

Abstract
Voltage-gated potassium channels related to the Shal gene of Drosophila (Kv4 channels) mediate a subthreshold-activating current (ISA) that controls dendritic excitation and the backpropagation of action potentials in neurons. Kv4 channels also exhibit a prominent low voltage–induced closed-state inactivation, but the underlying molecular mechanism is poorly understood. Here, we examined a structural model in which dynamic coupling between the voltage sensors and the cytoplasmic gate underlies inactivation in Kv4.2 channels. We performed an alanine-scanning mutagenesis in the S4-S5 linker, the initial part of S5, and the distal part of S6 and functionally characterized the mutants under two-electrode voltage clamp in Xenopus oocytes. In a large fraction of the mutants (>80%) normal channel function was preserved, but the mutations influenced the likelihood of the channel to enter the closed-inactivated state. Depending on the site of mutation, low-voltage inactivation kinetics were slowed or accelerated, and the voltage dependence of steady-state inactivation was shifted positive or negative. Still, in some mutants these inactivation parameters remained unaffected. Double mutant cycle analysis based on kinetic and steady-state parameters of low-voltage inactivation revealed that residues known to be critical for voltage-dependent gate opening, including Glu 323 and Val 404, are also critical for Kv4.2 closed-state inactivation. Selective redox modulation of corresponding double-cysteine mutants supported the idea that these residues are involved in a dynamic coupling, which mediates both transient activation and closed-state inactivation in Kv4.2 channels.
INTRODUCTION
Voltage-dependent potassium (Kv) channels are critically involved in the control of neuronal excitability and discharge behavior (Hille, 2001). Depending on the voltage range and the kinetics of their activation, Kv channels may prevent the generation of action potentials and/or shape the time course of action potential decay. Most Kv channels undergo a process of inactivation; i.e., the membrane depolarization, which activates them, also drives them into a nonconducting refractory state from which they can only recover during a phase of re- or hyperpolarization (Kurata and Fedida, 2005). Thus, in addition to their activation properties, inactivation kinetics and the voltage dependence of steady-state inactivation determine the availability of Kv channel conductances and their relative contribution to neuronal signaling.
A-type potassium conductances, first characterized in molluscan neurons (Hagiwara et al., 1961), show rapid activation followed by rapid inactivation, which results in transient current flow. Some of the potassium channel genes initially cloned from Drosophila, including Shaker and Shal, encode Kv channels with such typical A-type features (Wei et al., 1990). From these two Drosophila A-type channels, Shaker has been in use for many years as a model system to study potassium channel inactivation. It has been found that Shaker inactivation is mainly based on two distinct but functionally coupled mechanisms, termed N- and C-type inactivation, respectively (Choi et al., 1991; Hoshi et al., 1991). To induce considerable inactivation in Shaker channels, strong depolarization has to be applied, which leads to channel opening. Once opened, an N-terminal inactivation domain occludes the channel pore from the cytoplasmic side (Hoshi et al., 1990). Shaker C-type inactivation, on the other hand, represents a general dynamic rearrangement of the external channel mouth (Liu et al., 1996). Interestingly, N-type inactivation favors the entry of Shaker channels into the C-type inactivated state (Baukrowitz and Yellen, 1995) in which they accumulate during prolonged depolarizations. The recovery from C-type inactivation is usually rather slow (many seconds) and cannot be accelerated much by membrane hyperpolarization.
Shal gene–related (Kv4) A-type channels are closely related to Shaker but show a different inactivation behavior. In Kv4 channels both N- and C-type–related inactivation mechanisms have been identified; however, they seem to play a minor role in the gating of these channels (Gebauer et al., 2004; Kaulin et al., 2008). N-terminal truncation, which completely removes Shaker N-type inactivation (Hoshi et al., 1990), only moderately slows Kv4 channel inactivation (Jerng and Covarrubias, 1997; Bähring et al., 2001a); and high external potassium concentrations, which counteract Shaker C-type inactivation (Baukrowitz and Yellen, 1995), actually accelerate Kv4 channel inactivation rather than slow it (Jerng and Covarrubias, 1997; Bähring et al., 2001a; Kaulin et al., 2008). Furthermore, neither internal nor external tetraethylammonium, which interferes with Shaker N- or C-type inactivation, respectively (Choi et al., 1991), has any effect on Kv4 channel inactivation (Jerng and Covarrubias, 1997). Unlike Shaker channels, the Shal gene–related Kv4 channels exhibit a prominent, low voltage–induced closed-state inactivation (Jerng et al., 1999; Bähring et al., 2001a). Even during strong depolarization, when Kv4 channels do open and mediate A-type currents, they finally accumulate in the closed-inactivated state (Jerng et al., 1999; Bähring et al., 2001a; Wang et al., 2005) from which they rapidly recover (tens to hundreds of milliseconds) in a highly voltage-dependent manner. For numerous experimental manipulations of Kv4 channel gating, such as the use of rubidium instead of potassium as charge carrier (Bähring et al., 2001a; Shahidullah and Covarrubias, 2003), the coexpression of accessory subunits (Barghaan et al., 2008), the deletion of N-terminal domains (Barghaan et al., 2008), or the insertion of point mutations in the S4-S5 linker and the distal S6 segment (Jerng et al., 1999), it has been shown that the effects on tail current deactivation kinetics directly correlate with the effects on macroscopic inactivation kinetics. These findings support a model of preferential closed-state inactivation at all physiologically relevant membrane potentials due to a tight deactivation–inactivation coupling in Kv4 channels.
Although the existence of closed-inactivated states and the kinetic deactivation–inactivation coupling have been unequivocally shown for Kv4 channels, the structural correlates of closed-state inactivation have remained elusive. Here, we studied closed-state inactivation of Kv4.2 channels, the molecular substrate of the somatodendritic A-type potassium current. We hypothesized that, similar to the mechanism previously suggested for a hyperpolarization-activated cyclic nucleotide-gated (HCN) channel (Shin et al., 2004), temporary uncoupling at the interface between voltage sensor and cytoplasmic gate may underlie closed-state inactivation in Kv4.2 channels. We tested our hypothesis experimentally by applying a detailed thermodynamic analysis of low-voltage inactivation parameters using Kv4.2 channels with point mutations in the S4-S5 linker, the initial part of S5, and the distal S6 segment. Our results support a dynamic coupling between voltage sensor and gate, and they complement previous data showing that the voltage sensors are critically involved in Kv4.2 closed-state inactivation (Dougherty et al., 2008).
MATERIALS AND METHODS
Channel Constructs and Heterologous Expression
All constructs were based on a human Kv4.2 clone (Zhu et al., 1999) in a pGEM-HE bacterial expression vector. We performed an alanine-scanning mutagenesis in the S4-S5 linker region, including the initial part of S5 and in the distal part of S6 (see Figs. 3 and 4 A). These regions will, hereafter, be referred to as S4S5 and S6, respectively. Single- or double-point mutations were introduced with overlap PCR using Pfu-Turbo DNA polymerase (Agilent Technologies) in full-length Kv4.2 channels or N-terminally truncated Kv4.2Δ2-10 and Kv4.2Δ2-40 channels (Bähring et al., 2001b), and verified by automated DNA sequencing. In parallel to our mutant scan, we also studied the S4S5 single mutation C320S and the S6 double mutation V402I:V404I. These Kv4.2 mutations (Gebauer et al., 2004; Pourrier et al., 2004) or mutations at homologous sites in Kv4.1 (Jerng et al., 1999) and Kv4.3 (Wang et al., 2002) have been used in previous studies in different contexts. In these studies, the type of amino acid substitution was based on a sequence alignment of all four Shaker-related Kv channel subfamilies, which revealed that a cysteine instead of serine in S4S5 is Shal specific, and isoleucine instead of valine at two positions in S6 is only found in Shab channels (Jerng et al., 1999; see also Fig. 4 A).
Figure 3.

Voltage sensor gate uncoupling model and putative protein domains involved. (A) Conceptual gating scheme illustrating a working model for the closed-state inactivation of Kv4.2 channels (black letters). In each α-subunit, depolarization leads to a transition from “Resting” (R) to “Activated” (A). If all four subunits reach the activated state (AAAA), the channel can open (O; gray letters). However, the transition of a single subunit to an inactivated/desensitized (I/D) state is sufficient to prevent channel opening. Cartoons depict the three states of an individual α-subunit with its six-membrane–spanning domains (S1-S6; positive charge in S4) and cytoplasmic N and C termini. Resting (R), voltage sensor down, S6 gate closed; activated (A), voltage sensor up (vertical green arrow) and S6 gate open permissive (blue arrow pointing to the left); inactivated/desensitized (I/D), voltage sensor still up, but in a more stable conformation than before, and S6 gate closed again but uncoupled from the voltage sensor (green and blue arrow pointing to the right). “Inactivated” refers to the voltage sensor conformation, and “Desensitized” refers to the state of the uncoupled S6 gate. For simplicity, in the cartoon voltage sensor activation is depicted by an upward motion of S3S4 with no further conformational changes in S1-S4. (B) Ribbon representation of Kv1.2 crystal structure data (Long et al., 2005a). Two opposite α-subunits are shown from the side (angle of view parallel to the plasma membrane). Cytoplasmic portions preceding S1 have been omitted. The S4-S5 linker, including the initial S5 segment and the distal portion of S6 (orange in A and B), known to be critically involved in voltage-dependent gate opening, were examined here by scanning mutagenesis of Kv4.2.
Figure 4.

Scanning mutagenesis in Kv4.2 S4S5 and S6, and functional expression of mutants. (A) Alignment of Shaker, rat Kv1.2, rat Kv2.1, rat Kv3.1, and human Kv4.2 amino acid sequences in the S4-S5 linker, including the initial part of S5 (S4S5; left) and the distal S6 segment (S6; right). Complementary Shaker sequences in S4S5 and S6 shown by Lu et al. (2002) to be essential for voltage control over the gate are depicted in white letters. Kv4.2 scanning mutagenesis extended from Gly 309 to Phe 329 in S4S5 and from Val 379 to Tyr 413 in S6. Mutations of Gly 314, Leu 327, Phe 329, Ile 398, Pro 403, Ile 405, and Val 406 (red) to alanine produced channels with currents, which could not be distinguished from endogenous oocyte conductances. In previous studies, Cys 320 in Kv4.2 (triangle) or a homologous cysteine in other Kv4 channels has been mutated to serine, residues homologous to Val 402 and Val 404 in Kv4.2 (triangles) to isoleucine (see Materials and methods). The Kv4.2 residues Glu 323 in S4S5 and Val 404 in S6 (filled circles) are homologous to the Shaker residues Glu 395 and Val 476, which have been shown to be critically involved in voltage-dependent gate opening (Yifrach and MacKinnon, 2002). (B) Current families obtained for Kv4.2 wild-type (wt) and individual point mutants with voltage protocols as in Fig. 1 A. Here, only responses to test potentials between −80 and +60 mV in 20-mV increments are shown, according to the illustrated voltage steps (top left). Note the small amplitudes and apparent positive shift in voltage dependence of activation for L310A. (C) Current families obtained for individual S6 point mutants with the same voltage protocols.
Female Xenopus laevis frogs were anesthetized in ethyl 3-aminobenzoate methanesulfonate (1.2 g/l tap water; Sigma-Aldrich), and part of the ovary lobes were surgically removed. Mechanically dispersed oocytes were incubated under constant agitation for 3 h in a solution containing the following (in mM): 82.5 NaCl, 2 KCl, 1 MgCl2, 5 HEPES, and 1.3 mg/ml collagenase A (Sigma-Aldrich), pH 7.5, with NaOH. De-folliculated state V–VI oocytes were selected and injected the next day with cRNA (3–8 ng/50 nl) using a nanoliter 2000 microinjector (WPI). For some experiments, Kv4.2 channels were coexpressed with human Kv channel interacting protein (KChIP)2 or with KChIP2 and human dipeptidyl aminopeptidase-like protein (DPP)6. KChIP2 and Kv4.2 cRNAs were coinjected at a ratio of 10:1. For the experiments with ternary complexes, higher amounts of DPP6, KChIP2, and Kv4.2 cRNA were injected (200 ng–1 µg/50–100 nl) at an approximate 1:1:1 ratio. Injected oocytes were incubated at 18°C in a solution containing (in mM) 75 NaCl, 2 KCl, 2 CaCl2, 1 MgCl2, 5 HEPES, and 50 µg/ml gentamicin (Sigma-Aldrich), pH 7.5, with NaOH, and used for recordings after 1–4 d.
Data Acquisition and Analysis
Currents were recorded at room temperature (20–22°C) under two-electrode voltage clamp using TURBO TEC-10CX and TURBO TEC-03 amplifiers (npi electronic) controlled by PULSE software (HEKA). Glass microelectrodes were filled with 3 M KCl and the tip was sealed with Agar (2% in 3 M KCl) to minimize potassium leakage during extended voltage protocols. The electrode-to-bath resistances lay between 500 and 800 kΩ. Oocytes were constantly superfused with a solution containing the following (in mM): 91 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES, pH 7.4, with NaOH. Kv4.2 channels were activated by depolarizing voltage jumps (10-mV increments) from −110 mV (see Fig. 1 A). Steady-state inactivation was induced by conditioning prepulses, 30-s long, to different potentials between −120 and 0 mV (5-mV increments; Fig. 1 B). The onset of low-voltage inactivation was monitored with a protocol in which a prepulse potential was applied for variable durations between 25 ms to 51.2 s (see Fig. 2 A). For the measurement of the recovery kinetics, inactivation was induced at −55 mV for 20 s and the recovery was assayed after different amounts of time at the recovery potential (Fig. 2 C). Signals were filtered at 10 kHz with low-pass Bessel characteristics, amplified as required, and digitized at sample intervals between 300 µs and 1 ms. Capacitive current transients were not compensated, and a P/5 routine was used for leak subtraction.
Figure 1.

Activation and inactivation voltage dependence of Kv4.2 wild-type channels. (A) A-type currents in response to depolarizing voltage jumps from −110 mV to different test potentials between −90 and +90 mV in 10-mV increments. Currents were recorded under two-electrode voltage clamp in Xenopus oocytes. (B) Prepulse inactivation of outward currents at +40 mV induced by conditioning prepulses of 20-s duration to potentials between −120 and 0 mV in 5-mV increments. (C) Voltage dependence of steady-state inactivation (filled symbols; n = 9) and voltage dependence of relative peak conductance activation (open symbols; n = 7) reflect the channel availability. The inactivation curve represents a first-order Boltzmann function, the activation curve a fourth-order Boltzmann function.
Figure 2.

Kinetic inactivation parameters of Kv4.2 wild-type channels. (A) Onset of low-voltage inactivation. Outward currents obtained with brief voltage pulses to +40 mV, either directly from −110 mV (Icontrol) or after a conditioning prepulse of different length (between 25 ms and 51.2 s) to −65 mV (Itest). Note the time axis breaks and indicated durations for extremely long prepulses. (B) Relative current amplitudes (Itest/Icontrol) obtained with different prepulse durations at −65 mV (n = 8). A single-exponential fit to the data yields the time constant of onset of low-voltage inactivation (τL) and the fraction of non-inactivated channels (f). Single-exponential fitting curves for the data obtained with other prepulse potentials are shown in gray (see Fig. S2 A for complete datasets). (C) Recovery from low-voltage inactivation. After a voltage pulse from −100 to +40 mV, which elicits the control current, low-voltage inactivation is induced at −55 mV for 20 s. Then, after different amounts of time at −100 mV, the test current amplitude at +40 mV is measured. (D) Relative current amplitudes (test/control) obtained with different interpulse durations expressed as percent recovery from inactivation at −100 mV (n = 7). A single-exponential fit yields the time constant of recovery from low-voltage inactivation (τrec). Single-exponential fitting curves for the data obtained at other recovery potentials are shown in gray (see Fig. S2 B for complete datasets). Onset and recovery curves obtained at −80 mV in B and D, respectively (dark gray), represent equal time constants (see also Fig. S2 C).
The program PULSEFIT (HEKA) was used to analyze current traces, and the obtained data were further processed using Kaleidagraph (Synergy Software) and GraphPad Prism (GraphPad Software). Peak conductance–voltage (GP-V) relationships were determined based on the equation:
where IP is the peak current amplitude at the test potential V, and Vrev is the potassium reversal potential (−90 mV in our experiments). GP-V relationships were analyzed with a fourth-order Boltzmann function of the form:
where GP/GP,max is the normalized peak conductance at a test potential V. The voltage dependence is defined by V’ (6.5% of the maximal conductance) and the slope factor s. V1/2,act values in Table I are the potentials where the above function has a value of 0.5. The voltage dependence of steady-state inactivation was analyzed with a first-order Boltzmann function of the form:
where I/Imax is the relative current amplitude obtained with a prepulse potential V. V1/2,inact and s are the voltage of half-maximal inactivation and the slope factor, respectively. Inactivation kinetics (onset of low-voltage inactivation, τL; macroscopic A-type current decay, τA; and recovery from inactivation, τrec) were approximated by single- or double-exponential functions. Pooled data are presented as mean ± SEM.
TABLE I.
Activation and Inactivation Parameters of Wild-type and Mutant Kv4.2 Channels
| IP,+40 (µA) | f+40 | n | V1/2,act (mV) | sact (mV) | n | V1/2,inact (mV) | sinact (mV) | n | |
| wt | 3.8 ± 0.6 | 0.042 ± 0.005 | 7 | +7.5 ± 0.7 | 20.4 ± 0.7 | 7 | −66.9 ± 0.8 | 5.9 ± 0.3 | 9 |
| G309A | 11.5 ± 1.1 | 0.425 ± 0.020 | 5 | −1.7 ± 1.1 | 22.3 ± 0.1 | 5 | −74.7 ± 0.9 | 5.2 ± 0.2 | 5 |
| L310A | 1.4 ± 0.3 | 0.148 ± 0.025 | 9 | +131 ± 19 | 62.0 ± 7.3 | 4 | −19.7 ± 0.4 | 12.1 ± 0.6 | 3 |
| R311A | 3.9 ± 0.6 | 0.020 ± 0.011 | 5 | +26.1 ± 2.3 | 22.9 ± 0.8 | 5 | −48.2 ± 2.1 | 5.8 ± 0.1 | 5 |
| I312A | 13.9 ± 1.7 | 0.224 ± 0.018 | 5 | +13.8 ± 2.0 | 20.7 ± 0.3 | 5 | −72.3 ± 1.1 | 7.7 ± 0.4 | 5 |
| L313A | 3.3 ± 0.4 | 0.011 ± 0.007 | 5 | +12.0 ± 0.3 | 25.0 ± 0.2 | 5 | −77.5 ± 0.4 | 7.0 ± 0.1 | 5 |
| G314A | — | — | — | — | — | — | |||
| Y315A | 10.1 ± 1.3 | 0.055 ± 0.009 | 5 | +8.7 ± 1.8 | 22.9 ± 0.5 | 5 | −62.8 ± 1.1 | 5.5 ± 0.1 | 5 |
| T316A | 5.4 ± 1.0 | 0.006 ± 0.006 | 5 | +11.7 ± 2.3 | 22.9 ± 0.8 | 5 | −72.2 ± 0.6 | 6.8 ± 0.3 | 5 |
| L317A | 5.1 ± 0.7 | 0.066 ± 0.007 | 5 | +21.6 ± 1.2 | 23.9 ± 0.2 | 5 | −72.6 ± 0.9 | 7.6 ± 0.4 | 5 |
| K318A | 5.4 ± 0.5 | 0.030 ± 0.004 | 5 | +15.2 ± 0.5 | 24.1 ± 0.2 | 5 | −67.7 ± 0.4 | 5.4 ± 0.2 | 5 |
| S319A | 0.23 ± 0.03 | 0.390 ± 0.060 | 3 | — | — | — | — | ||
| C320A | 5.3 ± 0.7 | 0.033 ± 0.007 | 5 | +21.1 ± 0.9 | 24.5 ± 0.6 | 5 | −65.9 ± 1.2 | 5.0 ± 0.2 | 5 |
| C320S | 12.9 ± 1.8 | 0.309 ± 0.017 | 7 | +7.2 ± 1.7 | 31.2 ± 0.3 | 7 | −67.4 ± 1.3 | 4.9 ± 0.2 | 6 |
| A321V | 5.2 ± 0.6 | 0.178 ± 0.016 | 5 | −9.2 ± 1.1 | 21.1 ± 0.7 | 5 | −68.4 ± 0.7 | 5.0 ± 0.3 | 5 |
| S322A | 7.8 ± 2.3 | 0.042 ± 0.008 | 5 | +20.7 ± 1.9 | 20.2 ± 0.2 | 5 | −52.7 ± 2.1 | 5.8 ± 0.2 | 5 |
| E323A | 4.3 ± 0.3 | 0.000 ± 0.000 | 8 | −2.6 ± 0.4 | 19.2 ± 0.3 | 8 | −79.1 ± 0.5 | 6.8 ± 0.1 | 8 |
| L324A | 0.16 ± 0.03 | 0.693 ± 0.092 | 4 | — | — | — | — | ||
| G325A | 11.2 ± 1.3 | 0.046 ± 0.005 | 5 | +8.4 ± 1.7 | 20.4 ± 0.4 | 5 | −64.9 ± 0.9 | 6.3 ± 0.2 | 6 |
| F326A | 2.3 ± 0.3 | 0.111 ± 0.018 | 5 | +10.4 ± 4.9 | 21.0 ± 0.3 | 5 | −45.9 ± 3.0 | 6.9 ± 0.1 | 5 |
| L327A | — | — | — | — | — | — | |||
| L328A | 0.15 ± 0.01 | 0.477 ± 0.029 | 3 | — | — | — | — | ||
| F329A | — | — | — | — | — | — | |||
| V397A | 0.9 | 0.286 | 1 | +130 | 106 | 1 | — | — | |
| I398A | — | — | — | — | — | — | |||
| A399V | 0.2/0.2 | 0.23/0.22 | 2 | — | — | — | — | ||
| L400A | 1.3 ± 0.1 | 0.036 ± 0.036 | 4 | +2.9 ± 4.4 | 30.2 ± 2.1 | 4 | −92.1 ± 1.9 | 10.9 ± 0.4 | 5 |
| P401A | 0.4 ± 0.1 | 0.400 ± 0.176 | 4 | — | — | — | — | ||
| V402A | 2.4 ± 0.1 | 0.059 ± 0.007 | 5 | +0.9 ± 1.6 | 23.4 ± 0.6 | 5 | −59.2 ± 0.9 | 9.8 ± 0.2 | 5 |
| P403A | — | — | — | — | — | — | |||
| V404A | 4.8 ± 0.5 | 0.070 ± 0.026 | 4 | +13.6 ± 7.5 | 25.0 ± 2.3 | 4 | −95.2 ± 2.5 | 10.2 ± 2.5 | 6 |
| V402I:V404I | 18.4 ± 1.2 | 0.360 ± 0.015 | 6 | +10.8 ± 1.2 | 28.0 ± 0.5 | 6 | −54.4 ± 0.3 | 5.3 ± 0.2 | 6 |
| I405A | — | — | — | — | — | — | |||
| V406A | — | — | — | — | — | — | |||
| S407A | 9.6 ± 0.1 | 0.031 ± 0.004 | 9 | +7.6 ± 2.5 | 21.1 ± 0.5 | 9 | −61.0 ± 0.8 | 5.9 ± 0.4 | 6 |
| N408A | 2.7 ± 0.7 | 0.013 ± 0.008 | 5 | +28.9 ± 5.5 | 22.9 ± 1.5 | 5 | −56.2 ± 2.5 | 5.7 ± 0.3 | 5 |
| F409A | 0.5/0.4 | 0.020/0.170 | 2 | — | — | — | — | ||
| S410A | 4.1 ± 0.6 | 0.029 ± 0.010 | 5 | +12.7 ± 1.8 | 22.1 ± 0.5 | 5 | −65.3 ± 1.3 | 6.3 ± 0.2 | 5 |
| R411A | 5.3 ± 0.3 | 0.027 ± 0.010 | 5 | +8.5 ± 1.4 | 21.7 ± 2.2 | 5 | −63.2 ± 1.2 | 6.5 ± 0.7 | 8 |
| I412A | 4.4 ± 0.7 | 0.025 ± 0.003 | 6 | +8.5 ± 1.8 | 23.5 ± 0.4 | 6 | −80.2 ± 0.9 | 5.9 ± 0.2 | 9 |
| Y413A | 5.0 ± 1.7 | 0.089 ± 0.057 | 6 | +9.5 ± 0.8 | 22.0 ± 0.6 | 6 | −62.6 ± 1.2 | 7.0 ± 0.4 | 8 |
| E323A:V404A | 2.8 ± 0.2 | 0.042 ± 0.021 | 5 | +11.4 ± 3.2 | 25.3 ± 0.9 | 5 | −94.8 ± 4.5 | 7.1 ± 0.3 | 5 |
| E323A:S407A | 3.9 ± 0.6 | 0.060 ± 0.014 | 5 | +16.3 ± 2.5 | 24.1 ± 0.4 | 5 | −69.4 ± 0.9 | 6.3 ± 0.2 | 6 |
| E323A:N408A | 4.0 ± 0.3 | 0.006 ± 0.003 | 5 | +26.1 ± 2.2 | 26.4 ± 0.4 | 5 | −72.0 ± 0.7 | 6.3 ± 0.1 | 6 |
| E323A:I412A | 5.4 ± 0.7 | 0.000 ± 0.000 | 5 | +8.5 ± 2.3 | 25.0 ± 0.2 | 5 | −85.0 ± 0.8 | 6.9 ± 0.2 | 5 |
| S322A:V404A | 9.4 ± 1.2 | 0.115 ± 0.003 | 5 | +14.3 ± 1.3 | 25.5 ± 0.3 | 5 | −89.6 ± 0.5 | 5.2 ± 0.1 | 5 |
| S322A:S407A | 13.2 ± 0.4 | 0.116 ± 0.003 | 5 | +17.3 ± 0.3 | 21.5 ± 0.2 | 5 | −51.1 ± 0.4 | 5.4 ± 0.1 | 5 |
| S322A:N408A | 2.8 ± 0.4 | 0.046 ± 0.026 | 3 | +33.7 ± 2.3 | 22.3 ± 0.8 | 3 | −52.8 ± 1.1 | 5.9 ± 0.3 | 5 |
| S322A:I412A | 8.2 ± 1.2 | 0.019 ± 0.009 | 4 | +13.4 ± 1.6 | 20.7 ± 0.5 | 4 | −69.6 ± 1.4 | 6.6 ± 0.5 | 5 |
Peak current amplitudes at +40 mV (IP,+40) and the relative current remaining after 1.5 s (f+40) were measured for Kv4.2 wild-type (wt), single mutant, and double mutant channels. The mutations G314A, L327A, F329A, I398A, P403A, I405A, and V406A produced channels with currents indistinguishable from endogenous voltage-dependent oocyte conductances. From peak conductance–voltage relationships and steady-state inactivation curves, the voltages for half-maximal activation (V1/2,act) and inactivation (V1/2,inact), respectively, and the corresponding slope factors s were determined with Boltzmann analysis. Some mutants with poor expression and/or extreme voltage shift, including S319A, L324A, L328A, V397A, A399V, P401A, and F409A, were not further analyzed.
Thermodynamic Calculations and Homology Modeling
From the time constant of onset of low-voltage inactivation (τL) and the remaining non-inactivating current fraction (f; see also Fig. 2 B), apparent on- and off-rates for the closed-inactivated state were determined, according to the equations
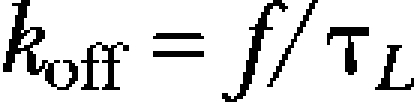 |
and
 |
Using the low-voltage inactivation rates kon and koff, the apparent affinity for the closed-inactivated state (Kci) was calculated for Kv4.2 wild-type and each mutant construct according to
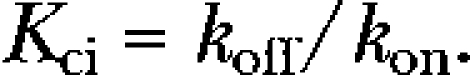 |
Mutation-induced changes in affinity were quantified as ln (Kci/Kci,wt), and the coupling coefficient for pairs of mutations was calculated as follows:
where wt:wt denotes the wild-type channel, S4S5mut:S6mut the double mutant, and S4S5:wt and wt:S6 single mutants with a point mutation in S4S5 or S6, respectively. We assumed linear propagation of independent errors through the above equation (see Table II). For Ω < 1 the reciprocal was used. All values are plotted as lnΩ.
TABLE II.
Thermodynamic Analysis of Kv4.2 Low-Voltage Inactivation
| τL,-65 (s) | f-65 | kon,-65 (s−1) | koff,-65 (s−1) | Kci,−65 | n | Ω (kT) | |
| wt | 1.28 ± 0.11 | 0.458 ± 0.032 | 0.448 ± 0.051 | 0.368 ± 0.030 | 0.898 ± 0.130 | 8 | |
| G309A | 0.60 ± 0.05 | 0.125 ± 0. 012 | 1.497 ± 0.129 | 0.208 ± 0.012 | 0.144 ± 0.016 | 5 | |
| R311A | 0.66 ± 0.26 | 0.922 ± 0.005 | 0.193 ± 0.077 | 2.260 ± 0.885 | 11.985 ± 0.821 | 5 | |
| I312A | 48.2 ± 8.47 | 0.428 ± 0.025 | 0.015 ± 0.005 | 0.011 ± 0.003 | 0.763 ± 0.085 | 5 | |
| L313A | 0.17 ± 0.03 | 0.167 ± 0.009 | 5.365 ± 0.671 | 1.085 ± 0.164 | 0.201 ± 0.013 | 5 | |
| Y315A | 1.10 ± 0.02 | 0.620 ± 0.032 | 0.348 ± 0.031 | 0.566 ± 0.028 | 1.713 ± 0.248 | 5 | |
| T316A | 0.41 ± 0.01 | 0.272 ± 0.007 | 1.791 ± 0.073 | 0.668 ± 0.015 | 0.375 ± 0.013 | 5 | |
| L317A | 0.36 ± 0.01 | 0.305 ± 0.025 | 1.971 ± 0.133 | 0.852 ± 0.048 | 0.446 ± 0.055 | 5 | |
| K318A | 0.91 ± 0.05 | 0.406 ± 0.017 | 0.657 ± 0.033 | 0.450 ± 0.034 | 0.687 ± 0.048 | 5 | |
| C320A | 1.10 ± 0.07 | 0.510 ± 0.049 | 0.452 ± 0.049 | 0.481 ± 0.075 | 1.150 ± 0.277 | 5 | |
| C320S | 5.13 ± 0.46 | 0.425 ± 0.066 | 0.122 ± 0.024 | 0.082 ± 0.009 | 0.859 ± 0.212 | 6 | |
| A321V | 3.50 ± 0.14 | 0.388 ± 0.039 | 0.174 ± 0.001 | 0.113 ± 0.015 | 0.661 ± 0.110 | 5 | |
| S322A | 1.32 ± 0.07 | 0.750 ± 0.039 | 0.187 ± 0.021 | 0.579 ± 0.063 | 3.401 ± 0.874 | 5 | |
| E323A | 0.16 ± 0.01 | 0.124 ± 0.007 | 5.478 ± 0.162 | 0.778 ± 0.053 | 0.142 ± 0.010 | 6 | |
| G325A | 1.26 ± 0.09 | 0.505 ± 0.028 | 0.404 ± 0.042 | 0.407 ± 0.028 | 1.047 ± 0.117 | 5 | |
| F326A | 0.68 ± 0.13 | 0.938 ± 0.010 | 0.100 ± 0.018 | 1.622 ± 0.440 | 15.940 ± 2.097 | 5 | |
| L400A | 0.39 ± 0.04 | 0.141 ± 0.016 | 2.341 ± 0.244 | 0.377 ± 0.049 | 0.167 ± 0.021 | 6 | |
| V402A | 0.56 ± 0.02 | 0.630 ± 0.027 | 0.664 ± 0.037 | 1.148 ± 0.088 | 1.768 ± 0.219 | 5 | |
| V404A | 0.14 ± 0.01 | 0.051 ± 0.010 | 6.783 ± 0.358 | 0.358 ± 0.055 | 0.054 ± 0.011 | 6 | |
| V402I:V404I | 17.42 ± 0.46 | 0.878 ± 0.008 | 0.007 ± 0.000 | 0.051 ± 0.002 | 7.384 ± 0.608 | 6 | |
| S407A | 1.74 ± 0.25 | 0.738 ± 0.031 | 0.165 ± 0.026 | 0.451 ± 0.044 | 3.079 ± 0.469 | 7 | |
| N408A | 0.72 ± 0.09 | 0.851 ± 0.037 | 0.215 ± 0.042 | 1.381 ± 0.303 | 7.759 ± 1.900 | 7 | |
| S410A | 0.72 ± 0.10 | 0.512 ± 0.033 | 0.735 ± 0.112 | 0.768 ± 0.124 | 1.092 ± 0.158 | 5 | |
| R411A | 0.73 ± 0.05 | 0.552 ± 0.019 | 0.629 ± 0.077 | 0.766 ± 0.036 | 1.240 ± 0.092 | 3 | |
| I412A | 0.60 ± 0.05 | 0.089 ± 0.006 | 1.579 ± 0.124 | 0.154 ± 0.016 | 0.097 ± 0.007 | 8 | |
| Y413A | 1.22 ± 0.20 | 0.561 ± 0.022 | 0.405 ± 0.098 | 0.529 ± 0.114 | 1.343 ± 0.132 | 8 | |
| E323A:V404A | 0.17 ± 0.01 | 0.068 ± 0.020 | 5.552 ± 0.351 | 0.402 ± 0.117 | 0.075 ± 0.024 | 5 | 8.74 ± 3.58a |
| E323A:S407A | 0.46 ± 0.03 | 0.360 ± 0.037 | 1.433 ± 0.170 | 0.784 ± 0.064 | 0.583 ± 0.090 | 5 | 1.19 ± 0.32 |
| E323A:N408A | 0.37 ± 0.03 | 0.222 ± 0.030 | 2.178 ± 0.262 | 0.591 ± 0.049 | 0.293 ± 0.049 | 5 | 4.19 ± 0.08 |
| E323A:I412A | 0.24 ± 0.01 | 0.056 ± 0.005 | 4.102 ± 0.163 | 0.225 ± 0.018 | 0.060 ± 0.006 | 5 | 3.81 ± 0.77 |
| S322A:V404A | 0.21 ± 0.01 | 0.022 ± 0.002 | 4.700 ± 0.308 | 0.105 ± 0.013 | 0.022 ± 0.002 | 5 | 9.27 ± 0.04 |
| S322A:S407A | 3.44 ± 0.21 | 0.918 ± 0.007 | 0.024 ± 0.003 | 0.271 ± 0.016 | 11.447 ± 0.896 | 5 | 1.02 ± 0.34 |
| S322A:N408A | 1.54 ± 0.23 | 0.897 ± 0.022 | 0.077 ± 0.022 | 0.623 ± 0.070 | 10.611 ± 2.279 | 5 | 2.77 ± 0.16 |
| S322A:I412A | 1.57 ± 0.28 | 0.434 ± 0.034 | 0.421 ± 0.093 | 0.296 ± 0.028 | 0.791 ± 0.104 | 5 | 2.15 ± 0.71 |
From the time constant of onset of low-voltage inactivation at −65 mV (τL,-65) and the fraction of non-inactivated channels, (f-65) on-rates (kon,-65) and off-rates (koff,-65) were calculated for wild-type (wt) and mutant channels to define their apparent affinity (Kci,-65) for the closed-inactivated state. With double mutant cycle analysis based on these affinity values, coupling coefficients (Ω) were determined for pairs of amino acids.
SEM values based on linear and independent error propagation.
Structural homology modeling was done with PyMol (DeLano Scientific), and coordinates of the Kv1.2 crystal structure (Long et al., 2005a) were used to study the orientation of side chains of mutated amino acids.
Online Supplemental Material
In Fig. S1, channel availability is plotted from 0–20% of the maximum. This shows the conductance windows under the activation and steady-state inactivation curves for Kv4.2 wild-type and the mutants C320S and V402I:V404I. Fig. S2 provides complete Kv4.2 wild-type datasets for the onset kinetics of low-voltage inactivation and the kinetics of recovery from inactivation tested at different membrane potentials. Fig. S2 also shows the bell-shaped voltage dependence of the inactivation time constants (onset and recovery) and the voltage dependence of backward rates (1/τrec and koff) of closed-state inactivation for Kv4.2 wild-type. Fig. S3 shows Pearson correlation analyses for 1/τrec and koff, for ΔV1/2,inact and ln (Kci/Kci,wt), and for ΔV1/2,inact and ΔV1/2,act, including the values from all tested channel constructs. Fig. S4 shows the results of double mutant cycle analysis at alternative membrane potentials between −75 and −50 mV. Fig. S5 shows individual current traces mediated by a Kv4.2 double-cysteine mutant during redox modulation. For comparison, native oocyte currents are shown under the same conditions. Figs. S1–S5 are available at http://www.jgp.org/cgi/content/full/jgp.200810073/DC1.
RESULTS
Activation and Low-Voltage Inactivation Parameters of Kv4.2 Channels
Kv4.2 channels were expressed in Xenopus oocytes and functionally characterized under two-electrode voltage clamp. The apparent activation threshold for macroscopic currents mediated by wild-type channels in response to depolarizing voltage jumps was around −50 mV. At more depolarized test potentials, wild-type channels mediated rapidly activating and inactivating A-type currents (Fig. 1 A), which showed a pronounced prepulse inactivation (Fig. 1 B). Analysis of peak conductance–voltage and steady-state inactivation curves for wild-type channels (Fig. 1 C) yielded mean V1/2 values of +7.5 (V1/2,act) and −66.9 mV (V1/2,inact), with mean slope factors s of 20.4 and 5.9 mV, respectively (Table I). The large separation of the two V1/2 values combined with the steep voltage dependence of steady-state inactivation results in an extremely small conductance window under the respective curves (Fig. 1 C and Fig. S1), indicative of inactivation coupled to closed states. Also, the pronounced inactivation of macroscopic currents at intermediate (between −40 and 0 mV), but considerably slower and incomplete decay at more positive test potentials (Fig. 1 A), reflects a maximal amount of steady-state inactivation at a certain potential and less inactivation at more positive potentials (see also Fig. S1). These features of Kv4 channel gating are caused by their preferential closed-state inactivation (Jerng et al., 1999; Bähring et al., 2001a; Beck and Covarrubias, 2001). We measured the onset kinetics of this low voltage–induced form of inactivation with a conditioning pulse protocol at different potentials below the apparent activation threshold (e.g., −65 mV in Fig. 2 A). For Kv4.2 wild-type channels, the onset of low-voltage inactivation at −65 mV (near V1/2,inact) occurred with a mean time constant τL of 1.28 s and showed a mean relative current amplitude f of 0.458, which represented the fraction of non-inactivated channels at steady state (Fig. 2 B and Table II; see also Fig. S2 A). The relative amplitudes f obtained at different onset potentials (Fig. 2 B) are a direct reflection of the steady-state inactivation curve.
The recovery from low-voltage inactivation (induced at −55 mV) was also measured at different membrane potentials (e.g., −100 mV in Fig. 2 C). At −100 mV, the mean recovery time constant τrec for Kv4.2 wild-type channels was 0.24 s. The data obtained at different recovery potentials reflect the strong voltage dependence of recovery from inactivation (Fig. 2 D and Fig. S2 B). At −80 mV, the mean recovery time constant was equal to the mean onset time constant. The time constants at −80 mV, together with the recovery time constants at more negative and the onset time constants at more positive potentials, showed a bell-shaped voltage dependence with a maximum at around −64 mV (Fig. S2 C). Combined with the voltage dependencies of activation and steady-state inactivation, these kinetic features reflect the prominent low voltage–induced Kv4.2 closed-state inactivation. The structural determinants involved in the gating transitions, which lead to refractoriness of closed states in Kv4.2 channels, have not been identified.
Mutational Analysis of S4S5 and S6 in Kv4.2
Kv4.2 closed-state inactivation may occur if the coupling between voltage sensor and cytoplasmic gate is temporarily lost (Fig. 3 A). To test this structural model of dynamic coupling experimentally, we examined the effects of mutations in the S4-S5 linker, including the initial part of S5 (S4S5) and mutations in the distal S6 segment (S6). These regions correspond to the major activation coupling interfaces previously identified in other Kv channels (Lu et al., 2002; Long et al., 2005b; Fig. 3, A and B).
We first ran our activation and inactivation protocols (Figs. 1, A and B, and 2 A) with two Kv4.2 constructs, which carry mutations in these regions and have been previously studied in different contexts (the S4S5 single mutant C320S and the S6 double mutant V402I:V404I; Jerng et al., 1999; Wang et al., 2002; Gebauer et al., 2004; Pourrier et al., 2004; see Materials and methods). C320S had no effect on the voltage dependence of activation or inactivation, whereas V402I:V404I induced a positive shift in the voltage dependence of both activation and steady-state inactivation (Table I and Fig. S1). Notably, both C320S and V402I:V404I caused a slowing of the onset of low-voltage inactivation at −65 mV (Table II). The data obtained with C320S and V402I:V404I indicated that introducing mutations in S4S5 or S6 may influence Kv4.2 channel closed-state inactivation to different extents, but obviously, the mutations do not influence voltage-dependent gating too much, so that activation and inactivation curves still show a large overlap with wild-type data.
We next performed a Kv4.2 mutagenesis scan that extended over S4S5 and S6. In S4S5 individual amino acids from Gly 309 to Phe 329 (Fig. 4 A, left) were mutated to alanine, and Ala 321 was mutated to valine. Likewise, in S6 individual amino acids from Val 397 to Tyr 413 (Fig. 4 A, right) were mutated to alanine and Ala 399 to valine. From a total of 38 mutants, 31 expressed voltage-dependent currents, which were clearly separable from endogenous oocyte conductances, albeit in some cases still too small for a detailed kinetic analysis (Table I). We suspect folding problems or extreme shifts in voltage dependence of gating to underlie the poor current expression of some mutants. 24 mutants (15 out of 21 in S4S5 and 9 out of 17 in S6) showed typical A-type behavior; i.e., rapid activation of macroscopic outward currents in response to depolarizing voltage jumps, followed by more or less rapid inactivation (Fig. 4, B and C). The mutant A-type currents differed in peak amplitude (between 1.4 and 14 µA at +40 mV; wild-type, 4 µA; Table I) and in the amount of macroscopic inactivation (relative current remaining at the end of a 1.5-s pulse to +40 mV between 0 and 0.425; wild-type, 0.042; Fig. 4, B and C, and Table I).
For the 24 A-type mutants, we determined activation and steady-state inactivation curves and compared them with wild-type data to judge mutation-induced shifts in either voltage dependence (results summarized in Table I). Then, for all of these mutants except L310A, the onset kinetics of low-voltage inactivation at −65 mV (summarized in Table II) and the recovery kinetics at −100 mV were measured. Fig. 5 shows such analyses for mutations in the two scanning regions, which either strongly shifted or had negligible effects on the voltage dependence of steady-state inactivation. Mutants with a large negative shift were E323A in S4S5 (ΔV1/2,inact = −12 mV) and V404A in S6 (ΔV1/2,inact = −28 mV; Fig. 5, A and B, left). Notably, the amino acid pair Glu 323 and Val 404 in Kv4.2 is homologous to the previously identified major activation coupling sites between S4S5 and S6 in Shaker channels (Yifrach and MacKinnon, 2002; Fig. 4 A). The largest positive shift in the voltage dependence of steady-state inactivation was found for L310A (ΔV1/2,inact = +47 mV), but significant positive shifts were also found for F326A in S4S5 (ΔV1/2,inact = +21 mV; incomplete inactivation at positive potentials) and for N408A in S6 (ΔV1/2,inact = +11 mV). In contrast, C320A in S4S5 (ΔV1/2,inact = +1 mV) and S410A in S6 (ΔV1/2,inact = +2 mV) left the voltage dependence of steady-state inactivation nearly unaffected (Fig. 5, A and B, left). Of the seven mutants shown in Fig. 5 the peak conductance–voltage relationship was maximally shifted negative for E323A (ΔV1/2,act = −10 mV) and positive for L310A (ΔV1/2,act = +151 mV), but a significant positive shift was also found for N408A (ΔV1/2,act = +21 mV). These activation shifts apparently matched the corresponding shifts in steady-state inactivation; however, a closer inspection of the mutation-induced shifts in V1/2,act and V1/2,inact revealed no clear correlation (Fig. S3 C).
Figure 5.

Analysis of voltage dependence of activation and inactivation, and inactivation kinetics for individual point mutants in S4S5 and S6. (A) Analysis for the S4S5 mutants E323A (triangles; n = 8), C320A (circles; n = 5), F326A (squares; n = 5), and L310A (inverted triangles; n = 3 for inactivation and n = 4 for activation). (Left) Steady-state inactivation (filled symbols; variable offset first-order Boltzmann function) and activation curves (open symbols; fourth-order Boltzmann function). The corresponding results for Kv4.2 wild-type (see also Fig. 1 C) are indicated as dotted lines without symbols. Note the large offset in steady-state availability for F326A and the extreme positive shift of the L310A activation curve. (Middle) Onset of low-voltage inactivation analyzed with a single-exponential function (n = 5–6 independent experiments). Wild-type data indicated as a dotted line without symbols. Inactivation kinetics were not analyzed for L310A. (Right) Recovery from inactivation analyzed with a single-exponential function. Wild-type data indicated as a dotted line without symbols. Note that in all shown S4S5 mutants, the recovery from inactivation was accelerated (n = 5–6). (B) Same analysis for the S6 mutants V404A (triangles; n = 4–7), S410A (circles; n = 5), and N408A (squares; n = 5–7). As in A, dotted lines without symbols represent wild-type data. Note that the strong negative shift of the V404A inactivation curve is accompanied by a positive shift of the V404A activation curve.
Kinetic analysis of low-voltage inactivation at −65 mV showed that both the steady-state amplitudes f and the corresponding time constants τL varied among mutant constructs. Steady-state amplitudes at −65 mV were of course very similar to wild-type for non-shifting mutants like C320A (f = 0.510) and S410A (f = 0.512; Fig. 5, A and B, middle, and Table II). Likewise, the time constant of low-voltage inactivation for C320A (τL = 1.10 s) was almost indistinguishable from wild-type. However, for both the non-shifting mutant S410A and the positively shifting mutant N408A (f = 0.851), τL was only 0.72 s. For F326A, the onset time constant was even smaller (τL = 0.68 s) but also accompanied by a large steady-state amplitude (f = 0.938), and therefore most likely reflecting both onset and recovery kinetics. However, for the two mutants with very small steady-state amplitudes, E323A in S4S5 (f = 0.124) and V404A in S6 (f = 0.051), the extremely small τL values of 0.16 and 0.14 s, respectively (Fig. 5, A and B, middle, and Table II), may indicate that the development of closed-state inactivation was accelerated. Notably, the recovery from inactivation at −100 mV was accelerated for all S4S5 mutants shown in Fig. 5 A (right; τrec between 0.11 and 0.15 s) and did not directly reflect the corresponding shifts in steady-state inactivation; i.e., faster recovery kinetics did not necessarily imply a positively shifted inactivation curve. In S6 the recovery kinetics showed a moderate acceleration for S410A (τrec = 0.16 s) and a stronger acceleration for N408A (τrec = 0.07 s), whereas recovery was dramatically slowed for V404A (τrec = 0.60 s; Fig. 5 B, right). Thus, for these three S6 mutants, the kinetics of recovery from inactivation directly reflected steady-state inactivation properties.
The above analysis examples, yielding macroscopic time constants, suggest that changes in onset kinetics and/or changes in recovery kinetics may determine the fraction of mutant Kv4.2 channels, which are inactivated at steady state at membrane potentials below the apparent activation threshold. Next, we took a thermodynamic approach to examine how a mutation in Kv4.2 influenced the likelihood of entering the closed-inactivated state. To this end, we calculated apparent on- and off-rates of closed-state inactivation (Fig. 6, A and B; see Materials and methods). For wild-type channels, kon at −65 mV was 0.45 s−1 (Fig. 6 A) and koff 0.37 s−1 (Fig. 6 B; see also Table II). The largest mutation-induced effects were observed for kon (10- to 15-fold increase), and they were obtained with L313A, E323A, and V404A (Fig. 6 A). A closer inspection of the values obtained for the individual mutants indicated that changes in steady-state inactivation at −65 mV may either be dictated by changes in kon (e.g., L313A, E323A, L400A, V404A, and S407) or may be due to similar but opposite changes in kon and koff (e.g., S322A, F326A, and N408A). In analogy, unchanged steady-state inactivation properties may either be due to unchanged rates (e.g., C320A and G325A) or parallel changes in kon and koff (e.g., A321V, S410A, and R411A). Based on previous data (Bähring et al., 2001a), we assumed that, after a prolonged period of depolarization, Kv4.2 wild-type and all studied mutants recover exclusively from closed-inactivated states when the membrane potential is repolarized. To test this assumption and to validate our rate calculations, we examined if our calculated off-rates (koff) corresponded to the measured recovery rates (1/τrec). For Kv4.2 wild-type koff and 1/τrec values obtained at different membrane potentials overlapped on a Boltzmann curve with a V1/2,off of −113 mV (Fig. S2 D; see Discussion); and calculated koff values at −65 mV for the mutants strongly correlated with the measured recovery rates at −100 mV (Fig. S3 A). Thus, we were confident that our calculated koff values thermodynamically quantified the off-kinetics of Kv4.2 closed-state inactivation in the voltage range relevant for the present study (i.e., between −50 and −80 mV).
Figure 6.

Transition rates and apparent affinity for the closed-inactivated state of wild-type and mutant Kv4.2 channels. From the time constants of low-voltage inactivation, τL, and the fractions of non-inactivated channels f at −65 mV, on-rates (kon,-65; A) and off-rates (koff,-65; B) were calculated for Kv4.2 wild-type (wt) and each individual point mutant of S4S5 and S6. Data obtained with the mutants C320S and V402I:V404I (extremely small kon,-65) are included for comparison. Vertical dotted lines in A and B represent wild-type data (wt). (C) The mutation-induced changes in apparent affinity for the closed-inactivated state in Kv4.2 channels relative to wild-type are expressed as ln (Kci/Kci,wt). Note that the effects of the C320A and the C320S mutation differ. Vertical dotted lines indicate ∣ ln (Kci/Kci,wt) ∣ = 1; n = 3–8 independent determinations.
Based on these conformities, we now calculated apparent Kci values, reflecting the affinity of wild-type and mutant Kv4.2 channels for the closed-inactivated state (see Materials and methods and Table II). The effect of each mutation on closed-state inactivation was then thermodynamically quantified as ln (Kci/Kci,wt), and the results are shown in Fig. 6 C. The data reflect the mutation-induced shifts in the voltage dependence of steady-state inactivation. Negative ln (Kci/Kci,wt) values, i.e., mutations that favor the closed-inactivated state, represent negative shifts, and positive ln (Kci/Kci,wt) values, i.e., mutations that inhibit closed-state inactivation, represent positive shifts in the voltage dependence of steady-state inactivation. This applied also to the previously studied mutants C320S and V402I:V404I, and a comparison of C320S with C320A shows that different amino acid exchanges at the same position can have different effects on closed-state inactivation (Fig. 6 C). A quantitative analysis including all studied mutants revealed the strong correlation between ln (Kci/Kci,wt) and ΔV1/2,inact values (Fig. S3 B), which is expected if the slope factors of the voltage dependence of steady-state inactivation do not change dramatically (Table I).
Kv4.2 Double Mutant Cycle Analysis Based on Low-Voltage Inactivation Parameters
The above considerations justified the use of our kinetic parameters to study thermodynamically a putative dynamic coupling between individual Kv4.2 amino acid residues in S4S5 and S6, respectively, during low-voltage inactivation. For this purpose we constructed several Kv4.2 double mutants, each of them containing one of the tested mutations in S4S5 and one in S6. The main selection criterion was a clear effect of the individual mutations; i.e., ∣ln (Kci/Kci,wt)∣ > 1 (Fig. 6 C). This condition was met by both E323A in S4S5 and V404A in S6. In addition to E323A, we chose S322A in S4S5 because it lies adjacent to Glu323 and its effect is opposite to the one induced by E323A (Fig. 6, A and C). In S6 we chose S407A, N408A, and I412A in addition to V404A (Fig. 6 C). Each of the resulting eight double mutants (E323A:V404A, E323A:S407A, E323A:N408A, E323A:I412A, S322A:V404A, S322A:S407A, S322A:N408A, and S322A:I412A) was functionally characterized in Xenopus oocytes. Like the corresponding single mutants, all double mutants expressed A-type currents (Fig. 7 A), which differed in peak amplitude (between 3 and 13 µA at +40 mV) and in the amount of macroscopic inactivation (relative current remaining at the end of a 1.5-s pulse to +40 mV between 0 and 0.116; Table I).
Figure 7.

Functional expression and low-voltage inactivation analysis of Kv4.2 double mutants. (A) Current families obtained for double point mutants, which contain either E323A (left) or S322A (right) in combination with V404A, S407A, N408A, or I412A (only responses to test potentials between −80 and +60 mV in 20-mV increments are shown). (B) Analysis of onset of low-voltage inactivation for the double mutants E323A:V404A (left; n = 5) and E323A:S407A (right; n = 5). Wild-type data are indicated by dotted lines, and data from corresponding individual mutants are shown in light and dark gray, as indicated. Note the apparent additivity of the effects induced by E323A and S407A.
Fig. 7 B shows the analysis of onset of low-voltage inactivation at −65 mV for the Kv4.2 double mutants E323A:V404A and E323A:S407A. It can be seen that the onset of low-voltage inactivation, which was accelerated by both E323A and V404A in the respective single mutants, was not further accelerated in the double mutant E323A:V404A (Fig. 7 B, left). In contrast, S407A, which reduced the amount of steady-state inactivation at −65 mV in the single mutant, exerted the same effect in the E323A:S407A double mutant, and both the time course and steady-state component of low-voltage inactivation lay exactly between wild-type and E323A data (Fig. 7 B, right).
In Fig. 8 A, ln (Kci/Kci,wt) values are shown for all eight double mutants, including their corresponding individual mutants. The data suggest that, in contrast to the other three S6 mutations, S407A causes additive effects both in combination with E323A and in combination with S322A in S4S5. This was confirmed by double mutant cycle analysis (see Materials and methods), which yielded extremely small coupling coefficients for S407A combined with E323A or S322A, whereas the coupling coefficients for the other pairs of point mutations were much larger. Notably, the largest coupling coefficients were obtained for E323A and S322A in combination with V404A (Fig. 8 B and Table II).
Figure 8.

Double mutant cycle analysis and putative spatial orientation of corresponding S4S5 and S6 amino acid side chains in Kv4.2. (A) Effects of the studied double mutations on the apparent affinity for the closed-inactivated state in Kv4.2 channels expressed as ln(Kci/Kci,wt) and depicted as black bars. Effects of the corresponding individual mutations are indicated as light and dark gray bars. Note that in strong contrast, especially to the combinations containing V404A, additive effects are apparent for the combinations containing S407A. (B) Thermodynamic coupling for the pairs of mutations is quantified as ln Ω. The double mutant cycle analysis indicates strong coupling for the amino acid pairs E323/V404 and S322/V404, whereas no coupling is seen for the amino acid pairs E323/S407 and S322/S407; n = 5–8 independent determinations. (C) Homology modeling of the amino acid residues involved in the double mutant cycle analysis based on Kv1.2 crystal structure data (Long et al., 2005a; zoomed view of the left α-subunit in Fig. 3 B). Orange, scanning regions; green, Kv4.2 S4S5 residues Glu 323 and Ser 322; dark blue, Kv4.2 S6 residues Val 404, Asn 408, and Ile 412. The Kv4.2 S6 residue Ser 407, which in accordance with its low coupling factors points in a different direction, is colored light blue.
For some double mutants, the steady-state amplitude f at −65 mV was either very small (e.g., E323A:V404A) or very large (e.g., S322A:S407A; see also Table II). Both may limit the validity of our double mutant cycle analysis. Therefore, we measured for selected pairs of mutations (double mutant and respective single mutants) the onset of low-voltage inactivation at more appropriate membrane potentials between −75 and −50 mV and compared the results with corresponding wild-type data (see Fig. 2 B and Fig. S4 A). The coupling coefficients obtained with double mutant cycle analysis at the alternative membrane potentials revealed no obvious differences to the data obtained at −65 mV (Fig. S4 B).
Homology modeling based on the Kv1.2 structure (Long et al., 2005a) of all residues involved in our double mutant cycle analysis (Fig. 8 C) suggests that the side chains of the Kv4.2 S6 amino acids Val 404, Asn 408, and Ile 412 point to the Kv4.2 S4S5 helix. Intimate contact seems likely between the S4S5 residue Glu 323 and the S6 residues Val 404 and Asn 408. The same applies to the S4S5 residue Ser 322 and the S6 residues Val 404 and Asn 408. In contrast, the side chain of the S6 amino acid Ser 407 points in a different direction and is unlikely to make contact with any amino acid in the S4S5 helix. Collectively, for both Glu 323 and Ser 322 in S4S5, the results of our double mutant cycle analysis involving the S6 residues Val 404, Ser 407, and Asn 408 are in agreement with homology modeling results.
Site-specific Redox Modulation of Coupling between Voltage Sensor and Gate in Kv4.2 Channels
Large coupling coefficients and the putative spatial orientation of amino acid side chains suggest that the Kv4.2 residues Glu 323 in S4S5 and Val 404 in S6 are close to each other and may functionally interact in a channel conformation related to closed-state inactivation. According to our model (Fig. 3 A), we think that dynamic coupling between Glu 323 and Val 404 is involved in both activation and inactivation of Kv4.2 channels. We decided to test for redox-dependent disulfide bonding between substituted cysteines at positions 323 and 404 to verify the close proximity between Glu 323 and Val 404 in the native Kv4.2 channel complex. We hypothesized that disulfide bonding between Cys 323 and Cys 404 may to some extent compromise activation but at the same time may affect inactivation. For these experiments, we introduced an E323C:V404C double mutation as well as the corresponding single mutations in a Kv4.2Δ2-10 background and coexpressed the mutants with KChIP2. The use of Δ2-10:E323C:V404C/KChIP2 channel complexes allowed us to study macroscopic currents in the absence of N-type inactivation (Barghaan et al., 2008). At the same time, the association with KChIP2 protected the channels to some extent against redox-dependent rundown. We tested the effects of the reducing agent dithiothreitol (DTT) and the oxidizing agent tert-butyl hydroperoxide (tbHO2) on current amplitude and decay kinetics (Fig. 9). Despite the presence of KChIP2, the currents mediated by the Δ2-10:E323C:V404C double mutant were rather small under control conditions (<1 µA; Fig. 9 A, left). These small currents also showed relatively slow inactivation kinetics (τA = 364 ms; Fig. 9 A, right and inset; see also Fig. S5). The application of 10 mM DTT caused both an increase in current amplitude (Fig. 9 A, left) and an acceleration of inactivation (τA = 177 ms; Fig. 9 A, right and inset; see also Fig. S5). Subsequent application of 2 mM tbHO2 caused a fast decline of the current amplitude (Fig. 9 A, left) but only a partial restoration of slow inactivation kinetics (τA = 238 ms; Fig. 9 A, right and inset; see also Fig. S5). Notably, none of these effects was observed for non-mutated Δ2-10 channels (Fig. 9 B), or for the single mutants Δ2-10:E323C (Fig. 9 C) and Δ2-10:V404C (Fig. 9 D). Also, the non-mutated channel constructs and single-point mutants expressed larger currents (some microamperes) with faster inactivation kinetics (τA ≤ 200 ms) than the Δ2-10:E323C:V404C double mutant already under control conditions. This indicates that, only in the double mutant, there were disulfide bonds under control conditions.
Figure 9.

Selective redox sensitivity of a Kv4.2 E323C:V404C double mutant. (A; left) Redox modulation of current amplitude and kinetics for the N-terminally truncated double-cysteine mutant Kv4.2Δ2-10:E323C:V404C coexpressed with KChIP2 in a Xenopus oocyte. Application of 10 mM DTT and 2 mM tbHO2 indicated by horizontal black and gray bars, respectively. (Inset) Current kinetics at time points 1, 2, and 3 during the experiment (normalized and averaged traces from five oocytes; gray shadings represent error bars; horizontal scale bar, 50 ms). (Right) Mean time constants of outward current decay before DTT application (point 1, control; τA = 364 ± 8 ms), 10–15 min after switching to DTT (point 2; τA = 177 ± 13 ms), and when the amplitude had returned to control level (indicated by horizontal dotted lines in A–D), usually after 5–10 min in tbHO2 (point 3; τA = 238 ± 25 ms; n = 5). (B) Redox experiment with an oocyte expressing Kv4.2Δ2-10 in the presence of KChIP2 (control, τA = 180 ± 10 ms; DTT, τA = 139 ± 4 ms; tbHO2, τA = 146 ± 2 ms; n = 3). (C) Redox experiment with an oocyte expressing the single-point mutant Kv4.2Δ2-10:E323C in the presence of KChIP2 (control, τA = 176 ± 11 ms; DTT, τA = 149 ± 19 ms; tbHO2, τA = 135 ± 19 ms; n = 3). (D) Redox experiment with an oocyte expressing the single-point mutant Kv4.2Δ2-10:V404C in the presence of KChIP2 (control, τA = 204 ± 8 ms; DTT, τA = 184 ± 6 ms; tbHO2, τA = 193 ± 4 ms; n = 3). Note that only the E323C:V404C double mutant shows selective redox modulation of current amplitude and kinetics, indicative of disulfide bridge formation between Cys 323 and Cys 404.
KChIP coexpression is known to modify Kv4 channel inactivation properties (Beck et al., 2002). Therefore, we conducted the same redox experiments with substituted cysteines at positions 323 and 404 in a Kv4.2Δ2-40 background and in the absence of KChIP2. The results were similar to the ones obtained with Δ2-10 mutants in the presence of KChIP2. However, the Δ2-40 currents were prone to considerable rundown (unpublished data), presumably due to redox modulation of native cysteines in the tetramerization domain (Wang et al., 2007).
The rundown of currents during our redox experiments could never be fully prevented but could be minimized in some cases by the coexpression of Kv4.2 mutants with both KChIP2 and DPP6 to favor ternary complex formation. In these experiments, we were able to run onset of low-voltage inactivation protocols under reducing (DTT 1) and oxidizing conditions (tbHO2), including also a recovery period (DTT 2) to study the reversibility of effects (Fig. 10). Again, fast and considerable current suppression when switching the bath solution from DTT to tbHO2 was observed with the Δ2-10:E323C:V404C double mutant (Fig. 10, A and B, left), but not with Δ2-10:E323C (Fig. 10 C, left) or Δ2-10:V404C (Fig. 10 D, left) single mutants. Notably, the onset kinetics of low-voltage inactivation measured at −65 mV substantially differed between the double mutant and the corresponding single mutants. Onset kinetics were well described by a single-exponential function in both Δ2-10:E323C (τL = 0.41 s in DTT; Fig. 10 C, right) and Δ2-10:V404C (τL = 0.56 s in DTT; Fig. 10 D, right). However, in the Δ2-10:E323C:V404C double mutant, onset kinetics clearly showed a double-exponential time course (τL,fast = 0.11 s in DTT and τL,slow in the order of tens of seconds; Fig. 10 B, right). We focused on the fast onset time constant and found that it was increased in the presence of tbHO2 (τL,fast = 0.27 s; Fig. 10 B, right). Furthermore, the data from two out of three tested oocytes indicated that this effect was reversible when switching the bath solution back to DTT (Fig. 10 B, right). In the single mutants, onset kinetics of low-voltage inactivation at −65 mV were not affected by switching between DTT- and tbHO2-containing bath solutions (Fig. 10, C and D, right).
Figure 10.

Redox modulation of low voltage–induced inactivation. Kv4.2 mutants with substituted cysteines were coexpressed with both KChIP2 and DPP6. All recordings were started in a DTT-containing bath solution. (A) Peak current amplitude increase (DTT 1), suppression (tbHO2), and recovery (DTT 2) for a Xenopus oocyte expressing Kv4.2Δ2-10:E323C:V404C + KChIP2 + DPP6. (B; left) Amplitude suppression after 10–15 min in tbHO2 and recovery for three oocytes expressing the E323C:V404C double mutant. (Right) Averaged data from three oocytes showing the onset of low-voltage inactivation at −65 mV in DTT 1 (black symbols) and tbHO2 (gray symbols). Fitting curves represent double-exponential functions (DTT 1, τL,fast = 0.11 ± 0.01 s; tbHO2, τL,fast = 0.27 ± 0.04 s; P = 0.0837, paired t test). Open symbols represent individual datasets from two out of three oocytes tested in DTT 2 (double-exponential onset kinetics indicated by dotted fitting curves; τL,fast = 0.13 and 0.16 s, respectively). (C; left) Peak current amplitudes during redox experiments, like the one shown in A, for seven oocytes expressing the E323C single mutant with KChIP2 and DPP6. (Right) Onset kinetics of low-voltage inactivation. Fitting curves represent single-exponential functions (DTT 1, τL = 0.41 ± 0.06 s; tbHO2, τL = 0.47 ± 0.07 s; DTT 2, τL = 0.43 ± 0.06 s; n = 7). (D) Peak current amplitudes and onset kinetics of low-voltage inactivation for eight oocytes expressing the V404C single mutant with KChIP2 and DPP6. (Right) Onset kinetics of low-voltage inactivation. Fitting curves represent single-exponential functions (DTT 1, τL = 0.56 ± 0.05 s; tbHO2, τL = 0.55 ± 0.07 s; DTT 2, τL = 0.50 ± 0.05 s; n = 8).
The results of our redox experiments clearly demonstrate selective disulfide bonding between substituted Cys 323 and Cys 404 in Kv4.2. The observed effects can be explained as follows: A rigid activation interface in the presence of the disulfide bridge may compromise activation gating, and the resulting low open probability allows only small current amplitudes. Breaking the disulfide bond under reducing conditions may lead to more efficient activation because the activation interface is now more flexible. At the same time, due to the increased flexibility, inactivation can occur.
Collectively, our findings show that the S4S5 residue Glu 323 and the S6 residue Val 404 functionally interact with each other in Kv4.2 channels. The observed effects support the view that this interaction is dynamic and involved in both Kv4.2 activation and closed-state inactivation.
DISCUSSION
Our scanning mutagenesis combined with double mutant cycle analysis shows that dynamic coupling between amino acid residues in S4S5 and S6, respectively, is involved in closed-state inactivation of Kv4.2 channels. The spatial arrangement of the respective amino acid side chains, as deduced from homology modeling based on the Kv1.2 structure, and the specific cysteine cross-linking at identified sites support this conclusion. The novel mechanism of Kv channel inactivation represents a “desensitization to voltage” and may be similar to the one suggested previously for the sea urchin HCN (spHCN) channel (Shin et al., 2004).
Studying Kv Channel Gating with Double Mutant Cycle Analysis
In Kv channels, functional coupling of the cytoplasmic gate to the voltage-sensing apparatus occurs mainly via interactions between S4S5 and S6. The crucial role of these two domains for voltage-dependent gate opening has been initially demonstrated with Shaker-KcsA chimeras in which voltage control over the gate was lost if S4S5 and S6 sequences did not match (Lu et al., 2002; see Fig. 4 A for essential complementary sequences). Functional coupling between S4S5 and S6 is also supported by the spatial orientation of these domains in the Kv1.2 crystal structure (Long et al., 2005b; see also Fig. 3 B). Although multiple residue interactions between S4S5 and S6 are thought to be involved in voltage-dependent gate opening (Lu et al., 2002), mutant scanning combined with double mutant cycle analysis has identified “hot spots” of this functional coupling between gating-sensitive residues in Shaker channels; namely Glu 395 in S4S5 and Val 476 in S6 with a coupling energy (ΔΔzFV1/2,act) of 3.32 kcal/mol (as compared with only 0.76 kcal/mol for the nearby Met 393 in combination with Val 476; Yifrach and MacKinnon, 2002). Based on these previous findings, we chose S4S5 and S6 for our Kv4.2 mutagenesis scan. We focused on Glu 323 and Val 404, which are homologous to the identified activation hot spots in Shaker, and on amino acid residues next to them. We also took a thermodynamic approach, but instead of studying activation, we studied closed-state inactivation with double mutant cycle analysis. Only few mutations in our scan caused a complete perturbation of channel function; rather, most mutations exerted specific effects on activation and inactivation, which differed quantitatively and depended on the type of amino acid exchange. This indicated that we were dealing with mutation-induced local effects rather than long-range conformational changes. Thus, the requirements for a detailed thermodynamic analysis were met.
Because too little is yet known about the nature of the interaction between S4S5 and S6, we formally defined an apparent affinity of the Kv4.2 channel for the closed-inactivated state as a thermodynamic parameter for the mutant cycle. For this purpose, instead of simply using the fraction f of non-inactivated channels as a measure of affinity, we directly calculated apparent affinities from experimentally determined on- and off-rates of low-voltage inactivation. The strong correlation between ln (Kci/Kci,wt) and ΔV1/2,inact (Fig. S3 B) implies similar analysis results if the fraction f were used; however, the calculation of on- and off-rates gave a more detailed picture and revealed how differently individual mutations affected closed-state inactivation. In particular, it became evident that both E323A and V404A, in addition to L313A, caused a large increase in the on-rate of closed-state inactivation (see Fig. 6 A). Moreover, apparent affinity calculations based on respective on- and off-rates (Kci values between 0.02 and 11.5 kT), instead of using f, also had the advantage of increasing the measuring range above unity (0 ≤ f ≤ 1), which made our thermodynamic calculations more sensitive.
Calculating the coupling energy (ΔΔG = RTlnΩ) for the Kv4.2 amino acid pair Glu 323 and Val 404 yields a value of 1.27 kcal/mol, which is in the same order but smaller than the value obtained previously with activation parameters for the homologous Shaker amino acid pair (Yifrach and MacKinnon, 2002). This deviation may be due to the use of different thermodynamic parameters for the mutant cycle. Alternatively, the strength of interaction, in a thermodynamic sense, at this specific site may be lower in Kv4.2 than in Shaker. It is worthy to note that in a domain-based double mutant cycle analysis of chimeric Kv2.1-Kv1.2 channels (Scholle et al., 2004), ΔΔG values between 1 and 2 kcal/mol were calculated based on activation parameters to characterize a voltage-dependent interaction between S4S5 and the cytoplasmic C terminus. Although in this study the chimeric replacement did not include the activation hot spot in S6, similar ΔΔG values to the ones obtained in our study are of interest because both Kv4.2 and Kv2.1 channels exhibit a pronounced closed-state inactivation (Klemic et al., 1998).
Our minimal and maximal coupling coefficients (lnΩ between 0.02 and 2.86 kT) compare well to the ones obtained in a previous study in which individual amino acid interactions between the N-terminal inactivation domain and the pore-lining face of S6 in Kv1.4 channels were examined with double mutant cycle analysis (Zhou et al., 2001). Certainly, the proposed dynamic coupling between S4S5 and S6, inferred from onset measurements of low-voltage inactivation, does not represent a first-order binding and unbinding reaction with a defined stoichiometry, like in the case of N-type inactivation (MacKinnon et al., 1993), but may be more complex (see below). Also, although the macroscopic onset kinetics of N-type inactivation directly reflect the association of protein domains, our calculated on-rates of closed-state inactivation merely represent a dissociation, which we call “uncoupling” in the absence of a precise knowledge about the nature of the interaction between S4S5 and S6. Nevertheless, the numbers obtained in the present study suggest that the functional significance of interactions between Kv4.2 amino acid residues Glu 323 or Ser 322 in S4S5 and Val 404 or Asn 408 in S6, respectively, are comparable to the functional significance of Shaker ball peptide binding to its receptor in the pore. In contrast, the coupling coefficients obtained with double mutant cycle analysis in a previous study on an N-type–related inactivation mechanism in Kv4.2 channels (Gebauer et al., 2004) were much lower (maximal lnΩ value of 0.85). Collectively, these results nicely illustrate that N-type inactivation plays a minor role, whereas closed-state inactivation, involving a dynamic coupling between S4S5 and S6, represents the major inactivation mechanism in Kv4.2 channels.
In general, homology modeling based on the Kv1.2 structure of thermodynamically identified Kv4.2 residues supports our dynamic coupling model. However, we also obtained significant coupling coefficients for the S6 residue Ile 412 in combination with Glu 323 or Ser 322 in S4S5, although homology modeling does not suggest a close spatial proximity of the respective amino acid side chains (see Fig. 8 C). Although this may indicate structural differences between Kv1.2 and Kv4.2, we favor an alternative explanation. The Kv1.2 crystal structure has captured the channel in the open state (Long et al., 2005a), and does not provide any information per se about the gating transitions leading to that state. Conformational changes during voltage-dependent gating transitions, including rotations and tilting motions within the voltage sensor apparatus (Long et al., 2007; Pathak et al., 2007), may allow functional interaction between Ile 412 and gating-sensitive residues in S4S5. In the absence of any experimental evidence, the finding that disulfide bonding compromises activation gating supports such conformational changes.
Uncoupling Model of Inactivation
Our working model of dynamic coupling between voltage sensor and gate is based on several untested assumptions, which rely on a high structural and functional similarity between Shaker (Kv1) and Kv4.2, aside a prominent N- and C-type inactivation found in the former and a preferential closed-state inactivation in the latter. We assumed that in both Shaker and Kv4.2, the distal S6 segment forms a gate and that the major activation interfaces between voltage sensor and gate are the same in the two channels. Furthermore, we assumed that Glu 323 and Val 404 in Kv4.2 represent hot spots of these interfaces for activation coupling. As yet, there is no Kv4.2 crystal structure available, which covers the whole channel complex including conduction pore, gate, and voltage sensors. Therefore, we were dependent on the Kv1.2 crystal structure (Long et al., 2005a) as a template, which may not reflect some special features of Kv4.2 channels. However, the previous finding that mutations in S6 critically influence Kv4 channel closure (Jerng et al., 1999) suggests that S6 also forms a gate in Kv4.2. Functional coupling between S4S5 and S6 has been shown previously for more distantly related voltage-dependent ion channels, including human ether-à-go-go–related gene (Ferrer et al., 2006) and HCN channels (Decher et al., 2004). This strongly indicates that the use of S4S5 and S6 as activation interfaces is a common architectural feature of all voltage-dependent ion channels.
Kv4.2 channel activation is always accompanied by inactivation, which cannot be circumvented, like in the case of Shaker, by the use of N-terminally truncated mutants (Jerng and Covarrubias, 1997; Zhu et al., 1999; Bähring et al., 2001a). In fact, not a single functional Kv4.2 mutant has been described to date, which lacks inactivation, and we cannot exclude that Kv4.2 S4S5 and S6 point mutations, which disrupted functional channel expression (see Table I and Fig. 4 A), report sites crucially involved in closed-state inactivation. P403A in Kv4.2 may be such a mutation. Intriguingly, mutating the homologous site to alanine in Kv4.3 (P400A) produces channels with no macroscopic inactivation (Bhattacharji et al., 2006). The strong coincidence of activation and inactivation gating in Kv4.2 channels, with no true steady-state of macroscopic currents, supports our uncoupling model, but it also precludes a detailed thermodynamic analysis of activation coupling for individual residues, as performed previously for Shaker channels (Yifrach and MacKinnon, 2002). Still, our finding that the specific cysteine cross-linking between Glu 323 and Val 404 compromised the activation of outward currents suggests that these residues are localized in close proximity and their functional interaction is critical for activation gating in Kv4.2 channels. Based on these experimental results and considering the high-sequence homology between Shaker and Kv4.2, especially in the gating-sensitive regions studied here (see Fig. 4 A), we think that Shaker and Kv4.2 are structurally and functionally very similar.
A further untested assumption, which underlies our working model, is that the uncoupled gate favors the closed conformation; i.e., we assume that, uncoupled from the voltage sensor, the Kv4.2 S6 gate slams shut. Notably, this is not what the data by Lu et al. (2002) suggest. In their experiments certain Shaker-KcsA chimeras, in which S4S5 and S6 lack essential complementary sequences (Fig. 4 A), mediate a strong voltage-independent leak current component, as if the default state of the uncoupled gate were open. Similar observations have been made with human ether-à-go-go–related gene and non-inactivating HCN2 channels mutated in S4S5 or S6 (Decher et al., 2004; Ferrer et al., 2006). Although not rigorously tested, a constitutive voltage-independent conductance after S4S5 or S6 manipulations was evident neither in the present study, nor in respective Kv2.1-Kv1.2 chimeras (Scholle et al., 2004) and Shaker point mutants (Yifrach and MacKinnon, 2002). Thermodynamic considerations with Shaker channels suggest that the pore is intrinsically more stable when closed, and the voltage sensors have to exert positive work to actually tear the S6 gates open (Yifrach and MacKinnon, 2002). Based on their high structural and functional similarity, we think that in both Shaker and Kv4.2 channels the default state of the gate is closed. The closed conformation may be thermodynamically even more favorable in Kv4.2 channels than in Shaker. Currently used theoretical models, which adequately describe Kv4 channel gating (Bähring et al., 2001a; Beck and Covarrubias, 2001; Beck et al., 2002; Barghaan et al., 2008) account for a more favorable closed state by a reverse-biased opening step or a reverse-biased transition to a pre-open closed state after the activation of all four voltage sensors (Kaulin et al., 2008). A strong tendency of the Kv4.2 S6 gate to adopt its closed conformation may support uncoupling from S4S5 during both strong and weak depolarizations.
Voltage Sensor Inactivation: Desensitization to Voltage
A model of uncoupling between voltage sensor and gate has been proposed previously for the spHCN channel, which, among other supporting features, including the trapping of the channel blocker ZD7288 in the inactivated state, no longer inactivates when “locked-open” with a Cd2+ bridge between engineered cysteines (Shin et al., 2004). Based on a presumed uncoupling between voltage sensor and gate during hyperpolarization, the authors referred to the spHCN inactivation as a “desensitization to voltage.” Support for such a desensitization to voltage in Kv4.2 channels, with a direct involvement of the voltage sensors themselves, comes from gating current measurements by Dougherty et al. (2008). Their data unequivocally show that, already during moderate depolarization, the Kv4.2 voltage sensors must undergo a slow conformational transition (see Fig. 3 A) that leads to considerable charge immobilization. As a consequence, S4 charge is “lost” during depolarization; i.e., <100% of the gating charge is available immediately after repolarization, and a recovery period at hyperpolarized potentials is necessary for the restoration of gating charge. The loss of gating charge may be referred to as “voltage sensor inactivation”; the consequence for the channel is a desensitization to voltage. Intriguingly, the kinetics of loss and recovery of gating charge, respectively, exactly match the time courses of onset and recovery from the macroscopic low-voltage inactivation (Dougherty et al., 2008). Thus, voltage sensor inactivation (assayed by the loss of gating charge) in Kv4.2 channels is a direct reflection of their closed-state inactivation. The V1/2,off value of −113 mV, defining the voltage dependence of closed-sate inactivation off-kinetics in the present study (koff and 1/τrec; see Fig. S2 D), may be a reflection of the exaggerated apparent V1/2 value describing the voltage dependence of gating charge recovery (−149 mV; Dougherty et al., 2008).
Mutating charged residues in the S4 voltage sensor of Kv4.3 has been shown to both shift the inactivation curve along the voltage axis and change the respective slope factors (Skerritt and Campbell, 2007). In contrast, mutations in S4S5 and S6 in the present study shifted the voltage dependence of inactivation without dramatically changing the steepness of the respective curves. Collectively, these findings are compatible with a mechanism of inactivation involving both the voltage sensors (inactivation) and the gate (desensitization). How is the Kv4.2 charge immobilization mechanistically connected to the dynamic coupling between voltage sensor and gate, demonstrated in the present study? At present, we do not know if the voltage sensor immobilization occurs as a consequence of uncoupling from the gate (i.e., uncoupling allows a certain stable voltage sensor conformation), or if the stable voltage sensor conformation actually leads to uncoupling from the gate. Tight binding of the activated voltage sensor to sites located on the outer pore shell (C-terminal portion of S5 and N-terminal portion of S6 including the pore loop) may underlie a stable voltage sensor conformation (Fig. 3 A). Although the Kv1.2 crystal structure suggests that the voltage sensors are relatively independent domains (Fig. 3 B; Long et al., 2005a), previous mutational analysis of Shaker channels has shown that interaction sites exist between the outer pore shell and the actual voltage sensor (S4; Li-Smerin et al., 2000; Lainé et al., 2003; Soler-Llavina et al., 2006). Shaker and Kv4.2 channels, although otherwise highly similar, may differ with respect to these interaction sites. Possibly, after the initial step of voltage sensor activation, distinct modes of interaction of the voltage sensor with the pore shell may correlate with distinct cumulative states of inactivation. Thus, depending on the final voltage sensor conformation, it may be either P/C-type inactivation, like in Shaker channels, or closed-state inactivation, like in Kv4.2 channels.
The scenarios described above are sequential in that they put voltage sensor immobilization and gate uncoupling in a causal relationship. Alternatively, a more general scenario may apply: The cumulative inactivated state may depend on how strong the interaction of the voltage sensor with the activation gate and the pore shell is. If both interactions are strong, P/C-type inactivation occurs. If both interactions are weak, preferential closed-state inactivation may occur.
Collectively, the findings of this study complement the previous discovery that the voltage sensors are directly involved in Kv4.2 channel closed-state inactivation (Dougherty et al., 2008). Our thermodynamic analysis of a model of dynamic coupling between S4S5 and S6 explains why Kv4.2 channels with inactivated voltage sensors are nonconducting. Future work will have to clarify in more detail the relationship between voltage sensor inactivation and gate desensitization, and the molecular mechanisms by which accessory β-subunits control inactivation gating in native Kv4.2 channel complexes.
Acknowledgments
We thank Christiane K. Bauer and Guiscard Seebohm for critical comments and Peter Bassalay, Telse Kock, and Andrea Zaisser for technical assistance.
This study was supported by grants BA 2055/1-1 and BA 2055/1-2 from the Deutsche Forschungsgemeinschaft to R. Bähring.
Edward N. Pugh Jr. served as editor.
Footnotes
Abbreviations used in this paper: DPP, dipeptidyl aminopeptidase-like protein; DTT, dithiothreitol; HCN, hyperpolarization-activated cyclic nucleotide-gated; KChIP, Kv channel interacting protein; Kv, voltage-dependent potassium; spHCN, sea urchin HCN; tbHO2, tert-butyl hydroperoxide.
References
- Bähring R., Boland L.M., Varghese A., Gebauer M., Pongs O. 2001a. Kinetic analysis of open- and closed-state inactivation transitions in human Kv4.2 A-type potassium channels.J. Physiol. 535:65–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähring R., Dannenberg J., Peters H.C., Leicher T., Pongs O., Isbrandt D. 2001b. Conserved Kv4 N-terminal domain critical for effects of Kv channel-interacting protein 2.2 on channel expression and gating.J. Biol. Chem. 276:23888–23894 [DOI] [PubMed] [Google Scholar]
- Barghaan J., Tozakidou M., Ehmke H., Bähring R. 2008. Role of N-terminal domain and accessory subunits in controlling deactivation-inactivation coupling of Kv4.2 channels.Biophys. J. 94:1276–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz T., Yellen G. 1995. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms.Neuron. 15:951–960 [DOI] [PubMed] [Google Scholar]
- Beck E.J., Covarrubias M. 2001. Kv4 channels exhibit modulation of closed-state inactivation in inside-out patches.Biophys. J. 81:867–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck E.J., Bowlby M., An W.F., Rhodes K.J., Covarrubias M. 2002. Remodelling inactivation gating of Kv4 channels by KChIP1, a small-molecular-weight calcium-binding protein.J. Physiol. 538:691–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharji A., Kaplan B., Harris T., Qu X., Germann M.W., Covarrubias M. 2006. The concerted contribution of the S4-S5 linker and the S6 segment to the modulation of a Kv channel by 1-alkanols.Mol. Pharmacol. 70:1542–1554 [DOI] [PubMed] [Google Scholar]
- Choi K.L., Aldrich R.W., Yellen G. 1991. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels.Proc. Natl. Acad. Sci. USA. 88:5092–5095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decher N., Chen J., Sanguinetti M.C. 2004. Voltage-dependent gating of hyperpolarization-activated, cyclic nucleotide-gated pacemaker channels: molecular coupling between the S4-S5 and C-linkers.J. Biol. Chem. 279:13859–13865 [DOI] [PubMed] [Google Scholar]
- Dougherty K., De Santiago-Castillo J.A., Covarrubias M. 2008. Gating charge immobilization in Kv4.2 channels: the basis of closed-state inactivation.J. Gen. Physiol. 131:257–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer T., Rupp J., Piper D.R., Tristani-Firouzi M. 2006. The S4-S5 linker directly couples voltage sensor movement to the activation gate in the human ether-à-go-go-related gene (hERG) K+ channel.J. Biol. Chem. 281:12858–12864 [DOI] [PubMed] [Google Scholar]
- Gebauer M., Isbrandt D., Sauter K., Callsen B., Nolting A., Pongs O., Bähring R. 2004. N-type inactivation features of Kv4.2 channel gating.Biophys. J. 86:210–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara S., Kusano K., Saito N. 1961. Membrane changes of Onchidium nerve cell in potassium-rich media.J. Physiol. 155:470–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. 2001. Ion Channels of Excitable Membranes. 3rd ed Sinauer Associates, Inc., Sunderland, MA [Google Scholar]
- Hoshi T., Zagotta W.N., Aldrich R.W. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation.Science. 250:533–538 [DOI] [PubMed] [Google Scholar]
- Hoshi T., Zagotta W.N., Aldrich R.W. 1991. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region.Neuron. 7:547–556 [DOI] [PubMed] [Google Scholar]
- Jerng H.H., Covarrubias M. 1997. K+ channel inactivation mediated by the concerted action of the cytoplasmic N- and C-terminal domains.Biophys. J. 72:163–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng H.H., Shahidullah M., Covarrubias M. 1999. Inactivation gating of Kv4 potassium channels: molecular interactions involving the inner vestibule of the pore.J. Gen. Physiol. 113:641–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaulin Y.A., De Santiago-Castillo J.A., Rocha C.A., Covarrubias M. 2008. Mechanism of the modulation of Kv4:KChIP-1 channels by external K+.Biophys. J. 94:1241–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemic K.G., Shieh C.C., Kirsch G.E., Jones S.W. 1998. Inactivation of Kv2.1 potassium channels.Biophys. J. 74:1779–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata H.T., Fedida D. 2005. A structural interpretation of voltage-gated potassium channel inactivation.Prog. Biophys. Mol. Biol. 92:185–208 [DOI] [PubMed] [Google Scholar]
- Lainé M., Lin M.C., Bannister J.P., Silverman W.R., Mock A.F., Roux B., Papazian D.M. 2003. Atomic proximity between S4 segment and pore domain in Shaker potassium channels.Neuron. 39:467–481 [DOI] [PubMed] [Google Scholar]
- Li-Smerin Y., Hackos D.H., Swartz K.J. 2000. A localized interaction surface for voltage-sensing domains on the pore domain of a K+ channel.Neuron. 25:411–423 [DOI] [PubMed] [Google Scholar]
- Liu Y., Jurman M.E., Yellen G. 1996. Dynamic rearrangement of the outer mouth of a K+ channel during gating.Neuron. 16:859–867 [DOI] [PubMed] [Google Scholar]
- Long S.B., Campbell E.B., MacKinnon R. 2005a. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel.Science. 309:897–903 [DOI] [PubMed] [Google Scholar]
- Long S.B., Campbell E.B., MacKinnon R. 2005b. Voltage sensor of Kv1.2: structural basis of electromechanical coupling.Science. 309:903–908 [DOI] [PubMed] [Google Scholar]
- Long S.B., Tao X., Campbell E.B., MacKinnon R. 2007. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment.Nature. 450:376–382 [DOI] [PubMed] [Google Scholar]
- Lu Z., Klem A.M., Ramu Y. 2002. Coupling between voltage sensors and activation gate in voltage-gated K+ channels.J. Gen. Physiol. 120:663–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon R., Aldrich R.W., Lee A.W. 1993. Functional stoichiometry of Shaker potassium channel inactivation.Science. 262:757–759 [DOI] [PubMed] [Google Scholar]
- Pathak M., Yarov-Yarovoy M.V., Agarwal G., Roux B., Barth P., Kohout S., Tombola F., Isacoff E.Y. 2007. Closing in on the resting state of the Shaker K+ channel.Neuron. 56:124–140 [DOI] [PubMed] [Google Scholar]
- Pourrier M., Herrera D., Caballero R., Schram G., Wang Z., Nattel S. 2004. The Kv4.2 N-terminal restores fast inactivation and confers KChlP2 modulatory effects on N-terminal-deleted Kv1.4 channels.Pflugers Arch. 449:235–247 [DOI] [PubMed] [Google Scholar]
- Scholle A., Zimmer T., Koopmann R., Engeland B., Pongs O., Benndorf K. 2004. Effects of Kv1.2 intracellular regions on activation of Kv2.1 channels.Biophys. J. 87:873–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidullah M., Covarrubias M. 2003. The link between ion permeation and inactivation gating of Kv4 potassium channels.Biophys. J. 84:928–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin K.S., Maertens C., Proenza C., Rothberg B.S., Yellen G. 2004. Inactivation in HCN channels results from reclosure of the activation gate: desensitization to voltage.Neuron. 41:737–744 [DOI] [PubMed] [Google Scholar]
- Skerritt M.R., Campbell D.L. 2007. Role of S4 positively charged residues in the regulation of Kv4.3 inactivation and recovery.Am. J. Physiol. Cell Physiol. 293:C906–C914 [DOI] [PubMed] [Google Scholar]
- Soler-Llavina G.J., Chang T.H., Swartz K.J. 2006. Functional interactions at the interface between voltage-sensing and pore domains in the Shaker Kv channel.Neuron. 52:623–634 [DOI] [PubMed] [Google Scholar]
- Wang G., Strang C., Pfaffinger P.J., Covarrubias M. 2007. Zn2+-dependent redox switch in the intracellular T1-T1 interface of a Kv channel.J. Biol. Chem. 282:13637–13647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Patel S.P., Qu Y., Hua P., Strauss H.C., Morales M.J. 2002. Kinetic properties of Kv4.3 and their modulation by KChIP2b.Biochem. Biophys. Res. Commun. 295:223–229 [DOI] [PubMed] [Google Scholar]
- Wang S., Bondarenko V.E., Qu Y.J., Bett G.C., Morales M.J., Rasmusson R.L., Strauss H.C. 2005. Time- and voltage-dependent components of Kv4.3 inactivation.Biophys. J. 89:3026–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei A., Covarrubias M., Butler A., Baker K., Pak M., Salkoff L. 1990. K+ current diversity is produced by an extended gene family conserved in Drosophila and mouse.Science. 248:599–603 [DOI] [PubMed] [Google Scholar]
- Yifrach O., MacKinnon R. 2002. Energetics of pore opening in a voltage-gated K+ channel.Cell. 111:231–239 [DOI] [PubMed] [Google Scholar]
- Zhou M., Morais-Cabral J.H., Mann S., MacKinnon R. 2001. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors.Nature. 411:657–661 [DOI] [PubMed] [Google Scholar]
- Zhu X.-R., Wulf A., Schwarz M., Isbrandt D., Pongs O. 1999. Characterization of human Kv4.2 mediating a rapidly-inactivating transient voltage-sensitive K+ current.Receptors Channels. 6:387–400 [PubMed] [Google Scholar]