

Abstract
Most mammalian cells exhibit transient delays in the G1 and G2 phases of the cell cycle after treatment with radiation or radiomimetic compounds. p53 is required for the arrest in G1, which provides time for DNA repair. Recently, a role of p53 in the G2/M transition has also been suggested. However, it has been reported that the presence of functional p53 does not always correlate with the induction of these checkpoints. To precisely assess the role of p53 in activating cell cycle checkpoints and in cell survival after radiation, we studied the response of two isogenic human fibrosarcoma cell lines differing in their p53 status (wild type or mutant). We found that when irradiated cells undergo a wild-type p53-dependent G1 arrest, they do not subsequently arrest in G2. Moreover, wild-type p53 cells irradiated past the G1 checkpoint arrest in G2 but do not delay in the subsequent G1 phase. Furthermore, in these cell lines, which do not undergo radiation-induced apoptosis, the wild-type p53 cell line exhibited a greater radioresistance in terms of clonogenic survival. These results suggest that the two checkpoints may be interrelated, perhaps through a control system that determines, depending on the extent of the damage, whether the cell needs to arrest cell cycle progression at the subsequent checkpoint for further repair. p53 could be a crucial component of this control system.
Mammalian cells respond to DNA-damaging agents by activating cell cycle checkpoints. These control mechanisms determine a temporary arrest at a specific stage of the cell cycle to allow the cell to correct possible defects (1, 2). At least two checkpoints monitor DNA damage: one at the G1/S transition and the other at the G2/M transition. The G1 checkpoint prevents replication of damaged DNA, whereas the G2/M transition is inhibited by damaged and/or incompletely replicated DNA (1, 2). Several findings have demonstrated that the product of the p53 tumor suppressor gene is responsible for the G1 checkpoint (3, 4). In response to genotoxic stress, the levels of p53 protein increase and this increase determines a transient arrest of cell cycle progression in the G1 phase (3, 4), or triggers apoptosis (5, 6). The arrest in G1 is thought to give the cells time to repair critical damage before DNA replication occurs, thereby avoiding the propagation of genetic lesions to progeny cells. The cell cycle can resume once the damage has been repaired or, if the damage is too extensive, the cell will undergo apoptosis. In this scenario, in cells with no or mutated p53, DNA replication proceeds in the presence of a damaged template, thereby generating clones of genetically aberrant cells from which malignant clones may arise. Loss of the G1 checkpoint also results in genomic instability, as seen by the increase in the frequency of gene amplification in p53-defective cells (7, 8). Based on such findings it has been proposed, and it is now largely accepted, that the main function of normal p53 is to preserve genome integrity by acting as the “guardian of the genome” (9).
Recent observations suggest that p53 also plays a role in regulating the G2/M transition (10–12). However, it has also been documented that the G2/M transition may be regulated independently from p53, since cells that are p53 nullizygous or with mutated p53 show a DNA damage-induced G2 arrest (3, 4).
Wild-type p53 is a sequence-specific transcription factor that activates the transcription of downstream effector genes. Among these genes is p21CIP1, the identification of which shed some light on the mechanisms by which p53 induction mediates cell cycle arrest. p21CIP1 inhibits kinase activity of various cyclin/cyclin-dependent kinase complexes, which are key elements in cell cycle progression (13–15), and by doing so it prevents the phosphorylation of the G1 cyclin/cyclin-dependent kinase substrate pRB, thereby preventing the transition from the G1 to the S phase of the cycle (16). Even though the role of p53 in mediating G1 arrest has been widely accepted, some discrepancies have been reported in that wild-type p53 cells do not always display a G1 arrest after radiation (17–19).
In contrast to the prediction based on the guardian of the genome role of wild-type p53, increased resistance to radiation has been reported in p53-defective cells (20–23). In contrast, other studies have shown no effect of p53 status on cell survival following radiation (24).
To reexamine p53 function in cell cycle control we have studied cell cycle progression in response to radiation in two isogenic human fibrosarcoma cell lines differing in their endogenous p53 status. Because these cell lines do not undergo apoptosis upon p53 overexpression (25), we have been able to address the issue of the role of p53 on cell survival without the unwanted effects of the apoptotic response. The results obtained suggest that wild-type p53 is a critical component of a cell cycle regulatory pathway(s) that controls the occurrence of both G1 and G2 arrest. p53 senses DNA damage at several stages of the cell cycle and accordingly determines whether the cell needs to arrest at the subsequent checkpoint to undergo DNA repair or proceed through it, in this way establishing an interrelation of the two checkpoints.
MATERIALS AND METHODS
Cell Cultures, Synchronization, and Radiation Treatment.
HT1080 fibrosarcoma cells (obtained from the American Type Culture Collection) and its derivative HT1080.6TG were cultured in Dulbecco’s modified Eagle’s medium (BioWhittaker) with 10% fetal calf serum (HyClone) in 5% CO2/95% air at 37°C. All radiation treatments were done exposing the cells to 4 Gy of γ-radiation from a 137Cs source at 2 Gy/min at room temperature. Synchronized cell populations were obtained by release from confluence/serum deprivation-induced G0 arrest. Cells were grown to high density in regular growth medium then switched to low serum medium (0.25% fetal calf serum) for 48 hr. They were then collected, counted, and 1 × 106 cells were replated in 25-cm2 flasks in regular growth medium.
Fluorescence-Activated Cell Sorter (FACS) Analyses.
Experiments with asynchronous cultures (see Fig. 1A) were done by plating 106 cells into 75-cm2 flasks. Two days later, cells were exposed to 4 Gy of radiation. At defined time intervals cells were incubated with 10 μM 5-bromodeoxyuridine (BrdUrd) for 30 min, collected, fixed by drop-wise addition of cold 70% ethanol, and stored at −20°C until ready to proceed. Cells were pelleted and resuspended in 2N HCl/0.5% Triton X-100 for 30 min, neutralized by washing in 0.1 M sodium borate and stained with fluorescein isothiocyanate-conjugated anti-BrdUrd antibody (Becton Dickinson), according to the manufacturer’s instructions. The cells were then resuspended in PBS containing 5 μg/ml of propidium iodide (PI). Two-color fluorescence analysis was done on a FACScan (Becton Dickinson) using the [scan]cellfit and lysis ii software and gating to exclude for cell doublets. When only PI staining was done (see Fig. 2), synchronized cells were collected, fixed, and incubated for 30 min at 37°C with 150 μg/ml RNase A in PBS prior to staining. Their cell cycle distributions were assessed using the cellfit software. The pulse-chase experiments with BrdUrd were done as follows. Exponentially proliferating cells were pulsed for 30 min with 10 μM BrdUrd, rinsed with fresh medium, and immediately irradiated with 4 Gy. They were then collected at various time intervals thereafter (see Fig. 3A). Alternatively, cells were synchronized to reenter the cycle by release from confluence/serum starvation and pulsed with 10 μM BrdUrd for 30 min when they were beginning to enter the S phase (5.5 hr after replating) (see Fig. 3B). They were then immediately irradiated or left untreated. In the BrdUrd pulse-chase experiments, cells were collected, processed as above, and two-color FACS analysis was performed, gating for the BrdUrd-positive cell population.
Figure 1.
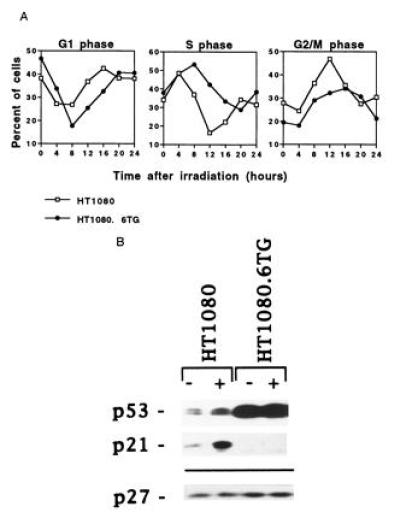
Response to ionizing radiation of exponentially growing cells. (A) Flow cytometric analysis of HT1080 (□) and HT1080.6TG (•) cells treated with 4 Gy γ-radiation. The number of cells in S phase was determined by BrdUrd incorporation. The number of cells in G1 and G2/M phases was estimated from the flow histograms using PI fluorescence. The experiments were repeated at least twice per cell line and the averages are shown. (B) Western blot analysis of p53, p21, and p27 protein levels in control and irradiated cells from each tumor cell line. The levels of these proteins were detected by immunoblotting 4 hr after treatment with 4 Gy γ-rays (+) or following no treatment (−). The membrane was probed for p27 to check for equal loading since its expression does not change in response to radiation.
Figure 2.
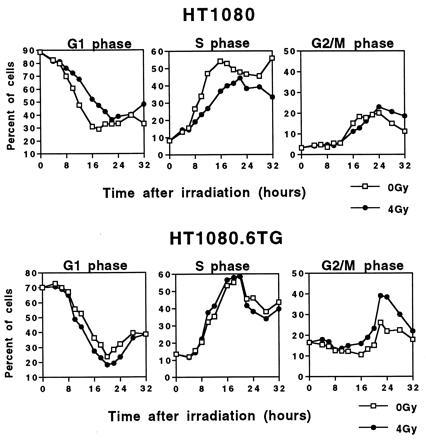
Cell cycle progression of synchronized cells irradiated in early G1 with 4 Gy of γ-radiation. (Upper) The percentage of control (0 Gy) or irradiated (4 Gy) synchronized HT1080 cells in the various phases of the cell cycle as a function of time after irradiation. (Lower) The percentage obtained for control (0 Gy) or irradiated (4 Gy) synchronized HT1080.6TG cells as a function of time.
Figure 3.
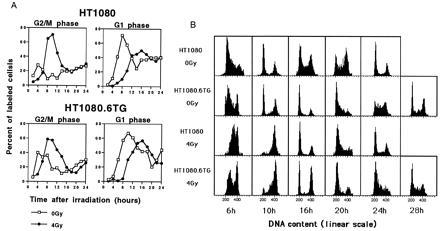
Progression of BrdUrd-labeled cells through the cell cycle following treatment with 4 Gy of γ radiation of asynchronously growing (A) or synchronously growing (B) cells. (A) (Upper) The percentage of BrdUrd-labeled control (0 Gy) or irradiated (4 Gy) HT1080 asynchronous cells in G2/M and G1 phases as a function of time after irradiation. (Lower) The percentage of BrdUrd-labeled control (0 Gy) or irradiated (4 Gy) for asynchronous HT1080.6TG cells. The cells were pulsed with BrdUrd, and then the BrdUrd-positive population was followed throughout the cell cycle by FACS analysis gating to consider only the BrdUrd-positive cells. The values reported are the means of two independent experiments. (B) Flow cytometric profiles of BrdUrd-positive synchronized HT1080 or HT1080.6TG cells irradiated in early S (4 Gy) or left untreated (0 Gy). Cells were pulsed with BrdUrd when they were beginning to enter into the S phase and immediately irradiated. Cells were collected every 2 hr over a period of 32 hr and two-color FACS analysis performed gating for BrdUrd-positive cells.
Western Blotting.
For protein analysis, cells were lysed in RIPA buffer (150 mM NaCl/1% Nonidet P-40/0.4% deoxycholate/0.1% SDS/50 mM Tris·HCl, pH 8/1 mM phenylmethylsulfonyl fluoride). Total proteins (50 μg) were separated on a SDS/12% polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Millipore). The membrane was probed with monoclonal (p53 and p21CIP1) and polyclonal (p27) antibodies (Santa Cruz Biotechnology) as recommended by the supplier and the signal was revealed by chemiluminescence (Amersham).
Clonogenic Assay.
The plating efficiencies of HT1080 and HT1080.6TG were in the ranges of 0.10 to 0.20 and 0.15 to 0.30, respectively. Appropriate numbers of cells were seeded and allowed to attach for 6 hr following which they were irradiated with graded doses of 137Cs γ-radiation. Following 10–12 days of incubation, colonies were stained, counted, and the surviving fraction calculated. Colony numbers ranged from 50 to 200.
RESULTS
Response to γ-Radiation.
We used the human fibrosarcoma cell line HT1080, which contains two wild-type p53 alleles (26) and an hypoxanthine phosphoribosyltransferase-deficient variant, HT1080.6TG, which has two mutant p53 alleles: Gly-245 → Arg-245 and Cys-277 → Phe-277 (27). Both cell lines are pseudodiploid and share the same marker chromosomes (ref. 27, E.J.S., unpublished observations). Both cell lines have similar in vitro growth characteristics and form aggressively growing tumors in nude mice (28). The cell cycle distributions of irradiated cells and unirradiated controls were determined by BrdUrd and PI incorporation using two-color FACS analysis. BrdUrd incorporation allowed an accurate estimate of the S-phase population. Cell cycle distributions were examined at regular intervals after irradiation for a period that covers the generation time of these cell lines (20–22 hr). This frequent sampling allowed us to measure more precisely the magnitude and the duration of the radiation-induced G1 and G2 arrests.
Asynchronously proliferating cells were exposed to 4 Gy of γ-radiation and harvested at various times thereafter (Fig. 1A). The p53 wild-type HT1080 cells showed temporary arrest at the G1 checkpoint as seen by the reduced number of cells exiting from G1 and progressing into S phase compared with the p53 mutant HT1080.6TG cells (Fig. 1A). The kinetics of transition through S phase also support the thesis of a temporary G1 arrest, because there is a greater depletion of the S-phase cells in HT1080 than in the p53 mutant cells. Both cell lines also displayed a transient arrest in G2 (Fig. 1A). Furthermore, the p53 and p21CIP1 protein levels increased only in irradiated p53 wild-type cells (Fig. 1B), underscoring the importance of elevated p21CIP1 expression in G1 arrest (29–32).
One significant finding emerging from the data in Fig. 1A is the marked difference between the two cell lines in the kinetics of passage through G2. The G2 arrest for HT1080 cells was quite short: it peaked at 12 hr after irradiation and was no longer apparent at 20 hr after irradiation. In contrast, HT1080.6TG cells showed a broader G2 delay: it peaked at 12–16 hr, but the percentage of G2/M cells did not return to baseline values until 24 hr after irradiation. The difference in the duration of the G2 arrest between the two cell lines could indicate that HT1080 cells that lie in early G1 at the time of radiation arrest at the G1 checkpoint, and that this event is not followed by a subsequent G2 arrest. Therefore, only the fraction of cells that are past the G1 checkpoint at the time of irradiation will display a G2 arrest and, ultimately, the transit through G2 should be faster than for HT1080.6TG cells, which can arrest their progression only in G2 to correct DNA damage.
p53-Mediated G1 Arrest Overrides a Subsequent G2 Block.
To assess more precisely the relationship between an initial G1 arrest and a subsequent G2 arrest for the same population of cells, we analyzed synchronously dividing cultures. Cells were maintained at high cell density in medium containing low serum to produce a G0 arrest. The cells were then released from G0, by replating at low cell density, and irradiated shortly (1 hr) after replating before they could reach the p53-mediated G1 checkpoint. Synchronized HT1080 (wild-type p53) and HT1080.6TG (mutant p53) cells were irradiated with 4 Gy or unirradiated (0 Gy) and samples were taken at serial time points for flow cytometry (Fig. 2). It can be seen in the irradiated HT1080 cells that there is a delay of approximately 4 hr in exiting G1 compared with the unirradiated controls. In contrast, no delay in exiting G1 is seen in the irradiated HT1080.6TG cells compared with the controls. However, the situation is reversed with respect to the G2 block. Irradiated HT1080 cells that have already experienced a G1 block do not undergo a subsequent G2 block. HT1080.6TG cells, which do not block in G1 following irradiation, experienced a significant G2 block (Fig. 2). Similar results were also obtained using cells synchronized in the M phase of the cell cycle by the mitotic inhibitor nocodazole (data not shown). These data clearly show that the G1 arrest overrides a subsequent G2 arrest.
Interrelation Between G1 and G2 Arrest.
Our observation of a lack of G2 arrest subsequent to a G1 arrest begs the question as to whether the reverse is true, a situation that would not be compatible with a second post-irradiation cycle G1 block.
To address this question, exponentially growing cells were pulse-labeled with BrdUrd for 30 min and irradiated immediately with 4 Gy or unirradiated and collected every 2 hr over a period of 24 hr. An analysis was made of the progression of the BrdUrd-labeled population through the cell cycle. These BrdUrd positive cells were in S phase at the time of radiation, so they were beyond the p53-dependent G1 checkpoint. The data in Fig. 3A clearly illustrate that both HT1080 and HT1080.6TG show evidence of a radiation-induced G2 block. In contrast to the PI analyses of asynchronous cell populations (Fig. 1A), the difference in the kinetics of G2 arrest between HT1080 and HT1080.6TG cells irradiated in the S phase was much less marked. Following arrest in G2, the kinetics of progression of the BrdUrd-positive subpopulation of cells through G1 were similar for both cell lines (Fig. 3A). In both cases the irradiated cells are delayed in entering the subsequent G1 phase compared with controls as a result of the transient G2 delay. However, there is no evidence of an increase in the percentage of G1 cells, or of a reduced rate of exit from G1, in irradiated samples compared with the unirradiated controls, indicating no subsequent G1 block.
To further confirm these findings a different strategy was used. Cells were synchronized by density inhibition, replated, then pulse-labeled with BrdUrd at the time when the majority of the cells were entering into the S phase (6 hr after replating), and then either immediately irradiated with 4 Gy or left untreated. This strategy allowed us to monitor a narrower window in time of cells that were irradiated in early S phase. This is in contrast to the previously described experiment where the whole S-phase population was followed (Fig. 3A). Samples were harvested for FACS analysis every 2 hr for up to 32 hr after irradiation. In Fig. 3B are shown profiles of BrdUrd-positive cells at relevant time points. First, consider the unirradiated control cells. At 10 hr after labeling (16 hr after plating), the cells from both cell lines have progressed from S into G2 and on to G1. At 16 hr after labeling, the HT1080 cells show significant further progression into S and through to G2. By 24 hr after labeling, a profile typical of an asynchronous population is seen. In contrast, HT1080.6TG cells take longer to transit through the first G1 (see profiles at 16 and 20 hr). As a result of this longer progression (≈4 hr), a profile typical of an asynchronous population is achieved by 28 hr. Now consider the irradiated populations. In contrast to the unirradiated controls, at 10 hr after irradiation there is a marked G2 block for both cell lines. Exit from this block is more rapid for the wild-type p53 cells (see 16-hr profiles). Once past the G2 checkpoint there is no evidence of a subsequent G1 arrest in either irradiated population and, correspondingly, there is clear evidence for progression from G1 to S in both HT1080 (20-hr profile) and HT1080-6TG (24-hr profile).
p53 Status and Radiosensitivity.
We also addressed the issue of the relationship between p53 status and radiation sensitivity. The sensitivity of exponentially growing cells to radiation-induced reproductive failure was measured using a colony-forming assay. Interestingly, the inactivation of p53 resulted in an increase in radiosensitivity (Fig. 4). HT1080 cells were found to be more radioresistant than the mutant p53 HT1080.6TG cells. The respective survival curve parameters were: Dq = 5.6 Gy and Do = 2.26 Gy for HT1080 and Dq = 3.2 Gy and Do = 2.17 Gy for HT1080.6TG. Therefore, the observed difference in radiosensitivity was completely due to a change in the shoulder of the survival curve (Dq) and not its terminal slope (1/Do).
Figure 4.
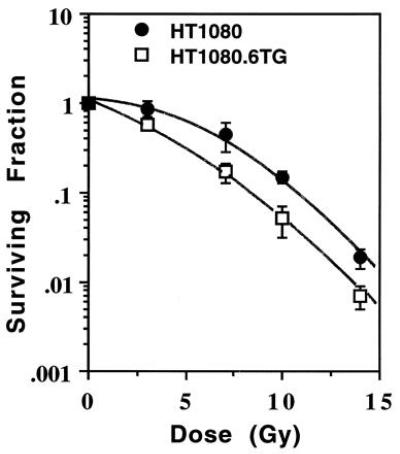
Surviving fraction as a function of dose for HT1080 (•) and HT1080.6TG (□) cells γ-irradiated in exponential growth phase. Points, means of five flasks per dose in two independent experiments; bars = standard deviation.
DISCUSSION
Despite the numerous studies done, conflicting claims as to the effects of wild-type p53 on cell cycle arrest and on cell survival upon treatment with γ-radiation have been made, perhaps because the majority of these studies employed cell lines of differing origins and growth characteristics for comparative purposes (17–19, 21–24). This study was designed to reexamine the role of p53 in the above mentioned issues, using isogenic cell lines having wild-type or mutant p53 alleles.
The first issue to be addressed was the induction and kinetics of the p53-dependent G1 arrest following treatment with γ-radiation. Even though p53 has been proven to be an essential factor in the induction of G1 arrest, it has recently been reported that some cell lines harboring wild-type p53 do not show G1 arrest upon radiation treatment (17). To verify the role of p53 in mediating a G1 arrest, HT1080 (wild-type p53) and HT1080.6TG (mutant p53) were treated with radiation. Irradiated wild-type p53 cells show a transient G1 arrest, probably mediated by induction of p21CIP1; this G1 arrest is not apparent in the mutant p53 cell line (Fig. 1).
The kinetics of the radiation-induced G1 arrest in asynchronous populations of wild-type p53 cells also have been questioned (18, 19). This is due, in part, because most observations have been based on the analysis of flow cytometric distributions at long times after irradiation (3, 4, 22, 23), where the possibility of contamination from second cycle cells was considered to be a likely confounding factor. This was not an unreasonable criticism. Using colcemid to block cell progression through mitosis, it has been shown that progression of irradiated human leukemic cells through the first cycle G1 is independent of p53 status, with no evidence of a G1 block (18, 19). The hypothesis formulated by the authors to reconcile these data with the bulk of the literature on p53-mediated G1 arrest is that the arrest previously observed by several groups was due to the fact that these cells delayed their progression through the G1 checkpoint in the second post-irradiation cell cycle (18). This would imply that cells that have undergone a G2 arrest experience a subsequent G1 block. Our data using G0 synchronized cell populations (Fig. 2) are in clear disagreement with the conclusion that progression through the first G1 cycle is independent of p53 status and are in agreement with those of Yount and colleagues who studied glioblastoma cells (33). The issue of which G1 phase is affected by radiation is essential for a better understanding of the molecular mechanisms of the p53-mediated radiation response in normal and cancer cells. Since mutant or deleted p53 alleles have been found in approximately 50% of all human tumors analyzed, the knowledge of how p53 affects the response to radiation may ultimately lead to more successful radiotherapeutic approaches in the treatment of such tumors.
Induction of the temperature sensitive p53Val135 mutant has been shown to arrest the cell cycle progression at G2/M in the rat cell line REF52 (12). Using an inducible expression system, it was reported that human p53 null fibroblasts show a G2 arrest upon induction of the exogenous p53 gene (10). These data, obtained in the absence of DNA damage and using transfected exogenous genes, suggested an involvement of p53 in regulating the G2/M transition. However, it has to be mentioned that the DNA damage-mediated G2 block certainly has a component that is p53 independent. In fact, arrest at the G2/M checkpoint has also been observed in p53-defective cells exposed to ionizing radiation (3, 4). Guillouf et al. (11) reported that the kinetics of γ-radiation-induced apoptosis in M1 leukemia cells correlates with the rapidity of exit from the G2 arrest, and that exit from G2 is accelerated by functionally wild-type p53. Our current data suggest that the duration of the G2 block following radiation may, at least in part, depend on p53, and that this is independent from induction of an apoptotic pathway since the cells analyzed do not undergo apoptosis (25). An intriguing possibility is that p53 may reduce the G2 delay because it stimulates a more efficient DNA repair process. The exact role of p53 in mediating DNA repair is under active investigation. To date the only firm data relate to nucleotide excision repair, which has a reduced efficiency in both heterozygous and homozygous mutant p53 Li-Fraumeni fibroblasts (34, 35). Interestingly, this reduced efficiency is seen only in terms of global DNA repair, and transcription-coupled repair is unaffected (35).
Studies of the influence of p53 status on cellular radiosensitivity have produced contradictory results. However, these studies were largely performed with non-isogenic cell lines (22–24, 36, 37) or cell lines transfected with p53 cDNA expression vectors (33). In contrast to what is expected, if wild-type p53 plays a role in promoting DNA repair, thereby avoiding accumulation of mutations, the majority of these studies reported an increased radioresistance in p53-defective cells compared with wild-type p53 cells. A possible explanation for these findings could be that cells having a mutated p53 were more resistant because of their inability to initiate apoptosis. The finding that HT1080 wild-type p53 cells exhibit a better survival than the mutant p53 cell line agrees with the prediction that cells with a normal p53 response pathway, and which do not undergo DNA damage-induced apoptosis, should be more resistant than p53-deficient cells. Moreover, the parameters of the survival curves (Dq and 1/Do) indicate that the principal difference between the two cell lines is their ability to repair potentially lethal radiation injury, further supporting the hypothesis that the presence of functional p53 may positively affect the repair capacity of these cells. Recently, it has been shown that loss of p53 can sensitize cells that do not undergo apoptosis to some drugs, including Taxol and cisplatin (38, 39).
By employing the isogenic HT1080 and HT1080.6TG cell lines we have documented the novel observation that a p53-mediated G1 block supercedes a subsequent G2 block following radiation damage. Furthermore, we have demonstrated that a G2 block is not subsequently followed by arrest in the next G1 phase of the cell cycle in p53 wild-type or mutant cells. The molecular mechanisms directing DNA repair at the G1 or at the G2 checkpoint are still unclear. However, one could speculate that the two control systems are able to metabolically interact with each other, perhaps via p53, since the activation of one checkpoint renders dispensable the activation of the subsequent checkpoint, assuming that the damage has been repaired.
Several questions concerning p53-mediated checkpoint control, following DNA damage, arise from these studies. In the guardian of the genome hypothesis (9) it is either implicitly or explicitly accepted that the G1 checkpoint is the critical distinguishing feature of wild-type p53-mediated cell cycle arrest, allowing for efficient DNA damage repair. Given that those cells in an asynchronously dividing population that are past the G1 checkpoint at the time of DNA damage arrest in G2, irrespective of p53 status, they would undergo the same DNA damage repair process. This, coupled with a lack of a subsequent G1 arrest in the succeeding cell cycle of the wild-type p53 cells, would presumably compromise a G1 arrest-based guardian of the genome effect. Alternatively, the G2 arrest seen in wild-type p53 HT1080 cells may, in part, be p53 mediated and therefore provide a more efficient DNA damage repair process compared with the p53-independent G2 arrest. This alternative explanation would invoke a guardian of the genome role for p53 in both G1 and G2 arrest. The kinetics of radiation survival undertaken in this asynchronous cell population do not allow us to distinguish between these possibilities. It is important to note that our studies were undertaken with isogenic cancer cell lines. Other authors have reported a prolonged G1 arrest in normal human fibroblasts, following DNA damage in G1 (32). To our knowledge the detailed analyses described in this study have not been reported for normal cells. Irrespective of the differences that may exist between normal fibroblasts. Irrespective of the differences that may exist between normal fibroblasts and the HT1080 isogenic cells we believe that the cell lines described in this paper should represent an excellent experimental model system to dissect the role of p53 in the relative effectiveness of G1 versus G2 arrest in determining the efficiency of DNA damage repair and the reproductive survival of irradiated cells.
Acknowledgments
We thank Sue DeMaggio of the Optical Biology Shared Resource of the Cancer Center Support Grant (CA62203) (University of California, Irvine), for expert assistance. This work was supported by grants from the National Institutes of Health. N.S.P. was a fellow of the Cancer Research Committee of the University of California.
Footnotes
Abbreviations: FACS, fluorescence-activated cell sorter; BrdUrd, bromodeoxyuridine; PI, propidium iodide.
References
- 1.Hartwell L H, Weinert T A. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 2.Hartwell L H, Kastan M B. Science. 1994;266:1821–1828. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 3.Kastan M B, Onyekwere O, Sidransky D, Vogelstein B, Craig R. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 4.Kuerbitz S J, Plunkett B S, Walsh W V, Kastan M B. Proc Natl Acad Sci USA. 1992;89:7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke A R, Purdie C A, Harrison D J, Morris R G, Bird C C, Hooper M L, Wyllie A H. Nature (London) 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 6.Lowe S W, Schmitt E M, Smith S W, Osborne B A, Jacks T. Nature (London) 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 7.Yin Y, Tainsky M, Bischoff F Z, Strong L C, Wahl G M. Cell. 1992;70:937–948. doi: 10.1016/0092-8674(92)90244-7. [DOI] [PubMed] [Google Scholar]
- 8.Livingstone L, White A, Sprouse J, Livanos E, Jacks T, Tlsty T. Cell. 1992;70:923–935. doi: 10.1016/0092-8674(92)90243-6. [DOI] [PubMed] [Google Scholar]
- 9.Lane D P. Nature (London) 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 10.Agarwal M L, Agarwal A, Taylor W R, Stark G R. Proc Natl Acad Sci USA. 1995;92:8493–8497. doi: 10.1073/pnas.92.18.8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guillouf C, Rosselli F, Krishnaraju K, Moustacchi E, Hoffman B, Liebermann D A. Oncogene. 1995;10:2263–2270. [PubMed] [Google Scholar]
- 12.Stewart N, Hicks G G, Paraskevas F, Mowat M. Oncogene. 1995;10:109–115. [PubMed] [Google Scholar]
- 13.Harper J W, Adami G R, Wei N, Keyomarsi K, Elledge S J. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 14.Xiong Y, Hannon G J, Zhang H, Casso D, Kobayashi R, Beach D. Nature (London) 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 15.Gu Y, Turck C W, Morgan D O. Nature (London) 1993;366:707–710. doi: 10.1038/366707a0. [DOI] [PubMed] [Google Scholar]
- 16.Slebos R J C, Lee M H, Plunkett B S, Kessis T D, Williams B O, Jacks T, Hedrick L, Kastan M B, Cho K. Proc Natl Acad Sci USA. 1994;91:5320–5324. doi: 10.1073/pnas.91.12.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li C-Y, Nagasawa H, Dahlberg W K, Little J B. Oncogene. 1995;11:1885–1892. [PubMed] [Google Scholar]
- 18.Nagasawa H, Li C-Y, Maki C G, Imrich A C, Little J B. Cancer Res. 1995;55:1842–1846. [PubMed] [Google Scholar]
- 19.Little J B, Nagasawa H, Keng P C, Yu Y, Li C-Y. J Biol Chem. 1995;270:11033–11036. doi: 10.1074/jbc.270.19.11033. [DOI] [PubMed] [Google Scholar]
- 20.Lowe S W, Ruley H E, Jacks T, Housman D E. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 21.O’Connor P M, Jackman J, Jondle D, Bhatia K, Magrath I, Kohn K W. Cancer Res. 1993;53:4776–4780. [PubMed] [Google Scholar]
- 22.McIlwrath A J, Vasey P A, Ross G M, Brown R. Cancer Res. 1994;54:3718–3722. [PubMed] [Google Scholar]
- 23.Fan S, El-Deiry W S, Bae I, Freeman J, Jondle D, Bhatia K, Fornace A J, Jr, Magrath I, Khon K W, O’Connor P M. Cancer Res. 1994;54:5824–5830. [PubMed] [Google Scholar]
- 24.Slichenmyer W J, Nelson W G, Slebos R J, Kastan M B. Cancer Res. 1993;53:4164–4168. [PubMed] [Google Scholar]
- 25.Pellegata N S, Cajot J-F, Stanbridge E J. Oncogene. 1995;11:337–349. [PubMed] [Google Scholar]
- 26.Tarunina M, Jenkins J R. Oncogene. 1993;7:1513–1523. [Google Scholar]
- 27.Anderson M J, Casey G, Fasching C L, Stanbridge E J. Genes Chromosomes Cancer. 1994;9:266–281. doi: 10.1002/gcc.2870090407. [DOI] [PubMed] [Google Scholar]
- 28.Plattner R, Anderson M J, Sato K Y, Fasching C L, Der C J, Stanbridge E J. Proc Natl Acad Sci USA. 1996;93:6665–6670. doi: 10.1073/pnas.93.13.6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dulic V, Kaufmann W K, Wilson S J, Tlsty T D, Lees E, Harper J W, Elledge S J, Reed S I. Cell. 1994;76:1013–1023. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 30.Bae I, Fan S, Bhatia K, Kohn K W, Fornace A J, Jr, O’Connor P M. Cancer Res. 1995;55:2387–2393. [PubMed] [Google Scholar]
- 31.Waldman T, Kinzler K W, Vogelstein B. Cancer Res. 1995;55:5187–5190. [PubMed] [Google Scholar]
- 32.Di Leonardo A, Linke S P, Clarkin K, Wahl G M. Genes Dev. 1994;8:2540–2551. doi: 10.1101/gad.8.21.2540. [DOI] [PubMed] [Google Scholar]
- 33.Yount G L, Haas-Kogan D A, Vidair C A, Haas M, Dewey W C, Isreal M A. Cancer Res. 1996;56:500–506. [PubMed] [Google Scholar]
- 34.Wang X W, Yeh H, Schaeffer L, Roy R, Moncollin V, Egly J-M, Wang Z, Friedberg E C, Evans M R, Taffe B G, Bohr V A, Weeda G, Hoeijmakers J H J, Forrester K, Harris C C. Nat Genet. 1995;10:188–195. doi: 10.1038/ng0695-188. [DOI] [PubMed] [Google Scholar]
- 35.Ford J M, Hanawalt P C. Proc Natl Acad Sci USA. 1995;92:8876–8880. doi: 10.1073/pnas.92.19.8876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brachman D G, Beckett M, Graves D, Haraf D, Vokes E, Weichselbaum R R. Cancer Res. 1993;53:3667–3669. [PubMed] [Google Scholar]
- 37.Smith M L, Chen I-T, Zhan Q, O’Connor P M, Fornace A J., Jr Oncogene. 1995;10:1053–1059. [PubMed] [Google Scholar]
- 38.Fan S, Smith M L, Rivet D J, II, Duba D, Zhan Q, Kohn K W, Fornace A J, Jr, O’Connor P M. Cancer Res. 1995;55:1649–1654. [PubMed] [Google Scholar]
- 39.Wahl A F, Donaldson K L, Fairchild C, Lee F Y F, Foster S A, Demers G W, Galloway D A. Nat Med. 1996;2:72–79. doi: 10.1038/nm0196-72. [DOI] [PubMed] [Google Scholar]