

Abstract
Sphingosine 1-phosphate (S1P), a bioactive lipid mediator, stimulates proliferation and contractility in hepatic stellate cells, the principal matrix-producing cells in the liver, and inhibits proliferation via S1P receptor 2 (S1P2) in hepatocytes in rats in vitro. A potential role of S1P and S1P2 in liver regeneration and fibrosis was examined in S1P2-deficient mice. Nuclear 5-bromo-2′-deoxy-uridine labeling, proliferating cell nuclear antigen (PCNA) staining in hepatocytes, and the ratio of liver weight to body weight were enhanced at 48 h in S1P2-deficient mice after a single carbon tetrachloride (CCl4) injection. After dimethylnitrosamine (DMN) administration with a lethal dose, PCNA staining in hepatocytes was enhanced at 48 h and survival rate was higher in S1P2-deficient mice. Serum aminotransferase level was unaltered in those mice compared with wild-type mice in both CCl4- and DMN-induced liver injury, suggesting that S1P2 inactivation accelerated regeneration not as a response to enhanced liver damage. After chronic CCl4 administration, fibrosis was less apparent, with reduced expression of smooth-muscle α-actin-positive cells in the livers of S1P2-deficient mice, suggesting that S1P2 inactivation ameliorated CCl4-induced fibrosis due to the decreased accumulation of hepatic stellate cells. Thus, S1P plays a significant role in regeneration and fibrosis after liver injury via S1P2.
Keywords: liver regeneration, liver fibrosis, hepatocyte, hepatic stellate cell, hepatic myofibroblast
Sphingosine 1-phosphate (S1P), which elicits a wide variety of cell responses (1), has emerged as a novel lipid intracellular mediator. S1P was shown to act as an intracellular second messenger of platelet-derived growth factor and serum in their mitogenic actions in cultured fibroblasts (2, 3), and furthermore, intracellular levels of S1P and ceramide were reported to determine cell survival or death (4, 5). However, evidence indicating that S1P also acts as an extracellular mediator has been reported; some of the diverse effects of S1P, such as stimulation of cell proliferation or contractility, are known to be sensitive to pertussis toxin (6) or ADP-ribosyltransferase C3 from Clostridium botulinum (7, 8), suggesting that S1P may activate a receptor coupled to G protein(s). Indeed, recent investigation has revealed that S1P acts through at least five high-affinity G protein-coupled receptors referred to as S1P1–5 (9, 10). Regarding the source of S1P in vivo, it is shown to be stored in platelets (11), and recent data using conditional knockouts of sphingosine kinases support release of S1P from erythrocytes (12, 13). These findings suggest that S1P has normal in vivo roles as well as potentially pathophysiological roles as a circulating paracrine mediator, a view further supported by the phenotypes of S1P receptor mutants (10, 14, 15).
S1P receptors are also expressed in the liver (14). To investigate the function of S1P in liver pathophysiology, we have determined the effect of S1P on liver cells in culture. We first demonstrated that S1P stimulates proliferation and contractility in rat hepatic stellate cells in culture; the stimulation of proliferation is pertussis toxin-sensitive, and the stimulation of contractility is C3 exotoxin-sensitive (16). This stimulatory effect of S1P on contractility in those cells was found to be via S1P2 with Rho activation (17). On the other hand, we revealed that S1P inhibits proliferation in cultured rat hepatocytes. With the use of a specific antagonist to S1P2 and C3 exotoxin, this inhibitory effect of S1P on hepatocyte proliferation in culture involved Rho activation via S1P2 (18). Furthermore, the administration of S1P in 70% hepatectomized rats indeed reduces a response of hepatocytes to synthesize DNA (18).
Irrespective of the insults, such as viruses, alcohol abuse, or drugs, the regenerative and wound-healing responses generally occur in the liver after the injury, and the persistence of these responses may result in liver fibrosis (19, 20). Hepatocytes play a major role in liver regeneration (19) as do hepatic stellate cells in the wound-healing response, and hence, liver fibrosis (20). The enhanced proliferation and contractility of hepatic stellate cells are among the main features of liver fibrosis (21, 22). Although our previous evidence was obtained mainly by in vitro studies and indicated a pharmacologically inhibitory effect of S1P on liver regeneration in vivo, it raised the possibility that S1P, and potentially S1P2, might play a pathophysiological role in the liver after the injury. Thus, we planned to extend our study using S1P2-deficient mice to clarify this point.
In this context, Serrier-Lanneau et al. (23) recently reported that the wound-healing response to acute liver injury elicited by carbon tetrachloride (CCl4) was reduced in S1P2−/− mice with reduced accumulation of hepatic myofibroblasts and that hepatic myofibroblasts isolated from S1P2−/− mice did not proliferate in response to S1P. There is controversy as to whether hepatic myofibroblasts are distinct from hepatic stellate cells. Hepatic myofibroblasts have been studied as the cells developed from hepatic stellate cells by transdifferentiation (24–27). In contrast, hepatic myofibroblasts have been reported to belong to a cell population different from hepatic stellate cells (28). Whatever the case, hepatic myofibroblasts have been assumed to be a principle matrix-producing cell of the diseased liver (29). On the other hand, the regenerative response after a single injection of CCl4 in S1P2−/− mice was not different from that in wild-type mice, suggesting that S1P2 inactivation did not affect hepatocyte regeneration (23). With these findings, we aimed to perform more-detailed examination of hepatocyte regeneration in acute liver injury elicited by dimethylnitrosamine (DMN) in addition to CCl4 and to determine whether reduced wound-healing response could lead to reduced liver fibrosis after chronic CCl4 administration in S1P2−/− mice.
MATERIALS AND METHODS
Animals
Heterozygous S1P2+/− mice were originally generated by our group on the (129/Sv × 129/J)F1 (an embryonic stem cell origin)/C57BL/6N (a blastocyst origin) mixed background (30). In this study, they were backcrossed onto the inbred C57BL/6N strain (Clea Japan, Tokyo, Japan) for 7–8 generations to achieve >99.2% genetic homogeneity, and the obtained S1P2+/− mice were bred to produce S1P2−/− mice. Deletion of S1P2 in these S1P2−/− mice has been repeatedly confirmed by the complete absence of s1p2 gene expression in tissues (e.g., adult lung, spleen, brain, heart, cochlea, and embryonic fibroblasts) in which s1p2 is normally expressed (15, 30, 31) as well as the specific appearance or disappearance of some S1P-mediated cellular signaling/systemic effects (15, 30–32). Age-matched wild-type C57BL/6N mice were used as controls. They were fed a standard pelleted diet and water ad libitum under normal laboratory conditions of 12 h light/dark cycles. All animals received humane care, and the research was conducted in conformity with Public Health Service policy on the humane care and use of laboratory animals. The experimental protocol was approved by the Animal Research Committee of the University of Tokyo and followed National Institutes of Health guidelines for the care and use of laboratory animals.
CCl4 treatment
To assess the responses to a single CCl4 administration, male and female wild-type and S1P2−/− mice, 8–10 weeks of age, were injected intraperitoneally with 0.5 ml/kg body weight of CCl4 dissolved in the same amount of olive oil (1:1) (23), in which liver injury, assessed by serum aminotransferase level, was nongender specific. To develop liver fibrosis, female wild-type and S1P2−/− mice, 8 weeks of age, were injected intraperitoneally with 1 ml/kg body weight of CCl4 dissolved in the same amount of olive oil (1:1), twice a week for 4 weeks (33, 34). Control mice received injection of the carrier (olive oil) alone. Each group consisted of four to five mice. For the experiment on chronic CCl4 treatment, the livers were harvested at 4 days after the end of the administration.
DMN treatment
Male and female wild-type and S1P2−/− mice, 8–10 weeks of age, were injected intraperitoneally with 15 mg/kg body weight DMN dissolved in HBSS (0.3%) (GIBCO) (35), in which liver injury, assessed by serum aminotransferase level, was nongender specific.
Measurement of nuclear labeling by 5-bromo-2′-deoxy-uridine
One hour before euthanization, 50 mg/4 ml/kg body weight of 5-bromo-2′-deoxy-uridine (BrdU) (Roche Molecular Biochemicals, Mannheim, Germany) was injected intraperitoneally, and then the mice were euthanized at 24, 48, 72, and 96 h after CCl4 administration. The liver was excised, immediately fixed in 10% formalin, and embedded in paraffin. The nuclear labeling was measured using BrdU labeling and detection kit II (Roche Molecular Biochemicals). The number of BrdU-positive hepatocytes was determined as the mean of five random areas at 400-fold magnification in each section.
Immunohistochemical analysis of proliferating cell nuclear antigen
Immunohistochemical staining for proliferating cell nuclear antigen (PCNA) was performed in liver tissue using a PCNA staining kit (Zymed Laboratories) in accordance with the protocol specified by the manufacturer. The ratio of PCNA-positive hepatocytes to all hepatocytes was determined with five random areas at 400-fold magnification in each section in CCl4-treated mice and with ten random areas in DMN-treated mice, because submassive hemorrhagic necrosis was focally found in DMN-treated livers as previously described (36). The percentage of PCNA-positive nonparenchymal cells was determined with five random areas at 400-fold magnification in each section in CCl4-treated mice.
Measurement of liver function
The serum levels of alanine aminotransferase (ALT) and alkaline phosphatase (ALP) were determined using an automated analyzer (Hitachi 7170; Hitachi Instruments Service Co., Ltd., Tokyo, Japan).
Histological analysis of fibrosis
Tissue sections (4 μm thick) of liver specimens fixed in formalin and embedded in paraffin were analyzed with Masson's trichrome staining (37). The microscopic fields (five per each slide) were blindly selected and captured with the aid of the Nikon Digital Camera DXM1200 (NIKON, Japan). A fibrous portion stained in blue was extracted, and the extent of liver fibrosis was quantified by the technique reported previously (38) by calculating the area of fibrosis/area of section using image analysis software of the public domain Scion Image (Scion Corporation).
Immunohistochemical analysis of smooth-muscle α-actin
Immunohistochemical analysis of smooth-muscle α-actin was performed in liver tissue fixed in formalin and embedded in paraffin using a Vector M.O.M. immunodetection kit (Vector Laboratories, Burlingame, CA) in accordance with the protocol specified by the manufacturer, with a 1:5 dilution of mouse monoclonal antibody to smooth-muscle α-actin (Sigma). The quantitation of the positive staining area for smooth-muscle α-actin was performed blindly on four or five liver fragments per animal using the computer software of the public domain Scion Image developed by the Scion Corporation.
Statistical analysis
Data are represented as mean ± SEM. An ANOVA followed by Tukey's honest significance differences test (Tukey's HSD) or Student's t-test were used to determine whether significant differences existed between groups. Fisher's exact test was performed when appropriate. Differences were considered to be significant at P values of <0.05.
RESULTS
Regenerative response of hepatocytes to CCl4-induced acute liver injury was enhanced in S1P2−/− mice
After a single CCl4 administration, DNA synthesis of hepatocytes determined by BrdU labeling peaked at 48 h in both wild-type and S1P2−/− mice (Fig. 1). As depicted in Fig. 1, BrdU-positive hepatocytes seemed more apparent in S1P2−/− mice than in wild-type mice (Fig. 1A) at 48 h after CCl4 administration, and in fact, the number of BrdU-positive hepatocytes at 48 h after CCl4 administration in S1P2−/− mice was significantly increased by 1.8-fold compared with wild-type mice (Fig. 1B). On the other hand, BrdU-positive hepatocytes were not found in both wild-type and S1P2−/− mice treated with olive oil alone. We next performed immunohistochemical analysis of PCNA in wild-type and S1P2−/− livers after a single CCl4 administration. As demonstrated in Fig. 1, PCNA-positive hepatocytes were more apparent in S1P2−/− mice than in wild-type mice (Fig. 1C) at 48 h after CCl4 administration. The number of PCNA-positive hepatocytes significantly increased by 1.7-fold in S1P2−/− mice compared with wild-type mice (Fig. 1D). On the other hand, it is of note that PCNA-positive nonparenchymal cells were less apparent in S1P2−/− mice compared with wild-type mice (Fig. 1C). The percentage of PCNA-positive nonparenchymal cells in S1P2−/− mice was reduced to 62% of that in wild-type mice (Fig. 1E). In both wild-type and S1P2−/− mice treated with olive oil alone, PCNA-positive cells were not determined. These results suggest that the regenerative response after CCl4 administration is enhanced in hepatocytes, but reduced in nonparenchymal cells in S1P2−/− mice.
Fig. 1.
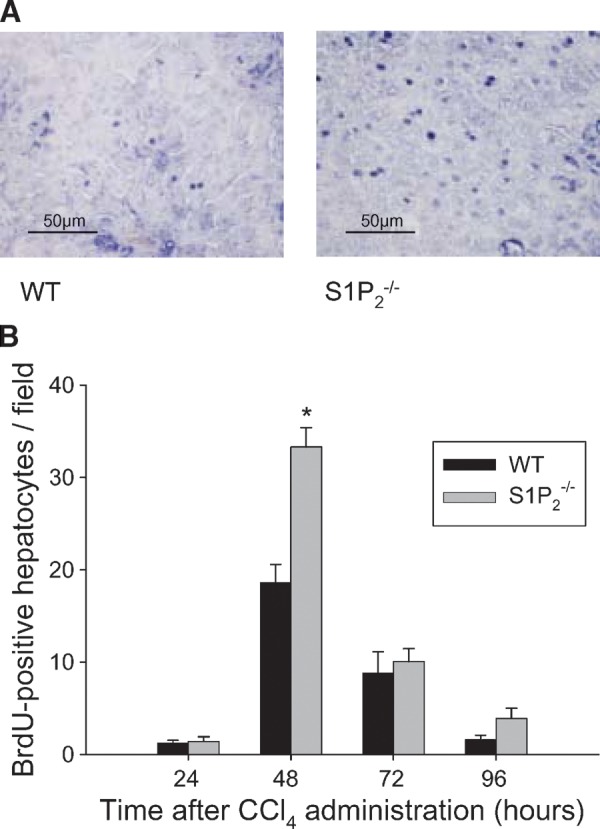
Regenerative response in hepatocytes and nonparenchymal cells of wild-type and S1P2−/− livers in acute injury induced by carbon tetrachloride (CCl4). A single injection of CCl4 was performed in wild-type (WT) and S1P2−/− mice. 5-Bromo-2′-deoxy-uridine (BrdU) was injected intraperitoneally at 1 h before euthanization at 24, 48, 72, and 96 h after CCl4 injection. The nuclear labeling in hepatocytes was determined using a BrdU labeling and detection kit II. Representative BrdU labeling in the livers of wild-type and S1P2−/− mice at 48 h after CCl4 injection is shown (A). Bar = 50 μm. The number of BrdU-positive cells was determined as the mean of five random areas at 400-fold magnification in each section (B). Columns and bars represent means ± SEM of four animals. Immunohistochemical analysis of proliferating cell nuclear antigen (PCNA) was also done using a PCNA staining kit. Representative photomicrographs of the liver of wild-type and S1P2−/− mice at 48 h after CCl4 injection are shown (C). Bar = 50 μm. Arrows indicate positive hepatocytes, and arrowheads, positive nonparenchymal cells. The percentages of PCNA-positive hepatocytes (D) and nonparenchymal cells (E) were determined with five random areas at 400-fold magnification in each section. Columns and bars represent means ± SEM of four animals. The asterisk indicates a significant difference from wild type in Student's t-test (P < 0.05).
We then analyzed both liver weight and body weight at euthanization after a single injection of CCl4. The ratio of liver weight to body weight was significantly higher in S1P2−/− mice than in wild-type mice at 48 h after CCl4 administration, but not different between wild-type and S1P2−/− mice at 24, 72, and 96 h (Fig. 2A). Both wild-type and S1P2−/− mice lost their body weight maximally at 24 h after CCl4 administration and then gradually gained weight up to 96 h, as shown in Fig. 2B. The alterations of body weight after CCl4 administration were not different between wild-type and S1P2−/− mice. These results suggest that the regenerative response in the liver after CCl4 administration is enhanced in S1P2−/− mice.
Fig. 2.


The ratio of liver weight to body weight, body weight loss, and liver function tests in wild-type and S1P2−/− mice in acute liver injury induced by CCl4. A single injection of CCl4 was performed in wild-type (WT) and S1P2−/− mice. The ratio of liver weight to body weight was measured in untreated wild-type and S1P2−/− mice from 24 to 96 h at euthanization after CCl4 injection (A). The alteration of body weight was determined by comparison of body weights before CCl4 injection with those at 24, 48, 72, and 96 h after CCl4 injection (B). Serum concentrations of alanine aminotransferase (ALT) (C) and alkaline phosphatase (ALP) (D) were measured in wild-type and S1P2−/− mice at 24, 48, 72, and 96 h after CCl4 injection. Columns and bars represent means ± SEM of four animals. The asterisk indicates a significant difference from wild type in Student's t-test (P < 0.05).
To examine the extent of liver damage elicited by CCl4 administration, serum levels of ALT and ALP were measured up to 96 h after treatment. Serum ALT level peaked at 24 h after CCl4 administration and was not significantly different between wild-type and S1P2−/− mice (Fig. 2C), as previously reported (23). Furthermore, serum level of ALP was not different between wild-type and S1P2−/− mice (Fig. 2D). These results suggest that the extent of liver damage caused by a single injection of CCl4 was not different in S1P2−/− mice compared with wild-type mice.
Enhanced regenerative response of hepatocytes to DMN-induced acute liver injury led to increased survival rate in S1P2−/− mice
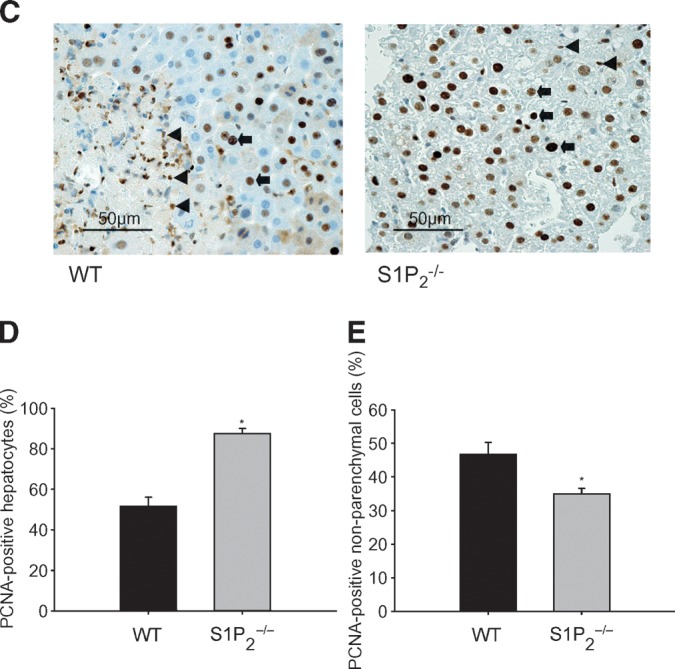
To determine whether the enhanced hepatocyte regeneration in S1P2−/− mice could be the specific phenomenon in CCl4-induced acute liver injury, the regenerative response of hepatocytes in S1P2−/− mice was also examined in DMN-induced acute liver injury (35). In this experiment, BrdU labeling was not suitable for determining the proliferative response, because significant amounts of ascites were found in both wild-type and S1P2−/− mice in which BrdU was administered intraperitoneally. Thus, we determined the proliferative response by PCNA staining. At 48 h after DMN administration, PCNA-positive hepatocytes were more apparent in S1P2−/− mice than in wild-type mice (Fig. 3A). The percentage of PCNA-positive hepatocytes increased by 1.5-fold in S1P2−/− mice compared with wild-type mice (Fig. 3B). On the other hand, PCNA-positive hepatocytes were not found in both wild-type and S1P2−/− mice treated with vehicle alone. This result suggests that the regenerative response is also enhanced after DMN administration in hepatocytes in S1P2−/− mice. At the same time point, the ratio of liver weight to body weight was not different between wild-type and S1P2−/− mice (Fig. 4A), and the trend of more body weight loss in wild-type mice was noted (P = 0.05116) (Fig. 4B). Serum levels of ALT (Fig. 4C) and ALP (Fig. 4D) were not different between wild-type and S1P2−/− mice at the same time point, suggesting that the extent of DMN-induced liver damage was not different in S1P2−/− mice compared with wild-type mice.
Fig. 3.
PCNA-positive hepatocytes of wild-type and S1P2−/− livers in acute liver injury induced by dimethylnitrosamine (DMN). A single injection of DMN was performed in wild-type (WT) and S1P2−/− mice. Immunohistochemical analysis of PCNA was done using a PCNA staining kit. Representative photomicrographs of the liver of wild-type and S1P2−/− mice at 48 h after DMN injection are shown (A). Bar = 50 μm. Arrows indicate positive hepatocytes. The percentage of PCNA-positive hepatocytes was determined with ten random areas at 400-fold magnification in each section (B). Columns and bars represent means ± SEM of four animals. The asterisk indicates a significant difference from wild type in Student's t-test (P < 0.05).
Fig. 4.


The ratio of liver weight to body weight, body weight loss, liver function test, and survival rate in wild-type and S1P2−/− mice in acute liver injury induced by DMN. A single injection of DMN was performed in wild-type (WT) and S1P2−/− mice. The ratio of liver weight to body weight (A), body weight loss (B), and serum concentration of ALT (C) and ALP (D) were measured at 48 h after DMN injection. Columns and bars represent means ± SEM of four animals. Survival rate was also determined in wild-type (n = 17) and S1P2−/− (n = 16) mice up to 120 h after DMN injection (E). Significantly higher survival rate in S1P2−/− mice was determined at 96 and 120 h after DMN injection in Fisher's exact test (P < 0.05).
Although we next planned to determine the regenerative response in hepatocytes in wild-type and S1P2−/− mice beyond 48 h after DMN administration, a substantial number of wild-type mice were found to be dead at 72 h after DMN administration. Thus, our experiment was changed to determine survival rate of wild-type and S1P2−/− mice with DMN intoxication. As shown in Fig. 4E, survival rate was significantly higher in S1P2−/− mice compared with wild-type mice at 96 to 120 h after DMN injection. Thus, increased regenerative response in hepatocytes after DMN intoxication may lead to increased survival rate in S1P2−/− mice.
Liver fibrosis induced by chronic CCl4 administration was reduced in S1P2−/− mice
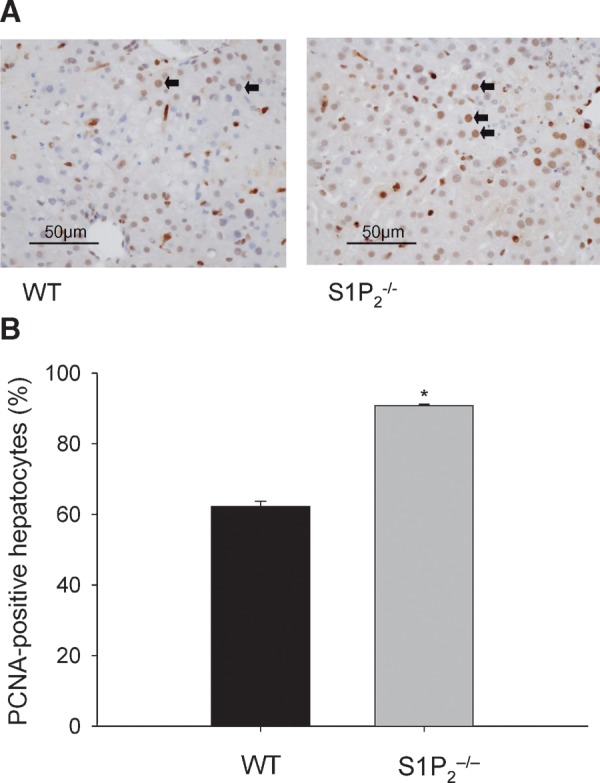
Next, the role of S1P2 receptor signaling in liver fibrosis was examined in chronic liver injury elicited by CCl4. In wild-type mice, chronic administration of CCl4 for 4 weeks caused an extensive accumulation of extracellular matrices with bridging fibrosis in the liver, whereas in S1P2−/− mice, such dense fibrotic septum development with nodule formation was not found (Fig. 5A). Quantitative analysis of fibrosis development revealed that the mean fibrotic area in the livers in wild-type mice was significantly larger than that in S1P2−/− mice treated with CCl4. The extent of liver fibrosis in S1P2−/− mice was approximately 40% that of wild-type mice (Fig. 5B). On the other hand, liver histology in S1P2−/− mice administered olive oil alone, i.e., without CCl4 treatment, was essentially unaltered compared with that of wild-type mice administered identically (Fig. 5B). In addition, serum ALT levels at the time of euthanization after chronic CCl4 administration for 4 weeks were essentially unaltered in wild-type and S1P2−/− mice; 58 ± 20 IU/l (n = 5) and 51 ± 30 IU/l (n = 5), respectively. These results suggest that S1P2 inactivation causes less liver fibrosis in response to chronic CCl4 administration.
Fig. 5.
Liver fibrosis in wild-type and S1P2−/− mice after chronic CCl4 administration. Wild-type (WT) and S1P2−/− mice were treated with CCl4 for 4 weeks. Representative liver histology with Masson's trichrome staining of wild-type and S1P2−/− mice is shown (A). Bar = 100 μm. Arrows indicate the formation of fibrotic septa. The extent of liver fibrosis in CCl4- and olive oil (vehicle)-treated wild-type and S1P2−/− mice was quantitated by calculating the ratio of the area of fibrosis to the area of section using Scion Image (B). Columns and bars represent means ± SEM of five animals. The asterisk indicates a significant difference between CCl4-treated wild-type and S1P2−/− mice in ANOVA followed by Tukey's honestly significant differences (Tukey's HSD) (P < 0.05).
We examined the ratio of liver weight to body weight at euthanization after chronic CCl4 administration. The trend of higher ratio of liver weight to body weight in S1P2−/− mice than in wild-type mice was noted (P = 0.1475) (Fig. 6A); 3.3% in wild-type and 4.8% in S1P2−/− mice. On the other hand, in mice treated with olive oil alone, the ratio of liver weight to body weight was not different with or without S1P2 inactivation (data not shown). Alterations of body weight in wild-type and S1P2−/− mice with chronic CCl4 administration were then examined by analysis of body weights at the first administration of CCl4 and at euthanization after chronic CCl4 administration. Repeated CCl4 administration for 4 weeks caused no increase in body weight in wild-type mice, but a 25.6% increase in S1P2−/− mice (Fig. 6B). This body weight gain in S1P2−/− mice after chronic CCl4 administration was comparable to that in both wild-type and S1P2−/− mice treated with olive oil alone for 4 weeks. Accordingly, only wild-type mice treated with CCl4 for 4 weeks had much less weight gain. These data may be compatible with the finding that CCl4-induced liver fibrosis was abrogated in S1P2−/− mice.
Fig. 6.

The ratio of liver weight to body weight and body weight loss in wild-type and S1P2−/− mice after chronic CCl4 administration. CCl4 injection was performed for 4 weeks in wild-type (WT) and S1P2−/− mice. The ratio of liver weight to body weight was measured at euthanization after CCl4 treatment (A). The alteration of body weight was determined by comparison of body weight at the first administration of CCl4 and at euthanization after the last administration of CCl4 (B). Columns and bars represent means ± SEM of five animals. The asterisk indicates a significant difference from wild type in Student's t-test (P < 0.05).
Both hepatic stellate cells and hepatic myofibroblasts are thought to be the principal matrix-producing cells with smooth-muscle α-actin expression, although controversy still exists as to whether hepatic myofibroblasts are distinct from hepatic stellate cells (28, 39). Thus, smooth-muscle α-actin expression in the liver is considered to be a marker of the matrix-producing cells in the liver. Chronic CCl4 administration caused a significant accumulation of smooth-muscle α-actin-positive cells in wild-type livers, whereas it was less apparent in S1P2−/− livers (Fig. 7A). Quantitative analysis revealed that the smooth-muscle α-actin-positive cell area was reduced by 78% in S1P2−/− livers compared with wild-type livers (Fig. 7B). Thus, these results suggest that S1P2 inactivation leads to decreased accumulation of the matrix-producing cells in the liver, namely hepatic stellate cells and/or hepatic myofibroblasts, in CCl4-induced liver fibrosis.
Fig. 7.

Smooth-muscle α-actin-positive cells in wild-type and S1P2−/− livers after chronic CCl4 administration. Immunohistochemical staining for smooth-muscle α-actin was performed in liver tissue as described in Materials and Methods, and representative results are shown in wild-type (WT) and S1P2−/− mice after chronic CCl4 administration (A). Bar = 100 μm. The extent of accumulation of smooth-muscle α-actin-positive cells (arrows) was quantitated using Scion Image (B). Columns and bars represent means ± SEM of five animals. The asterisk indicates a significant difference from wild type in ANOVA followed by Tukey's HSD (P < 0.05).
DISCUSSION
In the current study, liver regeneration was enhanced in S1P2−/− mice compared with wild-type mice after acute liver injury induced by a single injection of CCl4 or DMN, suggesting that S1P may be involved in liver regeneration, possibly as an inhibitory regulator via S1P2. This was determined by the increased nuclear labeling with BrdU and immunohistochemical staining of PCNA in hepatocytes and by the enhanced ratio of liver weight to body weight in S1P2−/− mice compared with wild-type mice in CCl4-induced acute liver injury. On the other hand, in DMN-induced acute liver injury, increased liver regeneration was determined by the increased immunohistochemical staining of PCNA in hepatocytes in S1P2−/− mice compared with wild-type mice, although the enhanced ratio of liver weight to body weight was not found in S1P2−/− mice. Because submassive hemorrhagic necrosis is characteristic of DMN-intoxicated liver (36), the resultant congestion in the liver may affect the exact weight determination of liver parenchyma. Furthermore, higher survival rate after DMN intoxication with a lethal dose (35) in S1P2−/− mice compared with wild-type mice was determined. This may be explained by increased liver regeneration, and the S1P2 antagonist merits consideration as a therapeutic agent for liver failure caused by impaired liver regeneration. Because the liver damage evaluated by liver function tests was not different between wild-type and S1P2−/− mice in both CCl4- and DMN-induced liver injury, the enhanced liver regeneration in S1P2−/− mice may not be due to a response to the enhanced liver damage. Collectively, the current evidence has suggested that S1P plays a physiological role in liver regeneration via S1P2 as a negative regulator.
A recent study by Serrier-Lanneau et al. (23) reported that the regenerative response after a single injection of CCl4 in S1P2−/− mice was not different from that in wild-type mice, where liver regeneration was assessed by Western blot analysis of PCNA expression in the liver. In our study, immunohistochemical analysis of PCNA revealed that PCNA-positive hepatocytes were increased in S1P2−/− livers by 1.7-fold compared with wild-type livers at 48 h after CCl4 administration. Another interesting finding was that PCNA-positive nonparenchymal cells were found in significant numbers in wild-type livers, but were much less apparent in S1P2−/− livers, which may be in line with the finding by Serrier-Lanneau et al. (23) that hepatic myofibroblasts from S1P2−/− livers were less regenerative compared with those from wild-type livers after a single CCl4 injection. It is likely that Western blot analysis of PCNA expression in the whole-liver extracts determined regeneration in both nonparenchymal cells and hepatocytes. We would also assume that a less than 2-fold increase may occasionally be difficult to detect by Western blot analysis, inasmuch as our Western blot analysis did not reveal significant differences in PCNA expression between wild-type and S1P2−/− livers (data not shown).
On the other hand, liver fibrosis induced by chronic CCl4 administration was less apparent in S1P2−/− mice than in wild-type mice. The reduced body weight loss in S1P2−/− mice after chronic CCl4 administration may be compatible with the reduction of CCl4-induced liver fibrosis in S1P2−/− mice. Such evidence indicates that S1P2 inactivation leads to reduced liver fibrosis induced by CCl4, suggesting that S1P and S1P2 may play a role in the development of liver fibrosis.
The reduction of liver fibrosis in S1P2 inactivation may not be explained by the alteration in the extent of liver damage by CCl4, because the liver damage was essentially the same in wild-type and S1P2−/− mice. Instead, the reduced accumulation of smooth-muscle α-actin-positive cells in the livers of S1P2−/− mice treated with CCl4 for 4 weeks may be a key to the reduced liver fibrosis in those mice, because smooth-muscle α-actin-positive cells in the liver are assumed to be the principal matrix-producing cells of the diseased liver, namely hepatic stellate cells and hepatic myofibroblasts (29).
We previously showed abundant mRNA expression of S1P1 and S1P2 in both hepatocytes (18) and hepatic stellate cells (16) in rats. On the other hand, increased mRNA expression of S1P2 and S1P3 and unaltered mRNA expression of S1P1 in the liver after a single CCl4 injection were reported in mice (23). Although our current evidence suggests that S1P2 may play a significant role in the response to liver injury, the significance of abundant expression of S1P1 in liver cells and upregulation of S1P3 expression in the liver after injury remains to be examined.
Recent evidence suggests that S1P receptors regulate important physiological functions of the vascular system, such as vascular morphogenesis and maturation, cardiac function, vascular permeability, and tumor angiogenesis (40–43). Our current evidence suggests that S1P2 may also play a significant role in liver pathophysiology.
Abbreviations
ALT, alanine aminotransferase
ALP, alkaline phosphatase
BrdU, 5-bromo-2′-deoxy-uridine
CCl4, carbon tetrachloride
DMN, dimethylnitrosamine
PCNA, proliferating cell nuclear antigen
S1P, sphingosine 1-phosphate
Published, JLR Papers in Press, October 27, 2008.
Footnotes
This work was supported by the Japanese Society of Laboratory Medicine Fund for the Promotion of Scientific Research (H.I.) and by National Institutes of Health Grants NS-048478 and R01 DA-019674 (J.C.).
References
- 1.Spiegel S., and A. H. Merrill, Jr. 1996. Sphingolipid metabolism and cell growth regulation. FASEB J. 10 1388–1397. [DOI] [PubMed] [Google Scholar]
- 2.Olivera A., and S. Spiegel. 1993. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature. 365 557–560. [DOI] [PubMed] [Google Scholar]
- 3.Rani C. S., F. Wang, E. Fuior, A. Berger, J. Wu, T. W. Sturgill, D. Beitner-Johnson, D. LeRoith, L. Varticovski, and S. Spiegel. 1997. Divergence in signal transduction pathways of platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) receptors. Involvement of sphingosine 1-phosphate in PDGF but not EGF signaling. J. Biol. Chem. 272 10777–10783. [DOI] [PubMed] [Google Scholar]
- 4.Cuvillier O., G. Pirianov, B. Kleuser, P. G. Vanek, O. A. Coso, S. Gutkind, and S. Spiegel. 1996. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 381 800–803. [DOI] [PubMed] [Google Scholar]
- 5.Edsall L. C., G. G. Pirianov, and S. Spiegel. 1997. Involvement of sphingosine 1-phosphate in nerve growth factor-mediated neuronal survival and differentiation. J. Neurosci. 17 6952–6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodemote K. A., M. E. Mattie, A. Berger, and S. Spiegel. 1995. Involvement of a pertussis toxin-sensitive G protein in the mitogenic signaling pathways of sphingosine 1-phosphate. J. Biol. Chem. 270 10272–10277. [DOI] [PubMed] [Google Scholar]
- 7.Tosaka M., F. Okajima, Y. Hashiba, N. Saito, T. Nagano, T. Watanabe, T. Kimura, and T. Sasaki. 2001. Sphingosine 1-phosphate contracts canine basilar arteries in vitro and in vivo: possible role in pathogenesis of cerebral vasospasm. Stroke. 32 2913–2919. [DOI] [PubMed] [Google Scholar]
- 8.Ohmori T., Y. Yatomi, M. Osada, F. Kazama, T. Takafuta, H. Ikeda, and Y. Ozaki. 2003. Sphingosine 1-phosphate induces contraction of coronary artery smooth muscle cells via S1P2. Cardiovasc. Res. 58 170–177. [DOI] [PubMed] [Google Scholar]
- 9.Ishii I., N. Fukushima, X. Ye, and J. Chun. 2004. Lysophospholipid receptors: signaling and biology. Annu. Rev. Biochem. 73 321–354. [DOI] [PubMed] [Google Scholar]
- 10.Gardell S. E., A. E. Dubin, and J. Chun. 2006. Emerging medicinal roles for lysophospholipid signaling. Trends Mol. Med. 12 65–75. [DOI] [PubMed] [Google Scholar]
- 11.Yatomi Y., F. Ruan, J. Ohta, R. J. Welch, S. Hakomori, and Y. Igarashi. 1995. Quantitative measurement of sphingosine 1-phosphate in biological samples by acylation with radioactive acetic anhydride. Anal. Biochem. 230 315–320. [DOI] [PubMed] [Google Scholar]
- 12.Pappu R., S. R. Schwab, I. Cornelissen, J. P. Pereira, J. B. Regard, Y. Xu, E. Camerer, Y. W. Zheng, Y. Huang, J. G. Cyster, et al. 2007. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 316 295–298. [DOI] [PubMed] [Google Scholar]
- 13.Chun J. 2007. Immunology. The sources of a lipid conundrum. Science. 316 208–210. [DOI] [PubMed] [Google Scholar]
- 14.Yang A. H., I. Ishii, and J. Chun. 2002. In vivo roles of lysophospholipid receptors revealed by gene targeting studies in mice. Biochim. Biophys. Acta. 1582 197–203. [DOI] [PubMed] [Google Scholar]
- 15.Herr D. R., N. Grillet, M. Schwander, R. Rivera, U. Muller, and J. Chun. 2007. Sphingosine 1-phosphate (S1P) signaling is required for maintenance of hair cells mainly via activation of S1P2. J. Neurosci. 27 1474–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikeda H., Y. Yatomi, M. Yanase, H. Satoh, H. Maekawa, I. Ogata, Y. Ozaki, Y. Takuwa, S. Mochida, and K. Fujiwara. 2000. Biological activities of novel lipid mediator sphingosine 1-phosphate in rat hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 279 G304–G310. [DOI] [PubMed] [Google Scholar]
- 17.Ikeda H., K. Nagashima, M. Yanase, T. Tomiya, M. Arai, Y. Inoue, K. Tejima, T. Nishikawa, N. Watanabe, M. Omata, et al. 2004. Sphingosine 1-phosphate enhances portal pressure in isolated perfused liver via S1P2 with Rho activation. Biochem. Biophys. Res. Commun. 320 754–759. [DOI] [PubMed] [Google Scholar]
- 18.Ikeda H., H. Satoh, M. Yanase, Y. Inoue, T. Tomiya, M. Arai, K. Tejima, K. Nagashima, H. Maekawa, N. Yahagi, et al. 2003. Antiproliferative property of sphingosine 1-phosphate in rat hepatocytes involves activation of Rho via Edg-5. Gastroenterology. 124 459–469. [DOI] [PubMed] [Google Scholar]
- 19.Michalopoulos G. K., and M. C. DeFrances. 1997. Liver regeneration. Science. 276 60–66. [DOI] [PubMed] [Google Scholar]
- 20.Friedman S. L. 2000. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 275 2247–2250. [DOI] [PubMed] [Google Scholar]
- 21.Minato Y., Y. Hasumura, and J. Takeuchi. 1983. The role of fat-storing cells in Disse space fibrogenesis in alcoholic liver disease. Hepatology. 3 559–566. [DOI] [PubMed] [Google Scholar]
- 22.Rockey D. C., C. N. Housset, and S. L. Friedman. 1993. Activation-dependent contractility of rat hepatic lipocytes in culture and in vivo. J. Clin. Invest. 92 1795–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serriere-Lanneau V., F. Teixeira-Clerc, L. Li, M. Schippers, W. de Wries, B. Julien, J. Tran-Van-Nhieu, S. Manin, K. Poelstra, J. Chun, et al. 2007. The sphingosine 1-phosphate receptor S1P2 triggers hepatic wound healing. FASEB J. 21 2005–2013. [DOI] [PubMed] [Google Scholar]
- 24.Adachi M., Y. Osawa, H. Uchinami, T. Kitamura, D. Accili, and D. A. Brenner. 2007. The forkhead transcription factor FoxO1 regulates proliferation and transdifferentiation of hepatic stellate cells. Gastroenterology. 132 1434–1446. [DOI] [PubMed] [Google Scholar]
- 25.Kojima N., M. Hori, T. Murata, Y. Morizane, and H. Ozaki. 2007. Different profiles of Ca2+ responses to endothelin-1 and PDGF in liver myofibroblasts during the process of cell differentiation. Br. J. Pharmacol. 151 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park E. J., Y. Z. Zhao, Y. C. Kim, and D. H. Sohn. 2007. Bakuchiol-induced caspase-3-dependent apoptosis occurs through c-Jun NH2-terminal kinase-mediated mitochondrial translocation of Bax in rat liver myofibroblasts. Eur. J. Pharmacol. 559 115–123. [DOI] [PubMed] [Google Scholar]
- 27.Watson M. R., K. Wallace, R. G. Gieling, D. M. Manas, E. Jaffray, R. T. Hay, D. A. Mann, and F. Oakley. 2008. NF-kappaB is a critical regulator of the survival of rodent and human hepatic myofibroblasts. J. Hepatol. 48 589–597. [DOI] [PubMed] [Google Scholar]
- 28.Knittel T., D. Kobold, B. Saile, A. Grundmann, K. Neubauer, F. Piscaglia, and G. Ramadori. 1999. Rat liver myofibroblasts and hepatic stellate cells: different cell populations of the fibroblast lineage with fibrogenic potential. Gastroenterology. 117 1205–1221. [DOI] [PubMed] [Google Scholar]
- 29.Saile B., N. Matthes, K. Neubauer, C. Eisenbach, H. El-Armouche, J. Dudas, and G. Ramadori. 2002. Rat liver myofibroblasts and hepatic stellate cells differ in CD95-mediated apoptosis and response to TNF-alpha. Am. J. Physiol. Gastrointest. Liver Physiol. 283 G435–G444. [DOI] [PubMed] [Google Scholar]
- 30.Ishii I., X. Ye, B. Friedman, S. Kawamura, J. J. Contos, M. A. Kingsbury, A. H. Yang, G. Zhang, J. H. Brown, and J. Chun. 2002. Marked perinatal lethality and cellular signaling deficits in mice null for the two sphingosine 1-phosphate (S1P) receptors, S1P(2)/LP(B2)/EDG-5 and S1P(3)/LP(B3)/EDG-3. J. Biol. Chem. 277 25152–25159. [DOI] [PubMed] [Google Scholar]
- 31.Goparaju S. K., P. S. Jolly, K. R. Watterson, M. Bektas, S. Alvarez, S. Sarkar, L. Mel, I. Ishii, J. Chun, S. Milstien, et al. 2005. The S1P2 receptor negatively regulates platelet-derived growth factor-induced motility and proliferation. Mol. Cell. Biol. 25 4237–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Means C. K., C. Y. Xiao, Z. Li, T. Zhang, J. H. Omens, I. Ishii, J. Chun, and J. H. Brown. 2007. Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 292 H2944–H2951. [DOI] [PubMed] [Google Scholar]
- 33.Simeonova P. P., R. M. Gallucci, T. Hulderman, R. Wilson, C. Kommineni, M. Rao, and M. I. Luster. 2001. The role of tumor necrosis factor-alpha in liver toxicity, inflammation, and fibrosis induced by carbon tetrachloride. Toxicol. Appl. Pharmacol. 177 112–120. [DOI] [PubMed] [Google Scholar]
- 34.Kanno K., S. Tazuma, and K. Chayama. 2003. AT1A-deficient mice show less severe progression of liver fibrosis induced by CCl(4). Biochem. Biophys. Res. Commun. 308 177–183. [DOI] [PubMed] [Google Scholar]
- 35.Morrison V., and J. Ashby. 1994. Reconciliation of five negative and four positive reports of the activity of dimethylnitrosamine in the mouse bone marrow micronucleus assay. Mutagenesis. 9 361–365. [DOI] [PubMed] [Google Scholar]
- 36.Jin Y. L., H. Enzan, N. Kuroda, Y. Hayashi, H. Nakayama, Y. H. Zhang, M. Toi, E. Miyazaki, M. Hiroi, L. M. Guo, et al. 2003. Tissue remodeling following submassive hemorrhagic necrosis in rat livers induced by an intraperitoneal injection of dimethylnitrosamine. Virchows Arch. 442 39–47. [DOI] [PubMed] [Google Scholar]
- 37.Bedossa P., and T. Poynard. 1996. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology. 24 289–293. [DOI] [PubMed] [Google Scholar]
- 38.Caballero T., A. Perez-Milena, M. Masseroli, F. O'Valle, F. J. Salmeron, R. M. Del Moral, and G. Sanchez-Salgado. 2001. Liver fibrosis assessment with semiquantitative indexes and image analysis quantification in sustained-responder and non-responder interferon-treated patients with chronic hepatitis C. J. Hepatol. 34 740–747. [DOI] [PubMed] [Google Scholar]
- 39.Rockey D. C., J. K. Boyles, G. Gabbiani, and S. L. Friedman. 1992. Rat hepatic lipocytes express smooth muscle actin upon activation in vivo and in culture. J. Submicrosc. Cytol. Pathol. 24 193–203. [PubMed] [Google Scholar]
- 40.Sanchez T., T. Estrada-Hernandez, J. H. Paik, M. T. Wu, K. Venkataraman, V. Brinkmann, K. Claffey, and T. Hla. 2003. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J. Biol. Chem. 278 47281–47290. [DOI] [PubMed] [Google Scholar]
- 41.Chae S. S., J. H. Paik, H. Furneaux, and T. Hla. 2004. Requirement for sphingosine 1-phosphate receptor-1 in tumor angiogenesis demonstrated by in vivo RNA interference. J. Clin. Invest. 114 1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.LaMontagne K., A. Littlewood-Evans, C. Schnell, T. O'Reilly, L. Wyder, T. Sanchez, B. Probst, J. Butler, A. Wood, G. Liau, et al. 2006. Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization. Cancer Res. 66 221–231. [DOI] [PubMed] [Google Scholar]
- 43.Sanna M. G., S. K. Wang, P. J. Gonzalez-Cabrera, A. Don, D. Marsolais, M. P. Matheu, S. H. Wei, I. Parker, E. Jo, W. C. Cheng, et al. 2006. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat. Chem. Biol. 2 434–441. [DOI] [PubMed] [Google Scholar]