upon cyanide addition to reduced enzyme must be viewed with caution. Although the 9% decrease in 2H modulation intensity that was measured supports the displacement of one inner sphere deuteron and, therefore, displacement of a (1−)OH ligand, the data were obtained by dividing normalized three-pulse ESEEM spectra where cross terms in the quotient add additional uncertainty to the measured amplitudes. In addition, our ESEEM data arise from EPR transitions between multiple electron spin manifolds on Mn(II). The simulation results shown in Figure 4C have amplitude changes of 9% and 16% for the loss of one and two deuterons, respectively. However, we have only simulated the inner sphere aqueous ligands, and not all spins within ≈8 Å of the Mn(II) center. For this reason the overall 2H-ESEEM amplitude was ≈50% of what we measured experimentally. This combination of experimental and theoretical data makes the interpretation of (1−)OH displacement over H2O displacement likely, but not proven.
Our CW-EPR results also show that azide addition to cytochrome c oxidase perturbs the Mn(II) binding site. Specifically, azide addition results in a broadening of the Mn(II) EPR spectra for both oxidized and reduced enzyme. Unlike cyanide addition, the line shape symmetries of the central fine structure transition are not affected, and the changes can be attributed to increased strain in the zero field splitting parameters without actual changes to D or E/D. It is possible that azide does not coordinate to Mn(II) but alters the protein structure in a fashion that increases the distribution of ligation structures at the Mn(II) site.
We have shown that R.s. oxidase binds azide in the fully oxidized state and the fully reduced oxidase can bind either azide or cyanide. Our data from R.s. oxidase are supported by observations made by Yoshikawa and Caughey in an IR spectroscopic investigation of bovine oxidase when they investigated the binding of both azide and cyanide to a common, nonredox, metal site (39). They observed that the unidentified site bound azide when the oxidase was either oxidized or reduced but that cyanide binding was only observed in the reduced oxidase. They optically characterized this unidentified binding site with the appearance of a band at 2033 cm−1 in the reduced oxidase plus azide, minus reduced oxidase plus cyanide, difference spectrum. Our data indicate that the center they were interrogating, with cyanide and azide, was Mg(II).
The ability of an inner sphere aqueous ligand to Mg(II) to cycle between hydroxide and water, upon CuA oxidation and reduction, could be involved in proton pumping. We suggest that CuA oxidation by heme a is linked to the release of this proton into the outer bulk phase and that the CuA–Mg site is what has been previously termed a proton loading site (40). The proton loading site is an acceptor site for pumped protons that is positioned above the heme a3 propionates, whose existence and position have been inferred on the basis of both electrostatic measurements (21, 40, 41) and theoretical studies (7). A comprehensive mechanism of proton pumping that incorporates the role of the Cu–−Mg site has recently been published (9).
Supplementary Material
Footnotes
This work was supported by National Institutes of Health Grants GM26916 (S.F.-M.) and GM54065 (J.M.), Michigan State University Research Excellence Funds REF Grant 03-016(S.F.-M.), and Michigan State University Foundation Strategic Partnership Grant: Quantitative Biology and Modeling Initiative (QBMI).
We will use the amino acid sequence of the bovine oxidase as our default. Residues from subunit II are identified with the notationII. Water molecules are identified by the prefix W(number) and the subscripted amino acid, heme, or metal that they are bonded to. If water molecules have been either identified or presumed to be protonated (hydronium) or deprotonated (hydroxide), they are further prefixed as H+− or as (1−)HO−, respectively. Thus, the protonated water that is bonded to the carboxylic acid group of residue E198 of subunit II in the structure 2EIJ, HOH2291, is rendered as H+−W1E198II.
The same Mn CW-EPR spectra are generated if the addition sequence of azide/cyanide and then dithionite is reversed and azide/cyanide is added to the reduced oxidase. This latter method was not used as it is possible to cause some oxidation of the sample on the addition of an aliquot of the ligand.
SUPPORTING INFORMATION AVAILABLE
CW-EPR spectra and spectral simulations for reduced cytochrome c oxidase and reduced cytochrome c oxidase and cyanide (Figure S1). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Wikstrom M, Bogachev A, Finel M, Morgan JE, Puustinen A, Raitio M, Verkhovskaya M, Verkhovsky MI. Mechanism of proton translocation by the respiratory oxidases–the histidine cycle. Biochim. Biophys. Acta. 1994;1187:106–111. doi: 10.1016/0005-2728(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 2.Wikstrom MKF. Proton pump coupled to cytochrome c oxidase in mitochondria. Nature. 1977;266:271–273. doi: 10.1038/266271a0. [DOI] [PubMed] [Google Scholar]
- 3.Wikstrom M, Verkhovsky MI. Mechanism and energetics of proton translocation by the respiratory heme-copper oxidases. Biochim. Biophys. Acta. 2007;1767:1200–1214. doi: 10.1016/j.bbabio.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Mills DA, Ferguson-Miller S. Understanding the mechanism of proton movement linked to oxygen reduction in cytochrome c oxidase: lessons from other proteins. FEBS Lett. 2003;545:47–51. doi: 10.1016/s0014-5793(03)00392-2. [DOI] [PubMed] [Google Scholar]
- 5.Belevich I, Bloch DA, Belevich N, Wikstrom M, Verkhovsky MI. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc. Natl. Acad. Sci. U.S.A. 2007;104:2685–2690. doi: 10.1073/pnas.0608794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konstantinov AA. Cytochrome c oxidase as a proton-pumping peroxidase: Reaction cycle and electrogenic mechanism. J. Bioenerg. Biomembr. 1998;30:121–130. doi: 10.1023/a:1020571930850. [DOI] [PubMed] [Google Scholar]
- 7.Fadda E, Yu CH, Pomes R. Electrostatic control of proton pumping in cytochrome c oxidase. Biochim. Biophys. Acta. 2008;1777:277–284. doi: 10.1016/j.bbabio.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Sharpe MA, Qin L, Ferguson-Miller S. Proton entry, exit and pathways in cytochrome oxidase: Insight from “conserved” water. In: Wikström M, editor. Biophysical and Structural Aspects of Bioenergetics. Royal Society of Chemistry; Cambridge: 2005. pp. 26–54. [Google Scholar]
- 9.Sharpe MA, Ferguson-Miller S. A chemically explicit model for the mechanism of proton pumping in heme-copper oxidases. J. Bioenerg. Biomembr. 2008 doi: 10.1007/s10863-008-9182-6. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toledo-Cuevas M, Barquera B, Gennis RB, Wikstrom M, Garcia-Horsman JA. The cbb3-type cytochrome c oxidase from Rhodobacter sphaeroides, a proton-pumping heme-copper oxidase. Biochim. Biophys. Acta. 1998;1365:421–434. doi: 10.1016/s0005-2728(98)00095-4. [DOI] [PubMed] [Google Scholar]
- 11.Puustinen A, Finel M, Virkki M, Wikström M. Cytochrome o (bo) is a proton pump in Paracoccus denitrificans and Escherichia coli. FEBS Lett. 1989;249:163–167. doi: 10.1016/0014-5793(89)80616-7. [DOI] [PubMed] [Google Scholar]
- 12.Verkhovskaya ML, Garcia-Horsman A, Puustinen A, Rigaud JL, Morgan JE, Verkhovsky MI, Wikstrom M. Glutamic acid 286 in subunit I of cytochrome bo3 is involved in proton translocation. Proc. Natl. Acad. Sci. U.S.A. 1997;94:10128–10131. doi: 10.1073/pnas.94.19.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papa S. Role of cooperative H+/e− linkage (redox Bohr effect) at heme a/CuA and heme a3/CuB in the proton pump of cytochrome c oxidase. Biochemistry (Moscow) 2005;70:178–188. doi: 10.1007/s10541-005-0099-y. [DOI] [PubMed] [Google Scholar]
- 14.Papa S, Capitanio N, Capitanio G, Palese LL. Protonmotive cooperativity in cytochrome c oxidase. Biochim. Biophys. Acta. 2004;1658:95–105. doi: 10.1016/j.bbabio.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 15.Medvedev DM, Medvedev ES, Kotelnikov AI, Stuchebrukhov AA. Analysis of the kinetics of the membrane potential generated by cytochrome c oxidase upon single electron injection. Biochim. Biophys. Acta. 2005;1710:47–56. doi: 10.1016/j.bbabio.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Siletsky SA, Belevich I, Jasaitis A, Konstantinov AA, Wikström M, Soulimane T, Verkhovsky MI. Time-resolved single-turnover of ba3 oxidase from Thermus thermophilus. Biochim. Biophys. Acta. 2007;1767:1383–1392. doi: 10.1016/j.bbabio.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 17.Zaslavsky D, Kaulen AD, Smirnova IA, Vygodina T, Konstantinov AA. Flash-induced membrane-potential generation by cytochrome c oxidase. FEBS Lett. 1993;336:389–393. doi: 10.1016/0014-5793(93)80843-j. [DOI] [PubMed] [Google Scholar]
- 18.Verkhovsky MI, Tuukkanen A, Backgren C, Puustinen A, Wikstrom M. Charge translocation coupled to electron injection into oxidized cytochrome c oxidase from Paracoccus denitrificans. Biochemistry. 2001;40:7077–7083. doi: 10.1021/bi010030u. [DOI] [PubMed] [Google Scholar]
- 19.Kotlyar A, Borovok N, Hazani M, Szundi I, Einarsdottir O. Photoinduced intracomplex electron transfer between cytochrome c oxidase and TUPS-modified cytochrome c. Eur. J. Biochem. 2000;267:5805–5809. doi: 10.1046/j.1432-1327.2000.01655.x. [DOI] [PubMed] [Google Scholar]
- 20.Gilderson G, Aagaard A, Gomes CM, Adelroth P, Teixeira M, Brzezinski P. Kinetics of electron and proton transfer during O2 reduction in cytochrome aa3 from A. ambivalens: an enzyme lacking Glu(I-286) Biochim. Biophys. Acta. 2001;1503:261–270. doi: 10.1016/s0005-2728(00)00195-x. [DOI] [PubMed] [Google Scholar]
- 21.Belevich I, Verkhovsky MI, Wikstrom M. Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature. 2006;440:829–832. doi: 10.1038/nature04619. [DOI] [PubMed] [Google Scholar]
- 22.Qin L, Hiser C, Mulichak A, Garavito RM, Ferguson-Miller S. Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 2006;103:16117–16122. doi: 10.1073/pnas.0606149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keilin D, Hartree EF. Cyanide compounds of ferroperoxidase and myoglobin and their reversible photodissociation. Biochem. J. 1955;61:153–171. doi: 10.1042/bj0610153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brunori M, Antonini G, Castagnola M, Bellelli A. Cooperative cyanide dissociation from ferrous hemoglobin. J. Biol. Chem. 1992;267:2258–2263. [PubMed] [Google Scholar]
- 25.Allocatelli CT, Cutruzzola F, Brancaccio A, Brunori M, Qin J, Lamar GN. Structural and functional characterization of sperm whale myoglobin mutants–Role of arginine(E10) in ligand stabilization. Biochemistry. 1993;32:6041–6049. doi: 10.1021/bi00074a015. [DOI] [PubMed] [Google Scholar]
- 26.Tang HL, Chance B, Mauk AG, Powers LS, Reddy KS, Smith M. Spectroscopic, electrochemical, and ligand-binding properties of the horse heart metmyoglobin His(64)-Tyr variant. Biochim. Biophys. Acta. 1994;1206:90–96. doi: 10.1016/0167-4838(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 27.Duah-Williams L, Hawkridge FM. The temperature dependence of the kinetics of cyanide dissociation from the cyanide complex of myoglobin studied by cyclic voltammetry. J. Electroanal. Chem. 1999;466:177–186. [Google Scholar]
- 28.Lin J, Merryweather J, Vitello LB, Erman JE. Metmyoglobin/azide: The effect of heme-linked ionizations on the rate of complex formation. Arch. Biochem. Biophys. 1999;362:148–158. doi: 10.1006/abbi.1998.0991. [DOI] [PubMed] [Google Scholar]
- 29.Jacobson T, Williamson J, Wasilewski A, Felesik J, Vitello LB, Erman JE. Azide binding to yeast cytochrome c peroxidase and horse metmyoglobin: comparative thermodynamic investigation using isothermal titration calorimetry. Arch. Biochem. Biophys. 2004;422:125–136. doi: 10.1016/j.abb.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 30.Mims WB, Davis JL, Peisach J. The accessibility of type-I Cu(II) centers in laccase, azurin, and stellacyanin to exchangeable hydrogen and ambient water. Biophys. J. 1984;45:755–766. doi: 10.1016/S0006-3495(84)84219-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mims WB. Envelope modulation in spin-echo experiments. Phys. Rev. B. 1972;5:2409–2419. [Google Scholar]
- 32.Astashkin AV, Raitsimring AM. Electron spin echo envelope modulation theory for high electron spin systems in weak crystal field. J. Chem. Phys. 2002;117:6121–6132. [Google Scholar]
- 33.Tan XL, Bernardo M, Thomann H, Scholes CP. Pulsed and continuous wave electron nuclear double-resonance patterns of aquo protons coordinated in frozen solution to high-spin Mn2+ J. Chem. Phys. 1993;98:5147–5157. [Google Scholar]
- 34.Dikanov SA, Tsvetkov YD. Electron Spin Echo Envelope Modulation (ESEEM) Spectroscopy. CRC Press; Boca Raton, FL: 1992. Chapter 2. [Google Scholar]
- 35.Espe MP, Hosler JP, Ferguson-Miller S, Babcock GT, McCracken J. A continuous-wave and pulsed EPR characterization of the Mn2+ binding site in Rhodobacter sphaeroides cytochrome c oxidase. Biochemistry. 1995;34:7593–7602. doi: 10.1021/bi00023a005. [DOI] [PubMed] [Google Scholar]
- 36.Kass H, MacMillan F, Ludwig B, Prisner TF. Investigation of the Mn binding site in cytochrome c oxidase from Paracoccus denitrificans by high-frequency EPR. J. Phys. Chem. B. 2000;104:5362–5371. [Google Scholar]
- 37.Van Gelder BF, Beinert H. Studies of the heme components of cytochrome c oxidase by EPR spectroscopy. Biochim. Biophys. Acta. 1969;189:1–24. doi: 10.1016/0005-2728(69)90219-9. [DOI] [PubMed] [Google Scholar]
- 38.Florens L, Schmidt B, McCracken J, Ferguson-Miller S. Fast deuterium access to the buried magnesium/manganese site in cytochrome c oxidase. Biochemistry. 2001;40:7491–7497. doi: 10.1021/bi0101188. [DOI] [PubMed] [Google Scholar]
- 39.Yoshikawa S, Caughey WS. Infrared evidence of azide binding to iron, copper, and non-metal sites in heart cytochrome c oxidase. J. Biol. Chem. 1992;267:9757–9766. [PubMed] [Google Scholar]
- 40.Kaila VRI, Verkhovsky MI, Hummer G, Wikstrom M. Glutamic acid 242 is a valve in the proton pump of cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 2008;105:6255–6259. doi: 10.1073/pnas.0800770105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lepp H, Svahn E, Faxen K, Brzezinski P. Charge transfer in the K proton pathway linked to electron transfer to the catalytic site in cytochrome c oxidase. Biochemistry. 2008;47:4929–4935. doi: 10.1021/bi7024707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.