

Abstract
DNA polymerases and aminoacyl-tRNA synthetases (ARSs) represent large enzyme families with critical roles in the transformation of genetic information from DNA to RNA to protein. DNA polymerases carry out replication and collaborate in the repair of the genome, while ARSs provide aminoacylated tRNA precursors for protein synthesis. Enzymes of both families face the common challenge of selecting their cognate small molecule substrates from a pool of chemically related molecules, achieving high levels of discrimination with the assistance of proofreading mechanisms. Here, the fidelity preservation mechanisms in these two important systems are reviewed, and similar features highlighted. Among the noteworthy features common to both DNA polymerases and ARSs are the use of multidomain architectures that segregate synthetic and proofreading functions into discrete domains; the use of induced fit to enhance binding selectivity; the imposition of fidelity at the level of chemistry; and the use of post-chemistry error correction mechanisms to hydrolyze incorrect products in a discrete editing domain. These latter mechanisms further share the common property that error correction involves the translocation of mis-incorporated products from the synthetic to the editing site, and that the accuracy of the process may be influenced by the rates of translocation in either direction. Fidelity control in both families can thus be said to rely on multiple elementary steps, each with its contribution to overall fidelity. The summed contribution of these “kinetic checkpoints” provides the high observed overall accuracy of DNA replication and aminoacylation.
Keywords: aminoacylation, aminoacyl-tRNA synthetases, DNA polymerases, fidelity
Despite challenges posed by reverse transcription of RNA and RNA editing, the paradigm of the central dogma that genetic information flows from DNA to RNA to protein remains intact. Accurate copying of a genome requires an array of DNA polymerases, repair enzymes, and accessory proteins that function as collaborative macromolecular assemblies to bring about DNA replication and repair. The selection for accurate replication is opposed by a need for genetic diversity, which is enhanced by multiple recombination pathways and the inherent error rate in the expression of genetic information. Individuals maintain flexibility by striking a balance between these opposing imperatives, thereby enhancing their responses to adverse environmental conditions (1, 2).
Here, two different families of enzymes that maintain the accuracy of genetic expression are compared, namely the DNA polymerases (3, 4) and the aminoacyl-tRNA synthetases (ARSs) (5-7). DNA polymerases replicate and repair the genome with varying degrees of processivity and accuracy. ARSs attach amino acids to their cognate tRNAs, thereby initiating protein synthesis. Each family catalyzes its respective reaction at sufficient speed and accuracy to maintain physiologically relevant growth rates, attempting to do so efficiently with respect to energy consumption. The enzymes in both families are characterized by complex, multi-domain architectures in which the synthetic and proofreading functions are segregated into different structural modules. For both systems, mistakes that occur at the synthetic active site are corrected in editing active sites, with an attendant cost in the consumption of NTP or dNTP equivalents. The parallel fashion in which polymerases and ARSs carry out these activities, and the similar architectures involved, has been recognized earlier (8).
While DNA polymerases and ARSs lack a significant evolutionary relationship, they nonetheless employ mechanistically similar error correction strategies. Both eliminate non-cognate substrates at multiple steps during the catalytic cycle, including during initial substrate binding, during pre-chemistry isomerization steps, during the chemical step itself, and by the application of post-synthetic editing mechanisms. Both employ kinetic checkpoints to eliminate non-cognate substrates, thereby reducing observed errors in replication and protein synthesis. In both systems, mis-acylated/mis-incorporated intermediates are translocated from the synthetic to editing site, where they undergo hydrolysis. While not convergent in the sense of employing identical catalytic architectures in unrelated structural frameworks, the shared mechanisms DNA polymerases and ARSs use to reduce errors illustrate how, in a broader sense, successful strategies for achieving fidelity recur broadly across the protein universe. As part of this review, a new model is proposed to account for the editing function by class I ARSs, building on the concept of a “double sieve” for editing by ARSs first elaborated by Fersht (9). Here, it will be proposed that the rate of partitioning by the CCA end of the tRNA between the synthetic and editing sites influences the choice between “pre-transfer” and “post-transfer” editing mechanisms. This revised role for translocation kinetics in editing is analogous to the role that kinetic partitioning plays in the mechanism of exonucleolytic proofreading in DNA polymerases.
The Multidomain Architecture of DNA Polymerase and Aminoacyl-tRNA Synthetase Families
DNA polymerases and ARSs possess multidomain architectures with separate modules for primary synthetic chemistry and critical editing functions (Figure 1). Notably, how conformational changes in, and collaboration between, these various subdomains regulates catalysis and overall fidelity remains a key question. DNA synthesis is performed by six different families (A, B, C, X, reverse transcriptase, and Y) that are specialized for genome replication, Okazaki fragment removal and re-synthesis, gap filling, error prone replication, and lesion bypass synthesis (3, 4, 10, 11). Polymerases are organized around the three principal domains originally identified in the Klenow fragment (KF) of DNA Pol I, colloquially referred to as the ‘palm, fingers, and thumb’, based on their superficial topological resemblance to a right hand (3, 4).
Figure 1.
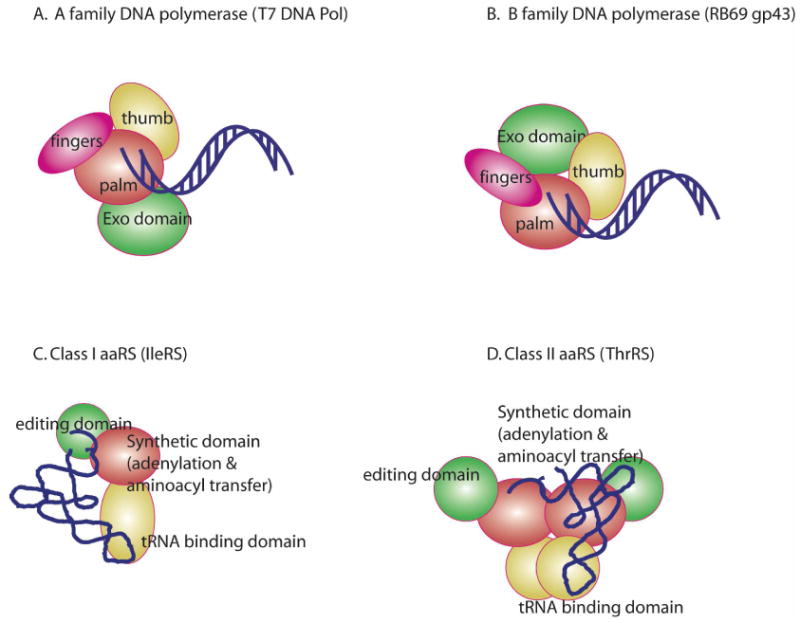
Cartoon representation of proofreading families of DNA polymerases and amninoacyl-tRNA synthetases. The N-terminal domain of RB69 DNA Pol in C. has been omitted for clarity
The palm domain is the most topologically conserved domain, and contains two highly conserved active site aspartate residues that coordinate the catalytic metal ions and provide a platform for the minor groove of the DNA duplex (3, 4). The α-helical rich fingers domain provides important interactions with incoming nucleotide and the complementary base in the template. The thumb domain is typically an antiparallel α helical or β strand hairpin motif; its interactions with the template increase the processivity of DNA synthesis. Many polymerases in the A- and B-families contain a separate domain that confers 3′-5′exonucleolytic proofreading activity (3). In the A- and B- families, the exo domain is located on either side of the polymerase active site (12, 13), while in the major E. coli DNA Pol III replicative complex, the 3′-5′activity resides in its own dedicated subunit (MutD). In general, the higher the ratio of exonuclease/polymerase activity, the lower the observed mutation frequency. This review will focus on the high fidelity A- and B-family polymerases, which feature separate polymerization and exonucleolytic editing domains. Polymerases in the other families, which are either much more complex or do not possess error correction functions, are not discussed here in the interest of space. For these, see reviews (14, 15).
Aminoacyl-tRNA synthetases divide into two classes, each of which includes 10-12 families specific for one of the proteogenic amino acids. Class I ARSs share an α/β Rossmann/nucleotide binding fold, and possess HIGH and KMSKS peptide motifs that function in ATP binding (5, 7). In class Ia and Ib enzymes, tRNA recognition domains are appended to the C-terminal side of the catalytic domain (5). Other independently folded domains (Connection Peptides or CP-I and CP-II) are inserted in the Rossmann fold of Class Ia enzymes (7), and provide tRNA recognition and editing functions. Class II enzymes all contain an α + β catalytic domain first observed in SerRS (16), but also three degenerate sequence motifs (Motif 1,2, and 3) that define the dimer interface and ATP binding site (17). Class II enzymes also contain separate domains to assist in tRNA recognition, as well as editing functions (18, 19).
The Initial Selection of Substrates by Polymerases and tRNA Synthetases
The DNA polymerization reaction features addition of a nucleoside monophosphate (NMP) to the growing 3′ end of a DNA primer template complex:
In each incorporation cycle, the 3′ OH primer terminus attacks the α phosphate of the incoming dNTP to form a phosphodiester bond, producing pyrophosphate as product (3). In bacteria, the accuracy of replicative DNA polymerases reflects the cumulative contributions of simple base pairing, induced fit by the active site, and 3′-5′ exonucleolytic proofreading (2, 20). For T7 DNA polymerase, the summed effect of these mechanisms leads to the removal of 99.9-99.99% of all mis-incorporation events (2, 20, 21). The error rate of aminoacylation is also on the order of 10-6, which is nearly 100-1,000 times more accurate than overall protein synthesis (1, 22). Aminoacyl-tRNA synthesis is more complex than DNA polymerization, featuring two distinct half reactions (22):
| (1) |
| (2) |
The amino acid is activated by condensation with ATP to form an on-enzyme adenylate intermediate in the first reaction; this intermediate is attacked by the 2′OH (Class I) or 3′OH (Class II) of the tRNA's A76 nucleotide in a subsequent step, producing aminoacyl-tRNA and AMP.
DNA polymerases and ARSs exert specificity for the correct nucleotide/amino acid at the level of initial binding or other pre-chemistry steps, at the level of chemistry itself, or by virtue of editing reactions that hydrolyze mis-incorporated nucleotides or the mis-acylated amino acid (Figure 2). Initial selection of small molecule substrates (Figure 2, Step 2) provides the first step where polymerases and ARSs exhibit significant control over fidelity. With some variability, polymerases prefer correctly paired dNTPs over mis-matched dNTP by factors of 10 to 1,000, (2). Hydrogen bonding between the incoming dNTP and the template provides the first level of specificity because the nascent base pair must conform to duplex DNA geometry (2, 23, 24). Polymerases appear to further enhance differences between correct and incorrect base pairs by constraining the geometry and translational freedom of bound dNTPs (thereby reducing ΔS), and by excluding water (24). The ARSs lack the templating mechanism of DNA polymerases to select their cognate amino acids, but still achieve higher accuracies of amino acid selection than what would be predicted from theoretical considerations alone. The discrepancy has been attributed to an underestimate of the energy available from van der Waals interactions, or from the pre-formed nature of the amino acid binding pocket on the enzymes, which constitutes a “pre-payment” of some of the entropic costs associated with binding (9).
Figure 2.

Idealized kinetic schemes for synthesis and editing by replicative DNA polymerases (A) and aminoacyl-tRNA synthetases (B). In both schemes, Es refers to the enzyme with substrates bound in the synthetic site, and Ee refers to enzyme with substrates bound in the editing site. E in panel A refers to the DNA polymerase, while E in panel B refers to the ARS. Step 2 in panel A (polymerases) refers only to the binding step for the incoming dNTP, while Step 2 in panel B (ARSs) include both the amino acid and ATP binding step and adenylation reaction. The merging of the binding and adenylation chemistry step for the ARSs is depicted in this fashion to simplify the global comparison between the two families. Additional elementary steps that may follow the binding of substrates and precede chemistry have been omitted by clarity. These include domain closure and other conformational changes, which may be rate limiting in some systems. In Panel A, the translocation of the primer-template from the editing site to the synthetic site (Step 6) is shown for aesthetic reasons to be in equilibrium with the species Es:DNA(n+1), but this is formally incorrect. Technically, the true species is Es:DNA(n), which is the immediate product of Step 1.
For both polymerases and synthetases, formation of a closed complex occurs with correct substrates, consistent with the classical model of “induced fit”. Previously, it was argued that induced fit cannot contribute to specificity because all substrates are constrained to pass through the same transition state (25). If, however, the enzyme can favor correct geometry for cognate substrates and promote unfavorable geometries for non-cognates, than a substantially higher transition state barrier should result for mismatched substrates (26). Such effects can provide kinetic partitioning, whereby correct substrates proceed forward along the reaction path, and incorrect substrates experience significant kinetic barriers to further reaction.
In A- and B-family DNA polymerases, initial complex formation with primer template is promoted by domain closure by the thumb domain. Subsequent cognate dNTP binding to produce the “closed complex” results involves conformational closure by the fingers domain, which sandwiches the incoming nucleotide and template base between the duplex DNA and the protein (13). This orients the primer 3′OH for nucleophilic attack on the incoming dNTP α-phosphate, aided by coordinated metal ions and neighboring enzyme groups. In some polymerases (i.e., KF, T7 DNA Pol, HIV-RT, Pol η and Dpo4), a fast chemistry step is flanked by slower conformational changes that may be related to domain motions (27). In other enzymes (e.g. T4 DNA Pol and Pol β), the chemistry step is slower than the conformational changes, or the data are unclear.
While the early mechanistic studies on polymerases led to the initial suggestion that domain closure constitutes the rate-determining step, later work showed that domain closure is much faster than the rate of polymerization, and that multiple pre-chemistry steps involving additional conformational changes contribute to accurate nucleotide selection (27-29). The detection of mismatched nucleotides appears to involve a selection step that precedes domain closure, with the selection of dNTPs over rNTPs concurrent with domain closure (30). Thus, multiple elementary steps that follow the initial encounter of the dNTP with the polymerase may serve as “kinetic checkpoints” in the pathway, with their contribution to fidelity being a function of their free energy barrier height (27, 30). For T7 DNA Pol, selection between matched and mismatched nucleotides at the level of ground state binding is modest, but the net Kd's for these substrates, which incorporate the influence of domain closure and other conformational changes, differ by a factor of at least 103(31).
ARSs similarly use “induced fit” mechanisms, ranging from local conformational changes of secondary structure to entire rigid body motions to structure their amino acid binding pockets (32, 33). Such motions help to assemble a network of hydrogen bonds between active site residues and polar functional groups of the amino acid substrates, and position residues for chemistry (34). In the prolyl- system, non-cognate substrates like alanine may elicit incomplete conformational changes, rendering the adenylate susceptible to attack by solvent (35-37). In the glutaminyl- and glutamyl- systems, induced fit interactions with the tRNA CCA end help structure the amino acid binding pocket (38), or ensure a productive conformation of ATP (39). By contrast, ARSs that recognize non-polar amino acids (i.e., Tyr, Phe, Cys, Ile, Leu, and Val) are characterized by fairly rigid amino acid binding pockets that maximize van der Waals and stacking interactions with the R- groups of these amino acid substrates (7). For both DNA polymerases and ARSs, therefore, selectivity for the correct nucleotide and the correct amino acid can be increased by mechanisms that follow initial binding and precede chemistry, including induced fit and other first order conformational changes.
Fidelity Control at the level of chemistry in polymerases and ARSs
DNA polymerases and ARS both exploit the catalytic inefficiency associated with the sub-optimal geometry of non-cognate reactants as a common fidelity control mechanism. In DNA Pol β, use of the non-hydrolyzable ATP analog AMPCPP and Mn2+ allowed complexes of matched and mismatched nucleotides in a near transition state configuration to be captured (40). Despite the proper location and orientation of the incoming nucleotide in the dG-dAMPCPP complex, the primer template is shifted 3 Å upstream from its position in the complex with matched nucleotide. The resulting staggered conformation of the newly formed base prevents formation of a normal Watson-Crick (W/C) interaction. The primer terminus is rotated away from the incoming nucleotide, lengthening the distance between O3′ and the α-phosphate, and distorting the geometry from in-line attack. For Pol β, the ratio (kpol/Kd,app)corr/(kpol/Kd,app)incorr is on the order of 104-105 fold, suggesting that unfavorable geometry creates a high free energy barrier to product formation (40). This agrees with earlier predictions from a study involving calculations and modeling (41). Non-native transition states may similarly be important in enforcing fidelity in T7 DNA Pol. Here, binding of a mismatched nucleotide brings about a ‘misaligned’ state with fluorescence changes distinct from those accompanying the matched nucleotide (31). Currently, there is no direct structural evidence that mismatched nucleotides induce a different conformation in the T7 DNA Pol system. However, if the precedent established by complexes in the Pol β system holds, such misaligned ternary complexes are likely to be observed in the high fidelity A- and B-family polymerases as well.
Two examples of near-cognate ARS substrate complexes in ‘closed conformations’ have been reported where the sub-optimal orientation of key reacting groups imposes a block to chemistry. For GlnRS, the hydrogen-bonding interactions made by the enzyme to distinguish between the R groups of substrates glutamine and glutamate are not discriminating, involving a conserved tyrosine and bound water molecules with incomplete co-ordination spheres (42). Recently, structures of GlnRS, tRNAGln, AMPCPP, and glutamine or glutamate have been reported that provide a potential structural basis for amino acid discrimination (43). While the GlnRS active site can bind Gln and Glu using similar hydrogen bonding networks involving constrained water molecules and polar side chains, conserved Arg30 may contribute a polar/electrostatic interaction that may increase the distance between the carboxyl nucleophile glutamate and the α-phosphate by at least 1 Å. The resulting unfavorable geometry may account for at least part of the 107 -fold decrease in catalytic efficiency observed with glutamate relative to glutamine.
The tRNA fidelity of the E. coli AspRS system may similarly rely on suboptimal geometry of non-cognate reactants (44). The heterologous yeast tRNAAsp is aminoacylated 1000 times less efficiently by the prokaryotic AspRS than the cognate tRNA from the same species, with comparable Km parameters (44). In the cognate complex, both tRNAs make identical interactions with their respective protein monomers, and the 3′ OH is positioned within 3 Å of the scissile bond. In the first of the two tRNAs in the heterologous complex, the anticodon stem–loop of the tRNA is bound by the enzyme, but the acceptor stem is partially disordered, as if captured in the process of approaching the active site. The second tRNA is fully docked, and nearly all interactions with the synthetase are as observed in the homologous complex. The terminal A76 nucleotide, however, is located too far from the amino acid carboxylate, and possesses the wrong geometry for attack. Thus, in two examples where ARSs are confronted by near-cognate substrates, the mis-aligned geometry of the closed state reasonably explains the observed poor catalytic efficiency of the complex, and suggests that the fidelity of these systems depends in part on their ability to direct these substrates into non-optimal transition states.
These correlations between altered structure and reduced rate of chemistry serve as models for how tRNA discrimination is controlled kinetically. The discrimination of tRNAs by ARSs (referred to as “tRNA identity”) relies both on direct contacts between synthetases and tRNA functional groups (“direct readout”), as well as modulation of RNA-protein affinity by global features of tRNA structure (“indirect readout”) (45, 46). Pre-steady state kinetics studies in the GlnRS (47), HisRS (48), and TrpRS (49) systems indicate that mutation of key recognition determinants on the tRNAs lead invariably to significant decreases in the rate of aminoacyl transfer, which may be the direct consequence of mis-alignment of the tRNA for chemistry. If the view provided by the AspRS heterologous complex is accurate, one major consequence may be misalignment of the CCA, with a concomitant decrease in the rate of transfer.
Fidelity Control by post synthetic-chemistry mechanisms: the importance of editing functions
As shown by the evolution of 3′-5′ exonucleolytic and other proofreading functions, initial selection at the level of binding, induced fit, and selection during the chemistry step alone are insufficient to achieve the fidelity necessary for genome replication and protein synthesis. In polymerases, the deletion or inactivation of the exonuclease domain leads to an increase in error frequency in vitro, and in the increase in vivo error in Pol III (2, 20). In general, exo domains have been estimated to contribute no more than 100-fold to overall accuracy of DNA replication, and somewhat less for non-replicative polymerases like the KF (2, 20).
In the aminoacyl-tRNA synthetases, induced fit and preferential rates of chemistry are sufficient for discrimination of many of the polar amino acids, but discrimination between many of the non-polar amino acids requires specialized editing functions (6, 50, 51). The clearest phenotypic consequence of defects in ARS editing is increased sensitivity to amino acid analogs. Editing may thus reduce the ‘infiltration’ of non-proteogenic amino acids into proteins (52, 53). These mis-incorporations may also decrease protein stability. A mutation in the editing domain of alanyl-tRNA synthetase is genetically linked to the neurodegenerative phenotypes of ataxia and cerebellar Purkinje cell loss in the “sticky” mouse, and apparently increases the extent of protein misfolding in these animals (54).
Proofreading functions in polymerases and tRNA synthetases are localized in specialized domains that perform chemistry distinct from that of the respective synthetic domain. The editing domains of A- and B-family polymerases share only limited structural homology (55), but exhibit similar phosphodiesterase activities to remove the 3′ terminal nucleotides from the mismatched primer terminus (56, 57). In all cases, the 3′ to 5′exonuclease domain displays a preference for a single stranded 3′OH chain terminus of a base paired duplex, and must work coordinately with the polymerase domain to achieve efficient error correction. Notably, because a mismatch may involve any of the four dNTPs, the exo site possesses no specificity with respect to the nucleotide at the primer terminus. A mechanism must also be in place to limit the number of nucleotides cleaved off the 3′ end, thus avoiding the unnecessary removal of properly templated nucleotides. Exonuclease function is also not the formal reverse of DNA polymerization, which is the pyrophosphorolysis reaction. Pyrophosphorolysis requires a completely paired template primer, yet the exonucleolytic activity is inactive on these substrates.
Editing functions in ARSs are similarly localized to domains separate from the catalytic domain, and catalyze a deacylation reaction that is specific for the cognate tRNA, and which specifically excludes the cognate amino acid (6, 50, 51). The selectivity of the editing site represents the critical difference between editing by DNA polymerases and ARSs. In the editing ARSs, adenylates derived from non-cognate amino acids, and misacylated cognate tRNAs are both potentially important substrates. In Fersht's “double sieve” mechanism (9), aminoacyl transfer in the synthetic site is followed by translocation of the mis-acylated CCA end of the tRNA to a distinct editing site (the “fine sieve”) where amino acids smaller than the cognate are removed. Thus, an intrinsic feature of the model is that the editing site performs a substrate selection that is the inverse of the synthetic site. Consistent with the double sieve theory, expansion of the binding pocket by truncating side chains that constitute the walls causes wild type ARSs to edit the cognate amino acid substrate (58, 59). Significantly, the “double sieve model” fails to provide a complete picture of ARS editing, because some ARSs are able to actively edit amino acids that are either larger than cognate, or of similar size but different chemistry (51). Rather than a simple sieve, ARS editing domains must rely on a complex interplay of steric, hydrogen bonding, electrostatic and other forces to accept non-cognate amino acids and reject the cognate amino acids.
Kinetic partitioning mechanisms in editing
As shown in Figure 2A, a proofreading DNA polymerase: primer-template complex can undergo one of three possible fates upon synthesis of each new base pair: synthesis of the next base pair according to templating rules (Step 2(n+1)); translocation of the 3′ primer end to a exonucleolytic proofreading site (Step 4); or dissociation of the primer-template from the polymerase (Step 7). Depending on the nature of the 3′primer terminus and the strength of duplex base pairing, the rates of these events will differ. This “kinetic partitioning” determines the probability of the error being corrected. For all DNA polymerases that possess 3′-5′ exo activity, proofreading is triggered when a DNA duplex terminating in a mismatch undergoes strand separation, allowing three to five nucleotides of the primer end to be translocated from the polymerase to the exo site (60). This process is not strictly dependent on synthesis of a new base pair, because the branched pathway is available each time a proofreading polymerase binds a new primer-template molecule.
The T7 DNA Pol system provides a good particularly clear example of kinetic partitioning (21, 31). Owing its high (700 sec-1) rate of single stranded DNA degradation, the activity of the exonuclease domain is tightly controlled, principally through translocation kinetics. With a standard duplex with a correct W/C base pair at its primer terminus, the rate of incorporation of the next base pair (300 sec-1) exceeds the rate of either translocation to the editing site (∼2.3 sec-1) or dissociation of primer template (0.2 sec-1). If, however, the primer terminus contains one or more mismatches, the rate of successive polymerization can decrease precipitously from 300 to 0.3 sec-1, and the rate of translocation to the exo site will increase. Once the 3′ end occupies the exo site, single or multiple rounds of exonucleolytic degradation are theoretically possible. However, the relatively rapid (700 sec-1) translocation of the 3′primer terminus from the exo site to the polymerase site provides a kinetic check to minimize removal of multiple nucleotides. Thus, a mismatch at the primer terminus promotes proofreading in two ways. First, it brings about fraying of the end of the duplex, facilitating the transfer to the exo site and, secondly, it decreases the rate of the next polymerization event, providing the sufficient time to allow proofreading to occur. Proofreading by polymerases can also be “processive” or “distributive”, depending on the relative rates of translocation and dissociation of template primer. If the primer-template is released before the next polymerization step, editing can occur by re-binding of the mispaired 3′ end to the editing site (61).
The ARSs provide two different post-chemistry opportunities to eliminate non-cognate amino acids, namely after the formation of the non-cognate adenylate (Figure 2B, Step 2), and after the formation of the mis-acylated tRNA (Figure 2B, Step 3). Decomposition of the mis-activated amino acid before transfer to the tRNA is colloquially referred to as “pre-transfer” editing, while hydrolysis of the mis-acylated tRNA is referred to as “post-transfer” editing (6, 50, 51). Post-transfer editing most closely resembles proofreading by DNA polymerases, as it requires covalent attachment of the discriminated entity to the 3′ end of a nucleic acid macromolecule, a dedicated domain distinct from the synthetic site for deacylation, and a translocation mechanism in which the end of the tRNA traverses between synthetic and editing sites. Relatively short (3-5 nucleotides) single stranded oligonucleotide segments undergo translocation in both cases (8). Notably, the misacylated tRNA intermediate can be detected in rapid quench kinetics experiments, and its rate of formation suggests that it is kinetically competent for the editing (62).
There are, however, some important differences between editing performed by the two families. Firstly, the unpaired nature of the CCA end means that a major contributor to the free energy that helps DNA polymerases drive the 3′ end to the polymerase active site (i.e., re-hybridization of the DNA duplex) is absent, such that the aminoacylation site need not represent the lowest energy state for the CCA end. Secondly, ARSs must discriminate between correct and near/non-cognate amino acids at their editing sites, relying on shape and charge complmentarity determining interactions. However, the translocation process may contribute to editing specificity, because several ARSs (including LeuRS and AlaRS) exhibit active post-transfer editing against substrates that are larger than the cognate, and mutations have been identified that lie outside the editing domain and yet still influence specificity (63, 64). Significantly, it also remains to be established whether or not translocation between the aminoacylation and editing sites occurs for correctly acylated substrates. Thus, significant gaps remain in our understanding of the role of the post transfer editing translocation process and its kinetics.
In addition to being either tRNA dependent or tRNA independent, the pre-transfer mechanism potentially includes hydrolysis of the adenylate in the synthetic site, release and hydrolysis of the adenylate in solution, or more controversially, translocation to, and subsequent hydrolysis in, a discrete editing active site (6, 51). In the simplest pathways, non-cognate amino acids such homocysteine, homoserine, and ornithine can undergo “self-editing” tRNA-independent reactions that involve intramolecular attack of the side chain thiolate/hydroxyl on the amino acid carbonyl forming a thiolactone, lactone, or Orn-lactam (50). Mechanisms also exist by which the cognate tRNA can assist in the hydrolysis of the adenylate (after Step 2), but before the aminoacyl transfer step. In these mechanisms, the tRNA serves as an active co-factor, eliciting hydrolysis of the non-cognate adenylate without participating in aminoacyl transfer (6). In fashion not yet fully understood, DNA aptamers have been selected that can mimic this role (65).
The site on the enzyme where pre-transfer editing occurs remains to be resolved. In one model, the tRNA enhances the hydrolysis of the mis-activated adenylate, stabilizing an altered conformational state that allows hydrolytic water molecules to enter the active site (36). Consistent with this model, non-editing ARSs can engage in futile cycles of cognate adenylate synthesis and destruction, if the ability of their cognate tRNAs to be aminoacylated is compromised (48, 66). The non-cognate adenylate has also been proposed to undergo translocation to, and hydrolysis in, the editing site. This model would require an unstable intermediate to diffuse over a distance of some 35-40 Å between two active sites, without hydrolysis in solution. Data cited in support of this mechanism include crystallographic soaking experiments indicating that the sites for binding of pre-transfer and post-transfer editing analogs overlap (67-69). In addition, mutational data (59) and results from fluorescence resonance energy experiments (70) have also been invoked in support of the adenylate translocation model.
Historically, studies investigating ARSs editing have typically relied on two steady state kinetic assays, namely the “total editing” assay, which measures the consumption of ATP in the presence of cognate tRNA and non-cognate amino acid, and the “post transfer editing assay”, which directly assesses the ability of a given ARSs variant to deacylate the cognate tRNA with a near or non-cognate amino acid (71). While both assays may be qualitatively useful, they are limited in their ability to directly distinguish between pre-transfer and post-transfer editing models, and provide insights into the thermodynamic preference of misacylated tRNA for the synthetic and editing site. If studies on DNA polymerases are a useful guide, structural biology and rapid kinetics should prove to be useful in identifying elementary steps that exist between substrate binding and reaction chemistry, and determining their rates.
A new model for editing by aminoacyl tRNA synthetases that invokes a role for kinetic partitioning
A new model for class I ARS editing can be proposed that contains some of the features of editing by DNA polymerases. In this model (Figure 3), the tRNA serves as an important co-factor in the entire process, with a “default” or “resting” state in which the CCA end occupies (or directly abuts) the editing site. The ARS is proposed to exist in four discrete states: (1) an apo state without ligands; (2) a “resting state” tRNA complex with the CCA end bound in the editing site; (3) a “synthetic mode”, where adenylate forms in the synthetic site, and the CCA translocates from the editing to the synthetic site, allowing aminoacyl transfer; and (4) an “editing mode” where the aminoacylated-CCA end (with cognate or non-cognate amino acid) translocates back to the editing site, allowing inspection and hydrolysis. With a cognate amino acid, the binding of CCA is not stable, and the aminoacylated tRNA is released into solution or captured directly by EF-Tu. If the amino acid is a non-cognate that passes the shape/charge restriction test, it is accepted into the editing site, and undergoes deacylation. This returns the tRNA to the “resting state”.
Figure 3.
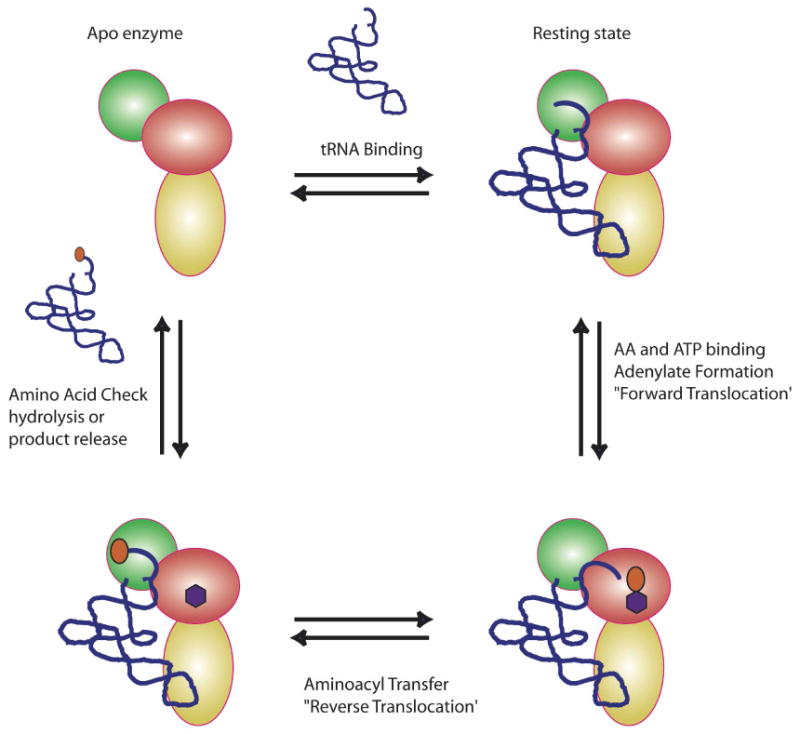
A new model to explain class I editing. The coloring scheme for the representation of a monomeric class I ARSs is as depicted in Figure 1C. The amino acid is depicted as an orange oval, and the ATP as a purple hexagon. Details of the model are provided in the text.
This model has precedents in the early studies of Yarus, who proposed that product release constitutes the rate-limiting step for the isoleucyl enzyme (72). Notably, this early study also included a model in which IleRS contains a tRNA entry/exit site that is distinct from the site of amino acid activation and aminoacyl transfer, an explicit feature of the model in Figure 3. In addition, in the crystallographic complexes of editing class I ARSs with their cognate tRNAs, the preferred orientation of the tRNA CCA end is in the editing site, and not in the synthetic site (8, 73, 74). In LeuRS, binding of the CCA end in the editing can be effectively competed by the post transfer editing analog Nva2AA, but not by a pre-transfer editing analog (74).
The editing site may thus constitute the thermodynamically preferred binding site for the CCA end in editing class I ARSs. Evidence that the incoming tRNA must sample the editing site is further provided by observations involving the boron based anti-LeuRS inhibitor, AN2690 (75). The mechanism of action for AN2690 involves the formation of a covalent adduct with the cis diols of A76, an interaction that is specific to the editing site. The non-competitive behavior of this inhibitor, and the tRNA dependent nature of that inhibition are consistent with the model that, even in the presence of leucine and ATP, the CCA end of the tRNA must sample the editing site before translocating to the synthetic site.
Many of the features of this model can readily be tested by measurements of the strength of the ARS:tRNA complex under various conditions, and by measurements of the effect of translocation kinetics on editing function. If the model is correct, then mutations in the editing domain that weaken interactions with the CCA end (or post transfer editing analog inhibitors) would be expected to reduce the affinity of tRNA for the ARS. Also, such mutations might well reduce pre-transfer editing by diminishing the residence time of the CCA end in the editing site, allowing translocation to the synthetic site and misacylation to occur before hydrolysis of the adenylate in the active site. Conversely, mutations that strengthen the CCA end-editing site interaction would be predicted to increase overall tRNA affinity, affect product release kinetics, and potentially strengthen pre-transfer editing. Finally, the model also raises the possibility that discrimination between cognate and non-cognate amino acids is influenced by the kinetics of translocation between sites, a prediction testable by identifying mutations that lie outside of the editing site, and yet affect discrimination between cognate and non-cognate amino acids.
Summary
DNA polymerases and ARSs both constitute complex, multidomain enzymes that synthesize macromolecular intermediates that are critical in biological information transfer. Both systems achieve rates of synthesis that support physiological growth rates, and yet preserve relatively high rates of fidelity (on the order of 1 in 106 mistakes per synthetic event). This balance is achieved through the use of distinct synthetic and proofreading domains, with antagonistic catalytic functions. The immediate product of the synthetic site (newly incorporated nucleotide and adenylate or acylated-tRNA, respectively) serves as an intermediate that undergoes partitioning between further biosynthesis (new synthesis or elongation factor capture) or processing in the editing site. Partitioning appears to involve the opportunity for physical translocation between synthetic and editing sites in both systems, and the degree to which the kinetics of translocation influence editing in both systems is a critical question.
In DNA polymerases, the editing domains apparently have no physical basis to discriminate between a correct incorporation and a mis-incorporation event. The exonucleolytic function is relatively non-specific, and multiple rounds are effectively suppressed by the fast kinetics of transfer from exo to synthetic site. In the case of the ARSs, which must make extremely fine distinctions against amino acid substrates that differ by no more than a single methyl group, the canonical view is that the specificity of the editing site is mirror image of the synthetic site, accepting smaller substrates but excluding the cognate and all larger substrates. Many ARSs apparently employ multiple checkpoints to exclude near/non-cognates, and apparently can be biased in favor of one mechanism or another by relatively subtle mutations. The role of translocation kinetics in synthetase error correction remains an interesting and largely unresolved question, with many implications for the design of family specific inhibitors. The significance of the apparent similarity in editing mechanism in the two families remains to be explored at a more mechanistic level, an effort that will surely benefit from further application of structural biology to trap additional predicted intermediates, and rapid kinetics to extract the elementary rate parameters for the aminoacyl synthesis and editing reaction.
Acknowledgments
The author wishes to thank Dr. Matthew, Hogg, Dr. Sylvie Doublié and Dr. Susan Martinis helpful discussions and comments.
Abbreviations
- ARS
aminoacyl-tRNA synthetase
- CP-I
connective polypeptide I
- DNAP
DNA polymerase
- dNTP
deoxyribonucleoside triphosphate
- GluRS
glutamyl-tRNA synthetase
- GlnRS
glutaminyl-tRNA synthetase
- HisRS
histidyl-tRNA synthetase
- KF
Klenow fragment of DNA polymerase I from Escherichia coli
- PPi
pyrophosphate
- ProRS
prolyl-tRNA synthetase
- SerRS
seryl-tRNA synthetases
- TrpRS
tryptophanyl-tRNA synthetase
References
- 1.Jakubowski H, Goldman E. Editing of errors in selection of amino acids for protein synthesis. Microbiol Rev. 1992;56:412–429. doi: 10.1128/mr.56.3.412-429.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunkel TA. DNA replication fidelity. J Biol Chem. 2004;279:16895–16898. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- 3.Steitz TA. DNA polymerases: structural diversity and common mechanisms. J Biol Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 4.Patel PH, Loeb LA. Getting a grip on how DNA polymerases function. Nat Struct Biol. 2001;8:656–659. doi: 10.1038/90344. [DOI] [PubMed] [Google Scholar]
- 5.Cusack S. Aminoacyl-tRNA synthetases. Curr Opin Struct Biol. 1998;7:881–889. doi: 10.1016/s0959-440x(97)80161-3. [DOI] [PubMed] [Google Scholar]
- 6.Hendrickson TL, Schimmel P. Transfer RNA-dependent amino acid discrimination by aminoacyl-tRNA synthetases. In: Lapointe, Brakier-Gingras L, editors. Translation Mechanisms. Kluwer/Plenum; New York: 2003. pp. 34–64. [Google Scholar]
- 7.Ibba M, Francklyn C, Cusack S. The Aminoacyl-tRNA Synthetases. Landes Bioscience; Georgetown, Texas: 2005. [Google Scholar]
- 8.Silvian LF, Wang J, Steitz TA. Insights into Editing from an Ile-tRNA Synthetase Structure with tRNAIle and Mupiricin. Science. 1999;285:1074–1077. [PubMed] [Google Scholar]
- 9.Fersht AR. Enzymatic editing mechanisms and the genetic code. Proc Royal Soc London Series B. 1981;212:351–379. doi: 10.1098/rspb.1981.0044. [DOI] [PubMed] [Google Scholar]
- 10.Beard WA, Wilson SH. Structural insights into the origins of DNA polymerase fidelity. Structure. 2003;11:489–496. doi: 10.1016/s0969-2126(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 11.Steitz TA. Visualizing polynucleotide polymerase machines at work. EMBO J. 2006;25:3458–3468. doi: 10.1038/sj.emboj.7601211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Sattar AK, Wang CC, Karam JD, Konigsberg WH, Steitz TA. Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]
- 13.Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 14.O'Donnell M. Replisome architecture and dynamics in Escherichia coli. J Biol Chem. 2006;281:10653–10656. doi: 10.1074/jbc.R500028200. [DOI] [PubMed] [Google Scholar]
- 15.Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc Natl Acad Sci USA. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cusack S, Berthet-Colominas C, Härtlein M, Nassar N, Leberman R. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.4 Å. Nature. 1990;347:249–265. doi: 10.1038/347249a0. [DOI] [PubMed] [Google Scholar]
- 17.Eriani G, Delarue M, Poch O, Gangloff J, Moras D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990;347:203–206. doi: 10.1038/347203a0. [DOI] [PubMed] [Google Scholar]
- 18.Sankaranarayanan R, Dock-Bregeon AC, Romby P, Caillet J, Springer M, Rees B, Ehresmann C, Ehresmann B, Moras D. The structure of threonyl-tRNA synthetase-tRNATyr complex enlightens its repressor activity and reveals an essential zinc ion in the active site. Cell. 1999;97:371–381. doi: 10.1016/s0092-8674(00)80746-1. [DOI] [PubMed] [Google Scholar]
- 19.Crepin T, Yaremchuk A, Tukalo M, Cusack S. Structures of two bacterial prolyl-tRNA synthetases with and without a cis-editing domain. Structure. 2006;14:1511–1525. doi: 10.1016/j.str.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 20.Kunkel TA, Bebenek K. DNA replication fidelity. Ann Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 21.Donlin MJ, Patel SS, Johnson KA. Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry. 1991;30:538–546. doi: 10.1021/bi00216a031. [DOI] [PubMed] [Google Scholar]
- 22.Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 23.Johnson KA. Conformational coupling in DNA polymerase fidelity. Ann Rev Biochem. 1993;62:685–713. doi: 10.1146/annurev.bi.62.070193.003345. [DOI] [PubMed] [Google Scholar]
- 24.Goodman MF. Hydrogen bonding revisited: geometric selection as a principal determinant of DNA replication fidelity. Proc Natl Acad Sci USA. 1997;94:10493–10495. doi: 10.1073/pnas.94.20.10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fersht AR. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. W.H. Freeman and Company; New York: 1999. [Google Scholar]
- 26.Post CB, Ray WJ., Jr Reexamination of induced fit as a determinant of substrate specificity in enzymatic reactions. Biochemistry. 1995;34:15881–15885. doi: 10.1021/bi00049a001. [DOI] [PubMed] [Google Scholar]
- 27.Joyce CM, Benkovic SJ. DNA polymerase fidelity: kinetics, structure, and checkpoints. Biochemistry. 2004;43:14317–14324. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 28.Purohit V, Grindley ND, Joyce CM. Use of 2-aminopurine fluorescence to examine conformational changes during nucleotide incorporation by DNA polymerase I (Klenow fragment) Biochemistry. 2003;42:10200–10211. doi: 10.1021/bi0341206. [DOI] [PubMed] [Google Scholar]
- 29.Rothwell PJ, Mitaksov V, Waksman G. Motions of the fingers subdomain of klentaq1 are fast and not rate limiting: implications for the molecular basis of fidelity in DNA polymerases. Mol Cell. 2005;19:345–355. doi: 10.1016/j.molcel.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 30.Joyce CM, Potapova O, Delucia AM, Huang X, Basu VP, Grindley ND. Fingers-closing and other rapid conformational changes in DNA Polymerase I (Klenow fragment) and their role in nucleotide selectivity. Biochemistry. 2008;47:6103–6116. doi: 10.1021/bi7021848. [DOI] [PubMed] [Google Scholar]
- 31.Tsai YC, Johnson KA. A new paradigm for DNA polymerase specificity. Biochemistry. 2006;45:9675–9687. doi: 10.1021/bi060993z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiu X, Janson CA, Blackburn MN, Chhohan IK, Hibbs M, Abdel-Meguid SS. Cooperative structural dynamics and a novel fidelity mechanism in histidyl-tRNA synthetases. Biochemistry. 1999;38:12296–12304. doi: 10.1021/bi990482v. [DOI] [PubMed] [Google Scholar]
- 33.Torres-Larios A, Sankaranarayanan R, Rees B, Dock-Bregeon AC, Moras D. Conformational movements and cooperativity upon amino acid, ATP and tRNA binding in threonyl-tRNA synthetase. J Mol Biol. 2003;331:201–211. doi: 10.1016/s0022-2836(03)00719-8. [DOI] [PubMed] [Google Scholar]
- 34.Arnez JG, Augustine JG, Moras D, Francklyn CS. The first step of aminoacylation at the atomic level in histidyl-tRNA synthetase. Proc Natl Acad Sci USA. 1997;94:7144–7149. doi: 10.1073/pnas.94.14.7144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yaremchuk A, Tukalo M, Grotli M, Cusack S. A succession of substrate induced conformational changes ensures the amino acid specificity of Thermus thermophilus prolyl-tRNA synthetase: comparison with histidyl-tRNA synthetase. J Mol Biol. 2001;309:989–1002. doi: 10.1006/jmbi.2001.4712. [DOI] [PubMed] [Google Scholar]
- 36.Hati S, Ziervogel B, Sternjohn J, Wong FC, Nagan MC, Rosen AE, Siliciano PG, Chihade JW, Musier-Forsyth K. Pre-transfer editing by class II prolyl-tRNA synthetase: Role of aminoacylation active site in “selective release” of noncognate amino acids. J Biol Chem. 2006;281:27862–27872. doi: 10.1074/jbc.M605856200. [DOI] [PubMed] [Google Scholar]
- 37.Splan KE, Ignatov ME, Musier-Forsyth K. Transfer RNA modulates the editing mechanism used by class II prolyl-tRNA synthetase. J Biol Chem. 2008;283:7128–7134. doi: 10.1074/jbc.M709902200. [DOI] [PubMed] [Google Scholar]
- 38.Sherlin LD, Perona JJ. tRNA-dependent active site assembly in a class I aminoacyl-tRNA synthetase. Structure. 2003;11:591–603. doi: 10.1016/s0969-2126(03)00074-1. see comment. [DOI] [PubMed] [Google Scholar]
- 39.Sekine S, Nureki O, Dubois DY, Bernier S, Chenevert R, Lapointe J, Vassylyev DG, Yokoyama S. ATP binding by glutamyl-tRNA synthetase is switched to the productive mode by tRNA binding. EMBO J. 2003;22:676–688. doi: 10.1093/emboj/cdg053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Structure of DNA Polymerase b with Active-Site Mismatches Suggest a Transient Abasic Site Intermediate during Misincorporation. Mol Cell. 2008;30:315–324. doi: 10.1016/j.molcel.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radhakrishnan R, Arora K, Wang Y, Beard WA, Wilson SH, Schlick T. Regulation of DNA repair fidelity by molecular checkpoints: “gates” in DNA polymerase beta's substrate selection. Biochemistry. 2006;45:15142–15156. doi: 10.1021/bi061353z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rath VL, Silvian LF, Beijer B, Sproat BS, Steitz TA. How glutaminyl-tRNA synthetase selects glutamine. Structure. 1998;6:439–449. doi: 10.1016/s0969-2126(98)00046-x. [DOI] [PubMed] [Google Scholar]
- 43.Bullock TL, Uter N, Nissan TA, Perona JJ. Amino acid discrimination by a class I aminoacyl-tRNA synthetase specified by negative determinants. J Mol Biol. 2003;328:395–408. doi: 10.1016/s0022-2836(03)00305-x. [DOI] [PubMed] [Google Scholar]
- 44.Moulinier L, Eiler S, Eriani G, Gangloff J, Thierry JC, Gabriel K, McClain WH, Moras D. The structure of an AspRS-tRNA(Asp) complex reveals a tRNA-dependent control mechanism. EMBO J. 2001;20:5290–5301. doi: 10.1093/emboj/20.18.5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giege R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26:5017–5035. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perona JJ, Hou YM. Indirect readout of tRNA for aminoacylation. Biochemistry. 2007;46:10419–10432. doi: 10.1021/bi7014647. [DOI] [PubMed] [Google Scholar]
- 47.Uter NT, Perona JJ. Long-range intramolecular signaling in a tRNA synthetase complex revealed by pre-steady-state kinetics. Proc Natl Acad Sci USA. 2004;101:14396–14401. doi: 10.1073/pnas.0404017101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guth E, Francklyn CS. Kinetic Discrimination of tRNA Identity by the Conserved Motif 2 Loop of a Class II Aminoacyl-tRNA Synthetase. Mol Cell. 2007;25:531–542. doi: 10.1016/j.molcel.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ibba M, Sever S, Praetorius-Ibba M, Soll D. Transfer RNA identity contributes to transition state stabilization during aminoacyl-tRNA synthesis. Nucleic Acids Res. 1999;27:3631–3637. doi: 10.1093/nar/27.18.3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jakubowski H. Accuracy of Aminoacyl-tRNA Synthetases: Proofreading of Amino Acids. In: Ibba M, Francklyn CS, Cusack S, editors. The Aminoacyl-tRNA Synthetases. Landes Bioscience; Georgetown, TX: 2005. pp. 384–396. [Google Scholar]
- 51.Mascarenhas A, Martinis S, An S, Rosen AE, Musier-Forsyth K. Fidelity Mechanisms in Aminoacyl-tRNA Synthetases. In: RajBhandary UL, Koehrer C, editors. Protein Engineering. Springer-Verlag; New York, NY: 2008. [Google Scholar]
- 52.Doring V, Mootz HD, Nangle LA, Hendrickson TL, de Crecy-Lagard V, Schimmel P, Marliere P. Enlarging the amino acid set of Escherichia coli by infiltration of the valine coding pathway. Science. 2001;292:501–504. doi: 10.1126/science.1057718. see comment. [DOI] [PubMed] [Google Scholar]
- 53.Karkhanis VA, Mascarenhas AP, Martinis SA. Amino acid toxicities of Escherichia coli that are prevented by leucyl-tRNA synthetase amino acid editing. J Bact. 2007;189:8765–8768. doi: 10.1128/JB.01215-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee JW, Beebe K, Nangle LA, Jang J, Longo-Guess CM, Cook SA, Davisson MT, Sundberg JP, Schimmel P, Ackerman SL. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. see comment. [DOI] [PubMed] [Google Scholar]
- 55.Reha-Krantz LJ. Are there highly conserved DNA polymerase 3′----5′ exonuclease motifs? Gene. 1992;112:133–137. doi: 10.1016/0378-1119(92)90315-g. [DOI] [PubMed] [Google Scholar]
- 56.Beese LS, Steitz TA. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brautigam CA, Sun S, Piccirilli JA, Steitz TA. Structures of normal single-stranded DNA and deoxyribo-3′-S-phosphorothiolates bound to the 3′-5′ exonucleolytic active site of DNA polymerase I from Escherichia coli. Biochemistry. 1999;38:696–704. doi: 10.1021/bi981537g. [DOI] [PubMed] [Google Scholar]
- 58.Mursinna RS, Lincecum TL, Jr, Martinis SA. A conserved threonine within Escherichia coli leucyl-tRNA synthetase prevents hydrolytic editing of leucyl-tRNALeu. Biochemistry. 2001;40:5376–5381. doi: 10.1021/bi002915w. [DOI] [PubMed] [Google Scholar]
- 59.Hendrickson TL, Nomanbhoy TK, de Crecy-Lagard V, Fukai S, Nureki O, Yokoyama S, Schimmel P. Mutational separation of two pathways for editing by a class I tRNA synthetase. Mol Cell. 2002;9:353–362. doi: 10.1016/s1097-2765(02)00449-5. [DOI] [PubMed] [Google Scholar]
- 60.Ollis DL, Brick P, Hamlin R, Xuong NG, Steitz TA. Structure of large fragment of Escherichia coli DNA polymerase I complexed with dTMP. Nature. 1985;313:762–766. doi: 10.1038/313762a0. [DOI] [PubMed] [Google Scholar]
- 61.Kuchta RD, Mizrahi V, Benkovic PA, Johnson KA, Benkovic SJ. Kinetic mechanism of DNA polymerase I (Klenow) Biochemistry. 1987;26:8410–8417. doi: 10.1021/bi00399a057. [DOI] [PubMed] [Google Scholar]
- 62.Fersht AR, Kaethner MM. Enzyme hyperspecificity. Rejection of threonine by the valyl-tRNA synthetase by misacylation and hydrolytic editing. Biochemistry. 1976;15:3342–3346. doi: 10.1021/bi00660a026. [DOI] [PubMed] [Google Scholar]
- 63.Bishop AC, Nomanbhoy TK, Schimmel P. Blocking site-to-site translocation of a misactivated amino acid by mutation of a class I tRNA synthetase. Proc Natl Acad Sci USA. 2002;99:585–590. doi: 10.1073/pnas.012611299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Williams AM, Martinis SA. Mutational unmasking of a tRNA-dependent pathway for preventing genetic code ambiguity. Proc Natl Acad Sci USA. 2006;103:3586–3591. doi: 10.1073/pnas.0507362103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hale SP, Schimmel P. Protein synthesis editing by a DNA aptamer. Proc Natl Acad Sci USA. 1996;93:2755–2758. doi: 10.1073/pnas.93.7.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gruic-Sovulj I, Uter N, Bullock T, Perona JJ. tRNA-dependent aminoacyl-adenylate hydrolysis by a nonediting class I aminoacyl-tRNA synthetase. J Biol Chem. 2005;280:23978–23986. doi: 10.1074/jbc.M414260200. [DOI] [PubMed] [Google Scholar]
- 67.Lincecum TL, Jr, Tukalo M, Yaremchuk A, Mursinna RS, Williams AM, Sproat BS, Van Den Eynde W, Link A, Van Calenbergh S, Grotli M, Martinis SA, Cusack S. Structural and mechanistic basis of pre- and posttransfer editing by leucyl-tRNA synthetase. Mol Cell. 2003;11:951–963. doi: 10.1016/s1097-2765(03)00098-4. [DOI] [PubMed] [Google Scholar]
- 68.Dock-Bregeon AC, Rees B, Torres-Larios A, Bey G, Caillet J, Moras D. Achieving error-free translation; the mechanism of proofreading of threonyl-tRNA synthetase at atomic resolution. Mol Cell. 2004;16:375–386. doi: 10.1016/j.molcel.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 69.Fukunaga R, Yokoyama S. Structural basis for substrate recognition by the editing domain of isoleucyl-tRNA synthetase. J Mol Biol. 2006;359:901–912. doi: 10.1016/j.jmb.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 70.Nomanbhoy TK, Hendrickson TL, Schimmel P. Transfer RNA-dependent translocation of misactivated amino acids to prevent errors in protein synthesis. Mol Cell. 1999;4:519–528. doi: 10.1016/s1097-2765(00)80203-8. [DOI] [PubMed] [Google Scholar]
- 71.Splan KE, Musier-Forsyth K, Boniecki MT, Martinis SA. In vitro assays for the determination of aminoacyl-tRNA synthetase editing activity. Methods (Duluth) 2008;44:119–128. doi: 10.1016/j.ymeth.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yarus M, Berg P. Recognition of tRNA by isoleucyl-tRNA synthetase. Effect of substrates on the dynamics of tRNA-enzyme interaction. J Mol Biol. 1969;42:171–189. doi: 10.1016/0022-2836(69)90037-0. [DOI] [PubMed] [Google Scholar]
- 73.Fukai S, Nureki O, Sekine S, Shimada A, Tao J, Vassylyev DG, Yokoyama S. Structural basis for double-sieve discrimination of L-valine from L-isoleucine and L-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell. 2000;103:793–803. doi: 10.1016/s0092-8674(00)00182-3. [DOI] [PubMed] [Google Scholar]
- 74.Tukalo M, Yaremchuk A, Fukunaga R, Yokoyama S, Cusack S. The crystal structure of leucyl-tRNA synthetase complexed with tRNALeu in the post-transfer-editing conformation. Nat Struct Mol Biol. 2005;12:923–930. doi: 10.1038/nsmb986. [DOI] [PubMed] [Google Scholar]
- 75.Rock FL, Mao W, Yaremchuk A, Tukalo M, Crepin T, Zhou H, Zhang YK, Hernandez V, Akama T, Baker SJ, Plattner JJ, Shapiro L, Martinis SA, Benkovic SJ, Cusack S, Alley MR. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]