

Abstract
Diabetic foot disease is a major health problem, which affects 15% of the 200 million patients with diabetes worldwide. Diminished peripheral blood flow and decreased local neovascularization are critical factors that contribute to the delayed or nonhealing wounds in these patients. The correction of impaired local angiogenesis may be a key component in developing therapeutic protocols for treating chronic wounds of the lower extremity and diabetic foot ulcers. Endothelial progenitor cells (EPCs) are the key cellular effectors of postnatal neovascularization and play a central role in wound healing, but their circulating and wound-level numbers are decreased in diabetes, implicating an abnormality in EPC mobilization and homing mechanisms. The deficiency in EPC mobilization is presumably due to impairment of eNOS-NO cascade in bone marrow (BM). Hyperoxia, induced by a clinically relevant hyperbaric oxygen therapy (HBO) protocol, can significantly enhance the mobilization of EPCs from the BM into peripheral blood. However, increased circulating EPCs failed to reach to wound tissues. This is partly a result of downregulated production of SDF-1α in local wound lesions with diabetes. Administration of exogenous SDF-1α into wounds reversed the EPC homing impairment and, with hyperoxia, synergistically enhanced EPC mobilization, homing, neovascularization, and wound healing. Antioxid. Redox Signal. 10, 1869–1882.
Chronic Wounds of the Lower Extremity and Diabetic Foot Ulcers
A chronic wound is any interruption in the continuity of the skin and integrity of the tissue that requires a prolonged time (>8 weeks) to heal, does not heal, or recurs (138). The most prevalent forms of chronic wounds are leg ulcers caused by vascular insufficiency (70, 123), and foot ulcerations associated with diabetic complications (120, 137). It has been estimated that up to 2 million Americans have non-healing lower-extremity wounds (75, 98). The economic costs associated with chronic wounds are enormous and include hospital costs, disability, decreased productivity, and loss of independence (103, 121).
Diabetes mellitus is increasing in incidence and represents a major health problem for the 21st century. Indeed, the total number of diabetic patients has been projected to increase from 171 million in 2000 to 366 million in 2030 (135). The annual incidence of foot ulcers among people with diabetes has been variously estimated at between 1% and 4.1%, and the annual incidence of amputation is 0.21–1.37% (9).
The pathophysiology of diabetic foot ulcers and delayed healing has been well described. Contributing factors include progressive development of a sensory, vasomotor, and autonomic neuropathy, leading to loss of protective sensation; joint and bone deformities that increase plantar foot pressure; and alterations in autoregulation of dermal blood flow. Diabetic patients show earlier development and progression of lower extremity peripheral arterial occlusive disease (PAD), with a predilection for blockages in the trifurcation level of vessels just distal to the knee.
The healing of a wound requires a well-orchestrated integration of the complex biologic and molecular events of cell migration, cell proliferation, and extracellular matrix (ECM) deposition. Normal wound healing requires proper circulation, nutrition, immune status, and avoidance of negative mechanical forces. The process usually takes 3–14 days to complete and has three phases: inflammation, proliferation, and remodeling with wound contraction (11, 30, 117) (Fig. 1). During the inflammatory phase, neutrophils and macrophages appear in the wounded area to phagocytize bacteria and debris. A functioning immune system and adequate supply of growth factors are necessary in this phase of wound healing. In the proliferative phase, fibroblasts produce a collagen matrix, new blood vessels invade the forming granulation tissue, and epidermal cells migrate across the wound surface to close the breach. During the remodeling phase, fibroblasts reorganize the collagen matrix and ultimately assume a myofibroblast phenotype to effect connective tissue compaction and wound contraction. Wounds gain ∼80% of their final strength in the first 3 weeks of normal wound healing through collagen deposition, remodeling, and wound contraction (117). When any of the components of the wound healing process is compromised, healing may be delayed. Chronic wounds are those that have failed to follow this sequence and do not achieve a sustained anatomic and functional result.
FIG. 1.
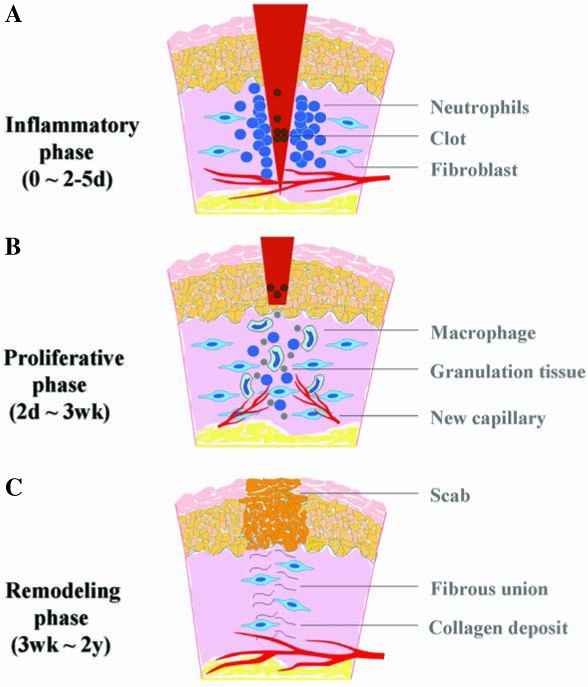
Schematic illustration of the three phases of acute wound healing. (A) Inflammatory phase: immediate to a few days. Immediately after the process of hemostasis, including vaso-constriction, platelet aggregation, and thromboplastin production, injured tissue undergoes vasodilation, and immune cells infiltrate and perform phagocytosis. (B) Proliferative phase: a few days to a few weeks. Fibroblasts lay a bed of collagen and fill the defect, while new capillaries grow (granulation). Injured tissue undergoes contraction. Wound edges pull together to reduce the defect. Afterward, epithelial cells migrate from the point of origin in all directions and cross the moist surface (epithelialization). (C) Remodeling phase: a few weeks to 2 years. During this phase, new collagen is formed and deposited, which increases tensile strength. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
In diabetes, myriad factors, including intrinsic factors (neuropathy, vascular problems, other complicating systemic effects due to diabetes) and extrinsic factors (wound infection, callus formation, and excessive pressure to the site) can impair wound healing. It is well known that peripheral vascular disease (macroangiopathy), along with diabetic neuropathy, plays a major role in diabetic foot ulceration. About 20% of diabetic lower extremity ulcers have arterial flow insufficiency as their primary etiology, ∼50% will have primary diabetic neuropathy, and ∼30% will have both conditions (105). Even after correction of large blood vessel dysfunction by open surgical or endovascular revascularization, only ∼47% of patients will heal in a span of 20 weeks with standardized treatment, including glycemic control, debridement of necrotic tissue, control of infection, use of moist dressings, protection from pressure or trauma related to ambulation, and adjuvant HBO therapy (2, 54, 58). Recently, microangiopathy has also been implicated in the pathogenesis of diabetic foot ulcers (67, 91). Some of these vascular complications in diabetes, as well as the healing defects, have been associated with a decrease in number and function of circulating endothelial progenitor cells (EPCs) (22, 59, 76, 77, 125, 132). In addition, impaired host responses to infection and other cellular dysfunctions contribute to the refractory nature of diabetic wounds.
Despite the existence of standard protocols and the adoption of novel biologic and cell therapies for the treatment of diabetic foot ulcers (15, 114), the effectiveness is limited, and the amputation rate remains high. Given the morbidity and mortality of amputations (2-year survival rate of 50–60%) (121), it is imperative to develop better therapies to treat diabetic ulcers. The development of novel, more efficacious modalities of treatment would have tremendous benefit to both individual patients and society.
Neovascularization and EPCs
Restoring blood flow to the site of injured tissue is a prerequisite for mounting a successful repair response. It is now well established that an essential part of normal healing for full-thickness cutaneous wounds is the formation of new blood vessels within the provisional wound matrix, which is referred to as granulation tissue. New blood vessel growth occurs in both physiologic and pathologic conditions. Until recently, neovascularization was thought to arise solely from preexisting vessels (angiogenesis). However, recent studies have proven that EPCs can contribute to neovascularization (vasculogenesis) as well (102). In 1997, Asahara et al. (5) first identified circulating EPCs contributing to neovascularization. Since then, increasing evidence has suggested that BM-derived EPCs can functionally contribute to neovascularization during wound healing, limb ischemia (52, 81, 122), postmyocardial infarction (65, 93, 94), endothelialization of vascular grafts (56, 115), atherosclerosis (109), retinal and lymphoid organ neovascularization (34, 95), vascularization during neonatal growth (140), and tumor growth (79, 106). It has been estimated that EPCs contribute up to 25% of endothelial cells in newly formed vessels in animal models (125).
Although reports have been conflicting regarding the importance of EPCs in neovascularization (33), a substantial amount of study in both animals and humans has provided strong evidence for the role of BM-derived EPCs in neovascularization (66). In particular, a very recent study showed that notwithstanding low numbers of EPC, recruitment of these EPCs is pivotal for the progression of avascular micrometastatic tumors to lethal macrometastatic ones (26). The lack of consensus may be due in part to heterogeneous phenotypic definitions of EPCs. In this aspect, two recent reports (89, 139) showed that the subpopulations of EPCs with distinct phenotypes give rise to different outcomes, indicating the importance of defining the correct EPCs for such studies. Overall, however, these researchers clearly demonstrated a significant contribution of EPCs to neovascularization. Other factors, such as types of ischemic models or tumors [in which the profiles of chemoattractant factors may vary (107)], time frame of the experiment, and method of in situ endothelial cell identification, may also contribute to some of the reported discrepancy. In general, it is now well accepted that both adjacent preexisting blood vessels and recruitment of BM-derived EPCs participate in tissue vascularization.
Multiple markers have been used for characterization of EPCs and matured endothelial cells (ECs). Platelet endothelial cell-adhesion molecule-1 (PECAM-1/CD31), vascular endothelial growth factor receptor 2 (VEGFR2, also known as KDR or Flk1), von Willebrand factor (vWF), and vascular endothelial cadherin (VE-cadherin) are commonly used for identification of endothelial cells (28, 44, 92, 101). The ability to take up Dil-labeled acetylated low-density lipoprotein (Dil-Ac-LDL) (5, 28) has been used as a marker for identification of endothelial precursors. CD34 is a marker for both endothelial and hematopoietic progenitor cells. In humans, EPCs (but not mature endothelial cells) express AC133 (CD133), a stem cell marker with as-yet-unrecognized functions (108). Purified populations of CD133+VEGFR2+ EPCs proliferate in vitro in an anchorage-independent manner and can be induced to differentiate into mature adherent CD133−VEGFR2+ endothelial cells. Expression of various markers is a dynamic process. It is likely that different markers are present on EPCs and matured endothelial cells at various points along their differentiation cascade from immature progenitors to mature endothelial cells. Expression of CD133 will be turned off with the maturation of endothelial cells. The corresponding marker for CD133 in murine EPCs has not yet been substantiated. Murine EPCs are characterized as Sca-1+/c-Kit+/Lin−/VEGFR2+/CXCR4+/Tie2+ (100). With maturation, expression of stem cell marker Sca-1 and hematopoietic marker c-Kit is turned off.
EPCs are embedded in a microenvironment (niche) of bone marrow and can be mobilized to the circulation (55). Under normal conditions, the number of circulating EPCs is relatively small but is increased in response to trauma or ischemia, which mobilizes these cells from the bone marrow and allows them to proliferate (29). The recruitment of EPCs from the bone marrow and circulation to homing sites of neovascularization is subject to regulation by many factors, including chemokines and growth factors (Fig. 2). The precise mechanism of EPC mobilization is not entirely elucidated and is still under investigation. Many chemokines and cytokines trigger stem/progenitor cell release by induction of matrix metallopeptidase 9 (MMP-9) in bone marrow. Nitric oxide (NO)-mediated signaling pathways have been previously proposed to be essential for EPC mobilization (3, 39, 40) (Fig. 3). By using vascular endothelial growth factor-A (VEGF-A) as a proximal stimulus, Aicher et al. (3, 4) demonstrated that endothelial nitric oxide synthase (eNOS) becomes activated in bone marrow stroma; NO then S-nitrosylates by paracrine mechanisms and activates MMP-9, which releases the stem cell–active cytokine, soluble Kit ligand. This agent shifts endothelial progenitor and hematopoietic stem cells from a quiescent to a proliferative niche and stimulates rapid stem cell mobilization to the peripheral blood (3, 39, 40, 90, 100, 116). It has been demonstrated that in the setting of trauma and ischemia, systemic VEGF-A levels increase, with a time course that mirrors the increase in circulating BM-derived EPCs. In addition, recent studies have suggested that EPCs may promote local neovascularization by secreting angiogenic growth factors in a paracrine manner. Transplantation of EPCs into ischemic tissues may emerge as a promising approach in the therapy of diseases associated with blood vessel disorders.
FIG. 2.

Overview of EPC mobilization and homing. Contribution of BM EPCs to wound healing involves two critical events. BM EPCs are released from their niche into the circulation (mobilization). The circulating EPCs are recruited (homing) to injured tissue and contribute to neovascularization and wound healing. Mobilization and homing are controlled by different mechanisms. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
FIG. 3.
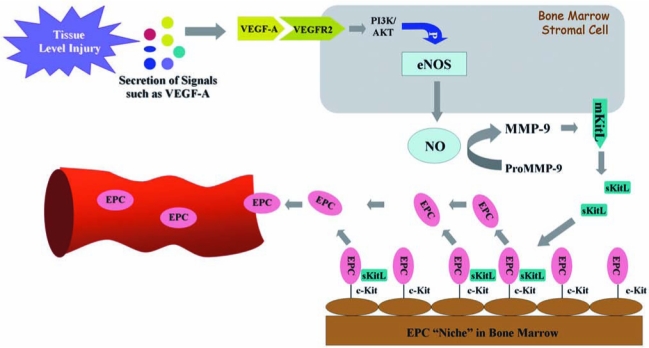
eNOS–NO–MMP-9–KitL cascade–mediated mobilization of bone marrow EPCs. Injured tissue releases signal molecules, such as VEGF-A, which can be sensed by bone marrow stromal cells. VEGF-A and other mobilizing stimuli might signal through their cognate receptors to activate the eNOS within the bone marrow stromal cells. eNOS induces production of NO, which stimulates and/or maintains MMP-9 activity, resulting in cleavage soluble kit ligand (sKitL) from the membrane-bound kit ligand (mKitL) to mobilize c-Kit+ EPCs from the bone marrow niche into the circulation. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
Impairment of EPC and eNOS Activity in Diabetes
The number and function of EPCs can be affected in diabetes mellitus. Reduced levels and impaired function of EPCs have been described in both type 1 and type 2 diabetic patients (77, 125). EPC recruitment for re-endothelialization after vascular injury is also impaired in diabetes (48). These alterations are likely to be involved in the pathogenesis of vascular disease in diabetes (22). Increasing evidence suggests that wound-healing mechanisms, in both the bone marrow and within the peripheral wound, are compromised by diabetes as a result of BM-derived EPC impairments (22, 59, 76, 77, 125, 132). Although cytokines such as granulocyte colony-stimulating factor (G-CSF) and growth factors such as VEGF-A can induce the release of progenitor cells from the bone marrow, the nonspecific effects on release of other white cells and platelets or the leaky-capillary effect has made these factors unsuitable to treat diabetic patients with nonhealing chronic wounds (24, 57, 72, 74, 130). eNOS is of paramount importance for the regulation of mobilization and function of EPCs (3, 133); however, evidence concerning the effect of eNOS deficiency on diabetic EPCs is not available. We have hypothesized that eNOS function is impaired in diabetics, thus preventing these cells from reaching the wound site in significant numbers. Data from our laboratory confirmed that the levels of biologically active phosphorylated eNOS protein were decreased in diabetic mice, although no changes in the amount of total eNOS protein were observed (25). In the experimental diabetic mouse model, it was found that the EPC and HSC populations remain constant, whereas the mesenchymal stromal cell and lymphocyte populations demonstrate a slight decrease (by ∼27% and 26%, respectively). Thus, it is possible that diabetes-induced changes in mesenchymal stromal cell and lymphocyte populations in the BM might be responsible for the observed downregulation of BM eNOS activation. However, the precise mechanism related to the impaired eNOS in diabetes mellitus remains unclear. Also, it is unknown whether NO-mediated signaling pathways are altered in the human diabetic bone marrow.
Enhancement of Mobilization of BM-Derived EPCs by Hyperoxia
Adult bone marrow is a rich reservoir of pluripotent stem cells and various lineage progenitor cells (37, 128). Emigration of stem/progenitor cells (in particular, hematopoietic progenitor cells) from the bone marrow is generally thought to occur after a period of cell proliferation within the marrow niche (19, 97, 136). However, the emerging evidence also indicates a rapid mobilization of progenitor/stem cells from bone marrow, which suggests that cell proliferation is not always necessary. In mice, infusion of soluble Kit ligand triggers mobilization of CD34+ cells in 1 h (90). A fourfold elevation in circulating progenitor cells occurred within 10 min when human volunteers were subjected to highly strenuous exercise (104). It was shown that a specialized microenvironment exists in the marrow where stem/progenitor cells exhibit different propensities for proliferation and mobilization and where MMP-9 activity mediates BM-derived EPC release (39, 40, 100). From our findings (25, 31, 128) and reports by Nakamura et al. (90) and Rehman et al. (104), it appears that a subpopulation of BM-derived EPCs exists within specialized bone marrow niches that are poised for rapid release to the circulation.
Oxygen plays a key role in wound healing. Low oxygen tension (hypoxia) around the wound is one of several critical factors that mutually enhance the progression of a chronic ulcer, whereas a plentiful supply of oxygen is essential for a variety of the healing processes. Oxygen tension is positively correlated with collagen production (46, 51, 112), bacterial killing (17, 42, 61), epithelialization (131), and angiogenesis (62, 63). All these components of wound healing increase greatly in well-oxygenated wounds. Of the available oxygen-delivery methods, HBO is noninvasive and appears to be the most potent choice. Systemic hyperoxia induced by HBO is a treatment approved by the United States Food and Drug Administration (FDA) as a safe, adjunctive therapy to stimulate wound healing in diabetic patients. Patients typically receive ≥20 treatments with pure oxygen at 2.0–2.4 atmospheres absolute (ATA), once or twice daily. Oxygen is transported by the blood in two different ways: (a) chemically bound to hemoglobin in erythrocytes, and (b) physically dissolved in plasma according to Henry's law. This law states that the degree to which a gas enters into physical solution in body fluids is directly proportional to the partial pressure of the gas to which the fluid is exposed (7). HBO treatment is known to accelerate healing in ischemic (43, 46) and refractory diabetic wounds (2, 8, 141).
Despite the beneficial actions of HBO therapy on chronic, nonhealing wounds, the molecular mechanisms of the action are incompletely understood. NO has been shown to play a central role in the bone marrow mobilization and release of EPCs (3). Based on the fact that the generation of NO results in EPC release from the BM and HBO has been shown to up-regulate NO production in cerebral cortex tissue, perivascular pulmonary tissue, and neutrophils via stimulation of NOS (126, 127, 129), it is likely that HBO benefits wound healing via NOS-NO cascade-mediated mobilization of progenitor cells from the bone marrow. Recent investigations from our laboratory and collaborators confirmed this possibility and demonstrated that EPC mobilization into circulation is triggered by HBO through induction of bone marrow NO, with resulting enhancement in ischemic limb perfusion and wound healing (31, 128). With ischemic and diabetic murine models, we recently determined that hyperoxia, induced by a clinically relevant HBO protocol, increases NO levels within femoral bone marrow, accelerates the spontaneous revascularization of surgically induced hindlimb ischemia, and increases the number of BM-derived EPCs in circulation and within cutaneous hindlimb ischemic incisional wounds and diabetic excisional wounds (25). These effects appear to be specific to the release of BM-derived EPCs, but not lymphocytes, and responsive to the cytokine milieu of the wound. In the ischemic and diabetic murine models that were used, therapeutic wound-healing effects of increased BM-derived EPC mobilization into circulation and recruitment into wounds were observed in association with enhancement of neovascularization of the wounds and spontaneous recovery of hindlimb perfusion.
Hyperoxia and the NOS-NO Cascade
NO is a small, pleiotropic, free radical that was discovered as the endothelium-derived relaxing factor responsible for the maintenance of vascular tone (47). Because of its highly hydrophobic characteristics (73), NO is a unique messenger in that it is produced in one cell and diffuses into adjacent target cells to activate cytosolic guanylate cyclase–bound heme to generate the NO-heme adduct of guanylate cyclase (84). NO plays a significant role in the intracellular signaling process in cardiovascular as well as in other systems.
Production of NO is controlled by NOSs, which catalyze a stepwise oxidation of l-arginine (Arg) to citrulline and NO (36, 82). In mammals, four NOS isoforms exist, including neuronal NOS (nNOS, type I), inducible NOS (iNOS, type II), endothelial NOS (eNOS, type III), and a more recently identified mitochondrial NOS (mtNOS), which differ in their primary sequence, posttranslational modifications, cellular location, and tissue expression (10, 20, 80, 85), consistent with their participating in a range of physiologic and pathologic systems.
eNOS is selectively expressed in vascular endothelial cells or surrounding stromal cells and therefore has been a focus of attention in vascular biology. eNOS plays an essential role in endothelial cell proliferation and is a central mediator of several endothelium growth stimulators, such as VEGF-A.
Among the four NOS enzymes, the activity of iNOS is controlled at the level of gene transcription, whereas the activities of nNOS and eNOS are controlled by intracellular calcium/calmodulin, several different phosphorylation mechanisms, and by binding of the molecular chaperone heat-shock protein 90 (HSP90) (13, 23, 27). It is believed that mtNOS is a constitutively active eNOS-like isoform (68). Studies with three major NOS enzymes (except mtNOS in limited studies) in vitro have shown that enzyme activity is influenced by the redox state and specifically by O2 tension. Elevated O2 tension influences NOS activity by hastening conversion of ferrous heme back to the native ferric conformation (Fig. 4).
FIG. 4.
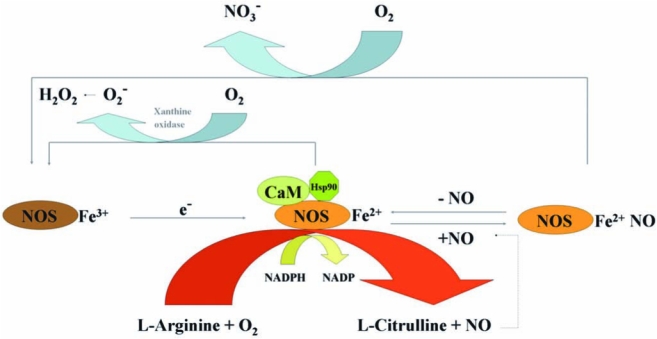
Model for the regulation of eNOS activity. The activities of eNOS (or nNOS) are controlled by intracellular calcium/calmodulin (CaM) and by binding of the molecular chaperone heat-shock protein 90 (HSP90). Addition of NADPH to CaM-bound NOSFe3+ reduces the flavins and enables transfer of electrons (e−) to the heme to form NOSFe2+, which then binds and activates O2 and catalyzes either the stepwise oxidation of l-arginine to NO and citrulline or superoxide production in the absence of l-arginine. When NO is formed, it can also bind to NOSFe2+ before leaving the catalytic site and form an inactive complex (NOSFe2+NO). NOSFe2+NO must either lose NO or react with O2 to re-form NOSFe3+ to participate in catalysis. NADPH oxidase may also be required for the generation of superoxide. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
In diabetic mice, the hyperoxia-induced increase of NO in BM was attenuated, likely as a result of impaired eNOS phosphorylation. However, with the induction of hyperoxic conditions, other NOS isoenzymes appear to compensate, leading to NO increases in the BM that are substantial and sufficient to reverse the defect in EPC mobilization in our diabetic mouse model.
An intriguing paradox exists. Based on our findings and the reported effects of hypoxia, it appears that both hyperoxia and hypoxia can result in NOS activation and a subsequent increase of NO level in BM and induce EPC release (25, 100, 124). The reasons for this seemingly paradoxic phenomenon are unclear. One explanation for this response involves the potential existence of an oxygen-tension sensor, triggered by any perturbation in the oxygen levels that results in NO production and EPC release. Another potential explanation is that both high and low local oxygen concentrations act via distinct mechanisms that ultimately converge on NOS activation (Fig. 5). Ischemic wounds signal for hypoxia by hypoxia-inducible factor (HIF)-dependent mechanisms. A functional HIF response requires stabilization of the α-subunit (e.g., HIF-1α), during hypoxia, and dimerization with HIF-1β, to drive target gene activation (60). Intriguingly, high concentrations of NO stabilize HIF-1α and thus mimic a hypoxic response under normoxia. Thus, it is likely that hyperoxia activates NOS activity and increases the BM NO level, which can result in HIF-1α stabilization. If this be the case, it supports a concept that both hyperoxia- and hypoxia-induced upstream biochemical events converge at the level of HIF-1α, and this oxygen-tension sensor will trigger a common signal to mobilize BM EPCs. Whatever the reason, it is clear that with hypoxia, systemic effects of VEGF-A via VEGFR2 mediate NOS activation, whereas with hyperoxia, NO levels increase within minutes, and the effects are quickly reversible on withdrawal of the stimulus, suggesting a direct activation of NOS by a change in oxygen tension. Further studies are needed more specifically to identify the underlying mechanisms.
FIG. 5.
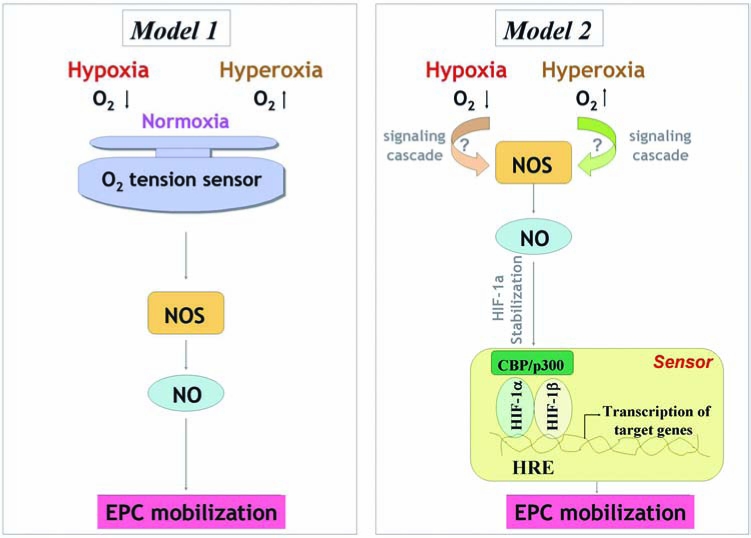
Two potential mechanisms of regulating EPC mobilization by hyperoxia versus hypoxia. Model 1: An oxygen-tension sensor may exist, calibrated by normoxia but can be triggered by any perturbation in the oxygen levels, either high or low, that results in NO production and EPC release. Model 2: Hyperoxia and hypoxia act via distinct mechanisms that ultimately converge on NOS activation, which catalyzes NO production. NO stabilizes HIF-1α, and this oxygen-tension sensor will trigger a common signal to mobilize BM EPCs. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
One of the advantages of HBO-induced EPC mobilization is that HBO specifically stimulates BM EPCs to be released into circulation without having a significant impact on the inflammatory cell numbers in circulation. In contrast, although G-CSF and other chemokines are able to increase circulating EPCs (99), an associated increase in leukocytes results in inflammation with the potential for enhanced acute coronary events. It, therefore, raises questions about the safety and clinical utility of these chemokines (53, 87). Thus, HBO appears to be a safe method in driving EPC mobilization.
Conversely, it should be pointed out that HBO-mediated enhancement of wound healing is multifactorial. HBO may have local tissue effects unrelated to EPC release that also enhance wound healing in selective wound environments. One of these effects may include increased tissue-level release of angiogenic factors such as VEGF-A (111, 113).
Oxygen-Dependent Redox-Sensitive Signaling Processes
It is known that hyperoxia induces the generation of excessive reactive oxygen species (ROS), including superoxide anions (O2−), hydroxyl radicals (OH−), and hydrogen peroxide (H2O2) (Fig. 6). ROS can affect the activity of signaling pathways by direct modification of regulatory proteins via disulphide bond formation, nitrosylation, carbonylation, or glutathionylation (21). ROS and the ROS-generating NADPH oxidases play important roles as signaling molecules in the vasculature (12, 41). Whether and how hyperoxia-induced ROS participate in the regulation of BM EPC mobilization remains unknown.
FIG. 6.

Generation of reactive oxygen radicals (ROS). Reactive oxygen species, such as H2O2, O2−, and OH−, are generated in cells by several pathways. O2− can be generated by xanthine oxidase or NADPH oxidase. Superoxide dismutase (SOD) converts O2− into H2O2, and then H2O2 is mostly degraded to H2O by glutathione (GSH) peroxidase and catalase. H2O2 produces a highly reactive radical OH− by the Fenton or Haber-Weiss reactions.
Although direct studies are lacking, several lines of evidence suggest that ROS may be involved in the control of EPC mobilization through modulating cell adhesion, regulating cytoskeleton and cell motility, and inducing VEGF-A production. For instance, it has been reported that ROS are capable of inducing conformational changes in integrins to change their binding affinity and function (35). In addition, ROS are important regulators of the actin cytoskeletal dynamics and cellular motility (86). Given the knowledge that the detachment of EPC from BM niche requires alteration of cell adhesion, and actin cytoskeleton is necessary for cell motility, it would be of interest to investigate whether hyperoxia–ROS–integrin (or other adhesion molecules) and hyperoxia–ROS interaction with the cell actin cytoskeleton are involved in this process. Moreover, at micromolar concentrations, H2O2 induces VEGF-A expression (111). Signaling studies identified a cascade comprising Ras–Raf–MEK1–ERK1/2 as the main pathway mediating H2O2-induced VEGF-A transcription (110). Besides, H2O2 also induces the expression by VEGFR2 by a NF-κB–dependent pathway (32). The consequence of VEGF-A–VEGFR2 interactions in the activation of bone marrow NOS, producing NO, which then stimulates MMP-9, enabling release of soluble Kit-ligand and liberating progenitor cells from the marrow, has been well-established (3, 38, (39). It may be interesting to understand whether BM EPCs respond to ROS in the same way as the matured endothelial cells in terms of VEGF-A and VEGFR2 induction, or which cell population(s) is the target(s) of the ROS and responsible for the activation of NOS–NO–MMP9-soluble Kit ligand cascade in the BM microenvironment.
Recruitment/Homing of EPCs to Diabetic Wound Tissues
The contribution of BM EPCs to neovascularization in wounds results from a multistep process. It involves sensing the ischemia signal from the remote tissue, releasing EPCs from the BM niche into circulation, homing of circulating EPCs to the target tissues, the integration of the EPCs into blood vessels, and the in situ differentiation/maturation of EPCs into matured, functional ECs. Our data demonstrate that hyperoxia selectively enhances EPC release, resulting in a small but significant improvement in diabetic wound healing, yet not having a significant impact on wound EPC homing (25). If EPCs are mobilized into circulation but fail to reach the injured tissue, the clinical usefulness of the HBO treatment becomes suboptimal. This may explain the variable clinical effects on wound healing reported with HBO treatment alone. Our findings suggest that homing factor(s) that controls recruitment of circulating EPCs into wounds may be decreased with diabetes. Therefore, a full understanding of impaired homing mechanisms in diabetic wounds is crucial for enhancing EPC recruitment and engraftment.
The process of EPC homing to sites of ischemia includes detachment from the BM niche, rolling into blood vessels and traveling within the circulation, adhesion to the endothelial cell monolayers, and incorporation into neovessels. Recent studies support the idea that EPC and progenitor cells use adhesion molecules for homing to sites of neovascularization, similar to the adhesion molecules engaged by leukocytes for recruitment to sites of inflammation (49, 71, 78). Chemokines play critical roles in the regulation of this trafficking of circulating EPCs from the bloodstream to ischemic tissues. Many of these factors are chemoattractants. Stromal cell–derived factor (SDF)-1α is the predominant chemokine that is upregulated in ischemic tissue and acts as a homing signal for EPCs (69). Inhibition of the SDF-1α–CXCR4 axis partially blocks the homing of progenitor/stem cells to the ischemic myocardium (1). Likewise, suppression of CXCR4 by anti-CXCR4 neutralizing antibodies significantly reduced SDF-1α–induced adhesion of EPC to endothelial cell mono-layers, the migration of EPCs in vitro (16), and the in vivo homing of myeloid EPCs to the ischemic limb in a model of hindlimb ischemia (134).
Although the role of SDF-1α in EPC homing to ischemic tissue is known, the effect of SDF-1α expression at the tissue level in diabetic wounds had not been previously studied. We recently demonstrated that the local concentration of SDF-1α in a diabetic wound is significantly decreased, and epithelial cells and myofibroblasts appeared to be responsible for the downregulation of SDF-1α in diabetic wounds (25) (Fig. 7). Moreover, the exogenous administration of SDF-1α to wounds of diabetic mice increases the wound-level EPC recruitment. More important, in combination with HBO therapy, which brings an increased amount of circulating EPCs, raising SDF-1α levels by local injection of recombinant SDF-1α protein significantly enhances EPC recruitment to wound tissues and improves wound healing in this diabetic animal model (Fig. 8).
FIG. 7.
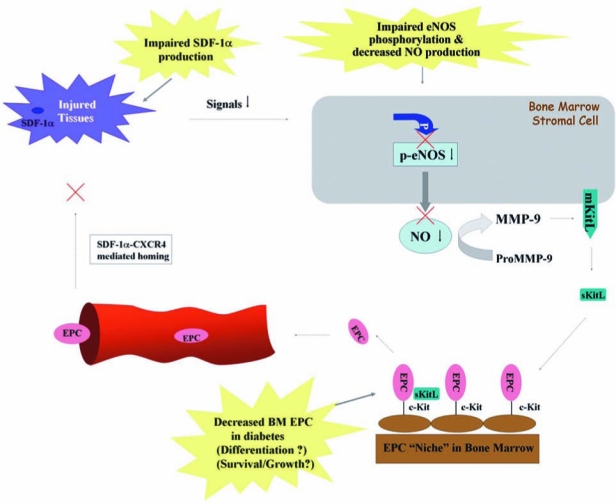
Impaired eNOS–NO cascade in the bone marrow and downregulated SDF-1α production at the injured tissues in diabetic mouse. Impaired eNOS phosphorylation results in decreased production of NO in bone marrow, which affects downstream pathway leading to EPC mobilization. In the injured tissues, production of SDF-1α is downregulated, resulting in decreased recruitment/homing of circulating EPCs. The EPC number in diabetic BM is diminished because of unknown mechanisms. These factors contribute to the impaired neovascularization and delayed or nonhealing wounds in diabetes. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
FIG. 8.
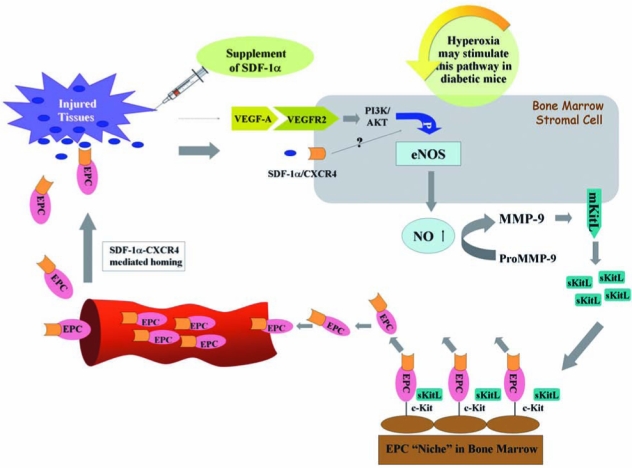
Synergistic effects of hyperoxia and exogenous SDF-1α on regulating EPC mobilization and homing. Hyperoxia (HBO) increases NO levels, likely through stimulating the NOS/NO pathway, in bone marrow to promote EPC mobilization. EPCs express CXCR4 on the cell surface. Local supplement of SDF-1α enhances EPC homing to the wound tissues via SDF-1α–CXCR4–mediated interaction. SDF-1α might also functions as a signal to stimulate bone marrow EPC mobilization. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
In addition to the SDF-1α/CXCR4 axis and VEGF, which is ischemia induced and well known for its role in recruiting EPCs, other factors may be potentially involved in the regulation of EPC homing to diabetic wounds but have not been studied. For instance, high-mobility group box-1 (HMGB-1) is a nuclear protein that is released extracellularly on activation of cells by inflammatory cytokines and during cell necrosis and acts as a chemoattractant for inflammatory cells, stem cells, and EPCs in vitro and in vivo (18, 96). IL-8 is an inflammatory chemokine that is able to stimulate angiogenesis (88). Local injection of IL-8 in the nonischemic myocardium increased the recruitment of CD34+ cells (64). Neutralizing anti-IL-8/Gro-α antibodies or antibodies against the IL-8 receptors, CXCR1 or CXCR2, reduced CD34+ cell-mediated improvement of neovascularization, establishing a role for CXC-chemokines (IL-8/Gro-α) for homing and neovascularization improvement by CD34+ cells. In addition, blocking CXCR2 inhibited the incorporation of human EPCs expressing CXCR2 at sites of arterial injury (45). Other elements, such as the integrin α4β1–VCAM pair, may also mediate EPC homing (50). It would be worthwhile to test whether these factors are impaired in chronic diabetic wounds and are responsible for mediating EPC homing. The identification of critical homing factor(s) will enhance the efficiency and specificity of EPC therapies and increase the efficacy of EPC-mediated neovascularization in diabetic wounds.
Summary and Perspective
Cell-based therapy is a promising therapeutic option for treating patients with diabetic, nonhealing wounds. Of various different types of stem or progenitor cells, the EPC is one type of cell that has been moved from experimental models to clinical trials. EPC has thus far been tested in patients with acute and chronic ischemic disease, and outcomes are very encouraging (6, 119). A few properties, such as its endogenous, BM-derived character, ability to home to sites of pathologic entities, and relative stability in terms of lineage specification in culture, which allows genetic and epigenetic manipulation, make EPC an ideal cell candidate to be tested in cell-based therapeutic applications for ischemic disorders.
The efficacy of cell therapies to augment neovascularization and healing depends not only on the sufficient amount of circulating EPCs, but also on the efficient recruitment of these cells to the target tissue. This is not surprising, as the contribution of EPC to neovascularization is a coordinated sequence of multiple steps, and each step is controlled by a specific mechanism(s). Several of these mechanisms are impaired in diabetes. Thus, a combination of therapeutic approaches to target individual impairments and to correct diabetes-related EPC deficits will likely synergize and lead to a more-successful treatment outcome for diabetic wounds.
The identification of hyperoxia as an efficient tool in the mobilization of BM-derived EPCs is of great significance, as HBO treatment is an FDA-approved, safe method. The biologic effect of hyperoxia on mobilizing EPCs is evident and somehow paradoxically similar to that produced by hypoxia. Although further study is required to elucidate the molecular and cellular mechanism underlying the biologic effect of HBO, from the therapeutic point of view, HBO is an acceptable approach and has actually been applied in the treatment of diabetic wounds with varying degrees of success. Understanding the limitations of HBO in modulating only the EPC mobilization step but not the homing of the increased circulating EPCs to the target tissue provides useful guidance for the development of new clinical protocols to treat diabetic wounds. In addition, development of more efficient and specific treatments targeting the eNOS-NO pathway, which may synergize with the HBO effects, would be a plus. Moreover, in addition to SDF-1α, other strategies for modifying the wound environment, such as PDGF-BB (118), fibroblasts delivered in an absorbable mesh (83), or in type 1 collagen (14), which, at least partially, function directly or indirectly through accelerating recruitment of EPCs, are under investigation. These strategies could be combined with HBO therapy, thus synergistically influencing EPC effects by targeting both homing and mobilization steps. Furthermore, elucidation of the mechanisms underlying the decreased number of BM EPCs in diabetes and development of a solution to correct this impairment and to increase the BM EPC number could provide a scientific basis for the establishment of combined treatment protocols. A better understanding of the molecular and cellular etiologies of nonhealing, diabetic wounds in combination with development of efficient approaches for correcting EPC deficits and functional impairments will eventually result in the development of clinically efficient and feasible therapies that prevent wound progression, eliminate amputations, and promote rapid healing in patients with diabetes.
Abbreviations
ATA, atmospheres absolute; BM, bone marrow; CaM, calmodulin; CXCR4, chemokine receptor 4; Dil-Ac-LDL, Dil-labeled acetylated low-density lipoprotein; eNOS, endothelial nitric oxide synthase; EPC, endothelial progenitor cell; ECM, extracellular matrix; G-CSF, granulocyte colony-stimulating factor; HSP90, heat-shock protein 90; HMGB-1, high-mobility group box-1; H2O2, hydrogen peroxide; OH−, hydroxyl radical; HIF, hypoxia inducible factor; HBO, hyperbaric oxygen; MMP-9, matrix metallopeptidase 9; NO, nitric oxide; O2, oxygen; PAD, peripheral arterial occlusive disease;:PECAM-1/CD31, platelet endothelial cell–adhesion molecule-1; ROS, reactive oxygen species; SDF, stromal cell–derived factor; O2−, superoxide anion; FDA, United States Food and Drug Administration; VEGF, vascular endothelial growth factor; VE-cadherin, vascular endothelial cadherin; VEGFR2, vascular endothelial growth factor receptor 2; vWF, von Willebrand factor.
References
- 1.Abbott JD. Huang Y. Liu D. Hickey R. Krause DS. Giordano FJ. Stromal cell-derived factor-1alpha plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation. 2004;110:3300–3305. doi: 10.1161/01.CIR.0000147780.30124.CF. [DOI] [PubMed] [Google Scholar]
- 2.Abidia A. Laden G. Kuhan G. Johnson BF. Wilkinson AR. Renwick PM. Masson EA. McCollum PT. The role of hyperbaric oxygen therapy in ischaemic diabetic lower extremity ulcers: a double-blind randomised-controlled trial. Eur J Vasc Endovasc Surg. 2003;25:513–518. doi: 10.1053/ejvs.2002.1911. [DOI] [PubMed] [Google Scholar]
- 3.Aicher A. Heeschen C. Mildner-Rihm C. Urbich C. Ihling C. Technau-Ihling K. Zeiher AM. Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–1376. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- 4.Aicher A. Zeiher AM. Dimmeler S. Mobilizing endothelial progenitor cells. Hypertension. 2005;45:321–325. doi: 10.1161/01.HYP.0000154789.28695.ea. [DOI] [PubMed] [Google Scholar]
- 5.Asahara T. Murohara T. Sullivan A. Silver M. van der Zee R. Li T. Witzenbichler B. Schatteman G. Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 6.Assmus B. Schachinger V. Teupe C. Britten M. Lehmann R. Dobert N. Grunwald F. Aicher A. Urbich C. Martin H. Hoelzer D. Dimmeler S. Zeiher AM. Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction (TOPCARE-AMI) Circulation. 2002;106:3009–3017. doi: 10.1161/01.cir.0000043246.74879.cd. [DOI] [PubMed] [Google Scholar]
- 7.Bakker DJ. Hyperbaric oxygen therapy: past, present and future indications. Adv Exp Med Biol. 1992;317:95–105. doi: 10.1007/978-1-4615-3428-0_8. [DOI] [PubMed] [Google Scholar]
- 8.Baroni G. Porro T. Faglia E. Pizzi G. Mastropasqua A. Oriani G. Pedesini G. Favales F. Hyperbaric oxygen in diabetic gangrene treatment. Diabetes Care. 1987;10:81–86. doi: 10.2337/diacare.10.1.81. [DOI] [PubMed] [Google Scholar]
- 9.Bartus CL. Margolis DJ. Reducing the incidence of foot ulceration and amputation in diabetes. Curr Diabetes Rep. 2004;4:413–418. doi: 10.1007/s11892-004-0049-x. [DOI] [PubMed] [Google Scholar]
- 10.Bates TE. Loesch A. Burnstock G. Clark JB. Mitochondrial nitric oxide synthase: a ubiquitous regulator of oxidative phosphorylation? Biochem Biophys Res Commun. 1996;218:40–44. doi: 10.1006/bbrc.1996.0008. [DOI] [PubMed] [Google Scholar]
- 11.Baum CL. Arpey CJ. Normal cutaneous wound healing: clinical correlation with cellular and molecular events. Dermatol Surg. 2005;31:674–686. doi: 10.1111/j.1524-4725.2005.31612. discussion 686. [DOI] [PubMed] [Google Scholar]
- 12.BelAiba RS. Djordjevic T. Bonello S. Artunc F. Lang F. Hess J. Gorlach A. The serum- and glucocorticoid-inducible kinase Sgk-1 is involved in pulmonary vascular remodeling: role in redox-sensitive regulation of tissue factor by thrombin. Circ Res. 2006;98:828–836. doi: 10.1161/01.RES.0000210539.54861.27. [DOI] [PubMed] [Google Scholar]
- 13.Bender AT. Silverstein AM. Demady DR. Kanelakis KC. Noguchi S. Pratt WB. Osawa Y. Neuronal nitric-oxide synthase is regulated by the Hsp90-based chaperone system in vivo. J Biol Chem. 1999;274:1472–1478. doi: 10.1074/jbc.274.3.1472. [DOI] [PubMed] [Google Scholar]
- 14.Brem H. Balledux J. Bloom T. Kerstein MD. Hollier L. Healing of diabetic foot ulcers and pressure ulcers with human skin equivalent: a new paradigm in wound healing. Arch Surg. 2000;135:627–634. doi: 10.1001/archsurg.135.6.627. [DOI] [PubMed] [Google Scholar]
- 15.Brem H. Sheehan P. Boulton AJ. Protocol for treatment of diabetic foot ulcers. Am J Surg. 2004;187:1S–10S. doi: 10.1016/S0002-9610(03)00299-X. [DOI] [PubMed] [Google Scholar]
- 16.Ceradini DJ. Kulkarni AR. Callaghan MJ. Tepper OM. Bastidas N. Kleinman ME. Capla JM. Galiano RD. Levine JP. Gurtner GC. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 17.Chang N. Mathes SJ. Comparison of the effect of bacterial inoculation in musculocutaneous and random-pattern flaps. Plast Reconstr Surg. 1982;70:1–10. doi: 10.1097/00006534-198207000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Chavakis E. Hain A. Vinci M. Carmona G. Bianchi ME. Vajkoczy P. Zeiher AM. Chavakis T. Dimmeler S. High-mobility group box 1 activates integrin-dependent homing of endothelial progenitor cells. Circ Res. 2007;100:204–212. doi: 10.1161/01.RES.0000257774.55970.f4. [DOI] [PubMed] [Google Scholar]
- 19.Cheng T. Rodrigues N. Dombkowski D. Stier S. Scadden DT. Stem cell repopulation efficiency but not pool size is governed by p27(kip1) Nat Med. 2000;6:1235–1240. doi: 10.1038/81335. [DOI] [PubMed] [Google Scholar]
- 20.Craven SE. Bredt DS. PDZ proteins organize synaptic signaling pathways. Cell. 1998;93:495–498. doi: 10.1016/s0092-8674(00)81179-4. [DOI] [PubMed] [Google Scholar]
- 21.England K. Cotter TG. Direct oxidative modifications of signalling proteins in mammalian cells and their effects on apoptosis. Redox Rep. 2005;10:237–245. doi: 10.1179/135100005X70224. [DOI] [PubMed] [Google Scholar]
- 22.Fadini GP. Miorin M. Facco M. Bonamico S. Baesso I. Grego F. Menegolo M. de Kreutzenberg SV. Tiengo A. Agostini C. Avogaro A. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol. 2005;45:1449–1457. doi: 10.1016/j.jacc.2004.11.067. [DOI] [PubMed] [Google Scholar]
- 23.Fontana J. Fulton D. Chen Y. Fairchild TA. McCabe TJ. Fujita N. Tsuruo T. Sessa WC. Domain mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circ Res. 2002;90:866–873. doi: 10.1161/01.res.0000016837.26733.be. [DOI] [PubMed] [Google Scholar]
- 24.Fukumoto Y. Miyamoto T. Okamura T. Gondo H. Iwasaki H. Horiuchi T. Yoshizawa S. Inaba S. Harada M. Niho Y. Angina pectoris occurring during granulocyte colony-stimulating factor-combined preparatory regimen for autologous peripheral blood stem cell transplantation in a patient with acute myelogenous leukaemia. Br J Haematol. 1997;97:666–668. doi: 10.1046/j.1365-2141.1997.842724.x. [DOI] [PubMed] [Google Scholar]
- 25.Gallagher KA. Liu ZJ. Xiao M. Chen H. Goldstein LJ. Buerk DG. Nedeau A. Thom SR. Velazquez OC. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J Clin Invest. 2007;117:1249–1259. doi: 10.1172/JCI29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao D. Nolan DJ. Mellick AS. Bambino K. McDonnell K. Mittal V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319:195–198. doi: 10.1126/science.1150224. [DOI] [PubMed] [Google Scholar]
- 27.Garcia-Cardena G. Fan R. Shah V. Sorrentino R. Cirino G. Papapetropoulos A. Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–824. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- 28.Garlanda C. Dejana E. Heterogeneity of endothelial cells: specific markers. Arterioscler Thromb Vasc Biol. 1997;17:1193–1202. doi: 10.1161/01.atv.17.7.1193. [DOI] [PubMed] [Google Scholar]
- 29.Gill M. Dias S. Hattori K. Rivera ML. Hicklin D. Witte L. Girardi L. Yurt R. Himel H. Rafii S. Vascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cells. Circ Res. 2001;88:167–174. doi: 10.1161/01.res.88.2.167. [DOI] [PubMed] [Google Scholar]
- 30.Goldman R. Growth factors and chronic wound healing: past, present, and future. Adv Skin Wound Care. 2004;17:24–35. doi: 10.1097/00129334-200401000-00012. [DOI] [PubMed] [Google Scholar]
- 31.Goldstein LJ. Gallagher KA. Bauer SM. Bauer RJ. Baireddy V. Liu ZJ. Buerk DG. Thom SR. Velazquez OC. Endothelial progenitor cell release into circulation is triggered by hyperoxia-induced increases in bone marrow nitric oxide. Stem Cells. 2006;24:2309–2318. doi: 10.1634/stemcells.2006-0010. [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez-Pacheco FR. Deudero JJ. Castellanos MC. Castilla MA. Alvarez-Arroyo MV. Yague S. Caramelo C. Mechanisms of endothelial response to oxidative aggression: protective role of autologous VEGF and induction of VEGFR2 by H2O2. Am J Physiol Heart Circ Physiol. 2006;291:H1395–H1401. doi: 10.1152/ajpheart.01277.2005. [DOI] [PubMed] [Google Scholar]
- 33.Gothert JR. Gustin SE. van Eekelen JA. Schmidt U. Hall MA. Jane SM. Green AR. Gottgens B. Izon DJ. Begley CG. Genetically tagging endothelial cells in vivo: bone marrow-derived cells do not contribute to tumor endothelium. Blood. 2004;104:1769–1777. doi: 10.1182/blood-2003-11-3952. [DOI] [PubMed] [Google Scholar]
- 34.Grant MB. May WS. Caballero S. Brown GA. Guthrie SM. Mames RN. Byrne BJ. Vaught T. Spoerri PE. Peck AB. Scott EW. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat Med. 2002;8:607–612. doi: 10.1038/nm0602-607. [DOI] [PubMed] [Google Scholar]
- 35.Gregg D. de Carvalho DD. Kovacic H. Integrins and coagulation: a role for ROS/redox signaling? Antioxid Redox Signal. 2004;6:757–764. doi: 10.1089/1523086041361604. [DOI] [PubMed] [Google Scholar]
- 36.Griffith OW. Stuehr DJ. Nitric oxide synthases: properties and catalytic mechanism. Annu Rev Physiol. 1995;57:707–736. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 37.Grzegorzewski K. Komschlies KL. Mori M. Kaneda K. Usui N. Faltynek CR. Keller JR. Ruscetti FW. Wiltrout RH. Administration of recombinant human interleukin-7 to mice induces the exportation of myeloid progenitor cells from the bone marrow to peripheral sites. Blood. 1994;83:377–385. [PubMed] [Google Scholar]
- 38.Gu Z. Kaul M. Yan B. Kridel SJ. Cui J. Strongin A. Smith JW. Liddington RC. Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 39.Heissig B. Hattori K. Dias S. Friedrich M. Ferris B. Hackett NR. Crystal RG. Besmer P. Lyden D. Moore MA. Werb Z. Rafii S. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heissig B. Werb Z. Rafii S. Hattori K. Role of c-kit/Kit ligand signaling in regulating vasculogenesis. Thromb Haemost. 2003;90:570–576. doi: 10.1160/TH03-03-0188. [DOI] [PubMed] [Google Scholar]
- 41.Herkert O. Djordjevic T. BelAiba RS. Gorlach A. Insights into the redox control of blood coagulation: role of vascular NADPH oxidase-derived reactive oxygen species in the thrombogenic cycle. Antioxid Redox Signal. 2004;6:765–776. doi: 10.1089/1523086041361695. [DOI] [PubMed] [Google Scholar]
- 42.Hohn DC. MacKay RD. Halliday B. Hunt TK. Effect of O2 tension on microbicidal function of leukocytes in wounds and in vitro. Surg Forum. 1976;27:18–20. [PubMed] [Google Scholar]
- 43.Hopf HW. Gibson JJ. Angeles AP. Constant JS. Feng JJ. Rollins MD. Zamirul Hussain M. Hunt TK. Hyperoxia and angiogenesis. Wound Repair Regen. 2005;13:558–564. doi: 10.1111/j.1524-475X.2005.00078.x. [DOI] [PubMed] [Google Scholar]
- 44.Hristov M. Erl W. Weber PC. Endothelial progenitor cells: mobilization, differentiation, and homing. Arterioscler Thromb Vasc Biol. 2003;23:1185–1189. doi: 10.1161/01.ATV.0000073832.49290.B5. [DOI] [PubMed] [Google Scholar]
- 45.Hristov M. Zernecke A. Bidzhekov K. Liehn EA. Shagdarsuren E. Ludwig A. Weber C. Importance of CXC chemokine receptor 2 in the homing of human peripheral blood endothelial progenitor cells to sites of arterial injury. Circ Res. 2007;100:590–597. doi: 10.1161/01.RES.0000259043.42571.68. [DOI] [PubMed] [Google Scholar]
- 46.Hunt TK. Pai MP. The effect of varying ambient oxygen tensions on wound metabolism and collagen synthesis. Surg Gynecol Obstet. 1972;135:561–567. [PubMed] [Google Scholar]
- 47.Ignarro LJ. Nitric oxide as a unique signaling molecule in the vascular system: a historical overview. J Physiol Pharmacol. 2002;53:503–514. [PubMed] [Google Scholar]
- 48.Ii M. Takenaka H. Asai J. Ibusuki K. Mizukami Y. Maruyama K. Yoon YS. Wecker A. Luedemann C. Eaton E. Silver M. Thorne T. Losordo DW. Endothelial progenitor thrombospondin-1 mediates diabetes-induced delay in reendothelialization following arterial injury. Circ Res. 2006;98:697–704. doi: 10.1161/01.RES.0000209948.50943.ea. [DOI] [PubMed] [Google Scholar]
- 49.Imhof BA. Aurrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nat Rev Immunol. 2004;4:432–444. doi: 10.1038/nri1375. [DOI] [PubMed] [Google Scholar]
- 50.Jin H. Aiyer A. Su J. Borgstrom P. Stupack D. Friedlander M. Varner J. A homing mechanism for bone marrow-derived progenitor cell recruitment to the neovasculature. J Clin Invest. 2006;116:652–662. doi: 10.1172/JCI24751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jonsson K. Jensen JA. Goodson WH., 3rd Scheuenstuhl H. West J. Hopf HW. Hunt TK. Tissue oxygenation, anemia, and perfusion in relation to wound healing in surgical patients. Ann Surg. 1991;214:605–613. doi: 10.1097/00000658-199111000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kalka C. Masuda H. Takahashi T. Kalka-Moll WM. Silver M. Kearney M. Li T. Isner JM. Asahara T. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci U S A. 2000;97:3422–3427. doi: 10.1073/pnas.070046397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kang HJ. Kim HS. Zhang SY. Park KW. Cho HJ. Koo BK. Kim YJ. Soo Lee D. Sohn DW. Han KS. Oh BH. Lee MM. Park YB. Effects of intracoronary infusion of peripheral blood stem-cells mobilised with granulocyte-colony stimulating factor on left ventricular systolic function and restenosis after coronary stenting in myocardial infarction: the MAGIC cell randomised clinical trial. Lancet. 2004;363:751–756. doi: 10.1016/S0140-6736(04)15689-4. [DOI] [PubMed] [Google Scholar]
- 54.Kantor J. Margolis DJ. Management of leg ulcers. Semin Cutan Med Surg. 2003;22:212–221. doi: 10.1016/S1085-5629(03)00043-9. [DOI] [PubMed] [Google Scholar]
- 55.Kaplan RN. Psaila B. Lyden D. Niche-to-niche migration of bone-marrow-derived cells. Trends Mol Med. 2007;13:72–81. doi: 10.1016/j.molmed.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 56.Kaushal S. Amiel GE. Guleserian KJ. Shapira OM. Perry T. Sutherland FW. Rabkin E. Moran AM. Schoen FJ. Atala A. Soker S. Bischoff J. Mayer JE., Jr Functional small-diameter neovessels created using endothelial progenitor cells expanded ex vivo. Nat Med. 2001;7:1035–1040. doi: 10.1038/nm0901-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawachi Y. Watanabe A. Uchida T. Yoshizawa K. Kurooka N. Setsu K. Acute arterial thrombosis due to platelet aggregation in a patient receiving granulocyte colony-stimulating factor. Br J Haematol. 1996;94:413–416. doi: 10.1046/j.1365-2141.1996.d01-1807.x. [DOI] [PubMed] [Google Scholar]
- 58.Kessler L. Bilbault P. Ortega F. Grasso C. Passemard R. Stephan D. Pinget M. Schneider F. Hyperbaric oxygenation accelerates the healing rate of nonischemic chronic diabetic foot ulcers: a prospective randomized study. Diabetes Care. 2003;26:2378–2382. doi: 10.2337/diacare.26.8.2378. [DOI] [PubMed] [Google Scholar]
- 59.Keswani SG. Katz AB. Lim FY. Zoltick P. Radu A. Alaee D. Herlyn M. Crombleholme TM. Adenoviral mediated gene transfer of PDGF-B enhances wound healing in type I and type II diabetic wounds. Wound Repair Regen. 2004;12:497–504. doi: 10.1111/j.1067-1927.2004.12501.x. [DOI] [PubMed] [Google Scholar]
- 60.Khanna S. Roy S. Maurer M. Ratan RR. Sen CK. Oxygen-sensitive reset of hypoxia-inducible factor transactivation response: prolyl hydroxylases tune the biological nor-moxic set point. Free Radic Biol Med. 2006;40:2147–2154. doi: 10.1016/j.freeradbiomed.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Knighton DR. Halliday B. Hunt TK. Oxygen as an antibiotic: the effect of inspired oxygen on infection. Arch Surg. 1984;119:199–204. doi: 10.1001/archsurg.1984.01390140057010. [DOI] [PubMed] [Google Scholar]
- 62.Knighton DR. Hunt TK. Scheuenstuhl H. Halliday BJ. Werb Z. Banda MJ. Oxygen tension regulates the expression of angiogenesis factor by macrophages. Science. 1983;221:1283–2285. doi: 10.1126/science.6612342. [DOI] [PubMed] [Google Scholar]
- 63.Knighton DR. Silver IA. Hunt TK. Regulation of wound-healing angiogenesis: effect of oxygen gradients and inspired oxygen concentration. Surgery. 1981;90:262–270. [PubMed] [Google Scholar]
- 64.Kocher AA. Schuster MD. Bonaros N. Lietz K. Xiang G. Martens TP. Kurlansky PA. Sondermeijer H. Witkowski P. Boyle A. Homma S. Wang SF. Itescu S. Myocardial homing and neovascularization by human bone marrow angioblasts is regulated by IL-8/Gro CXC chemokines. J Mol Cell Cardiol. 2006;40:455–464. doi: 10.1016/j.yjmcc.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 65.Kocher AA. Schuster MD. Szabolcs MJ. Takuma S. Burkhoff D. Wang J. Homma S. Edwards NM. Itescu S. Neo-vascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med. 2001;7:430–436. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- 66.Kopp HG. Ramos CA. Rafii S. Contribution of endothelial progenitors and proangiogenic hematopoietic cells to vascularization of tumor and ischemic tissue. Curr Opin Hematol. 2006;13:175–181. doi: 10.1097/01.moh.0000219664.26528.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.La Fontaine J. Harkless LB. Davis CE. Allen MA. Shireman PK. Current concepts in diabetic microvascular dysfunction. J Am Podiatr Med Assoc. 2006;96:245–252. doi: 10.7547/0960245. [DOI] [PubMed] [Google Scholar]
- 68.Lacza Z. Puskar M. Figueroa JP. Zhang J. Rajapakse N. Busija DW. Mitochondrial nitric oxide synthase is constitutively active and is functionally upregulated in hypoxia. Free Radic Biol Med. 2001;31:1609–1615. doi: 10.1016/s0891-5849(01)00754-7. [DOI] [PubMed] [Google Scholar]
- 69.Lapidot T. Mechanism of human stem cell migration and repopulation of NOD/SCID and B2mnull NOD/SCID mouse: the role of SDF-1/CXCR4 interactions. Ann N Y Acad Sci. 2001;938:83–95. doi: 10.1111/j.1749-6632.2001.tb03577.x. [DOI] [PubMed] [Google Scholar]
- 70.Leung PC. Diabetic foot ulcers: a comprehensive review. Surgeon. 2007;5:219–231. doi: 10.1016/s1479-666x(07)80007-2. [DOI] [PubMed] [Google Scholar]
- 71.Ley K. Laudanna C. Cybulsky MI. Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 72.Li B. Ogasawara AK. Yang R. Wei W. He GW. Zioncheck TF. Bunting S. de Vos AM. Jin H. KDR (VEGF receptor 2) is the major mediator for the hypotensive effect of VEGF. Hypertension. 2002;39:1095–1100. doi: 10.1161/01.hyp.0000018588.56950.7a. [DOI] [PubMed] [Google Scholar]
- 73.Li CQ. Wogan GN. Nitric oxide as a modulator of apoptosis. Cancer Lett. 2005;226:1–15. doi: 10.1016/j.canlet.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 74.Lindemann A. Rumberger B. Vascular complications in patients treated with granulocyte colony-stimulating factor (G-CSF) Eur J Cancer. 1993;29A:2338–2339. doi: 10.1016/0959-8049(93)90236-9. [DOI] [PubMed] [Google Scholar]
- 75.Lindholm C. Bjellerup M. Christensen OB. Zederfeldt B. A demographic survey of leg and foot ulcer patients in a defined population. Acta Derm Venereol. 1992;72:227–230. [PubMed] [Google Scholar]
- 76.Loomans CJ. De Koning EJ. Staal FJ. Rabelink TJ. Zonneveld AJ. Endothelial progenitor cell dysfunction in type 1 diabetes: another consequence of oxidative stress? Antioxid Redox Signal. 2005;7:1468–1475. doi: 10.1089/ars.2005.7.1468. [DOI] [PubMed] [Google Scholar]
- 77.Loomans CJ. de Koning EJ. Staal FJ. Rookmaaker MB. Verseyden C. de Boer HC. Verhaar MC. Braam B. Rabelink TJ. van Zonneveld AJ. Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. 2004;53:195–199. doi: 10.2337/diabetes.53.1.195. [DOI] [PubMed] [Google Scholar]
- 78.Luster AD. Alon R. von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- 79.Lyden D. Hattori K. Dias S. Costa C. Blaikie P. Butros L. Chadburn A. Heissig B. Marks W. Witte L. Wu Y. Hicklin D. Zhu Z. Hackett NR. Crystal RG. Moore MA. Hajjar KA. Manova K. Benezra R. Rafii S. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 80.MacMicking J. Xie QW. Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 81.Majka SM. Jackson KA. Kienstra KA. Majesky MW. Goodell MA. Hirschi KK. Distinct progenitor populations in skeletal muscle are bone marrow derived and exhibit different cell fates during vascular regeneration. J Clin Invest. 2003;111:71–79. doi: 10.1172/JCI16157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marletta MA. Hurshman AR. Rusche KM. Catalysis by nitric oxide synthase. Curr Opin Chem Biol. 1998;2:656–663. doi: 10.1016/s1367-5931(98)80098-7. [DOI] [PubMed] [Google Scholar]
- 83.Marston WA. Hanft J. Norwood P. Pollak R. The efficacy and safety of Dermagraft in improving the healing of chronic diabetic foot ulcers: results of a prospective randomized trial. Diabetes Care. 2003;26:1701–1705. doi: 10.2337/diacare.26.6.1701. [DOI] [PubMed] [Google Scholar]
- 84.Maulik N. Engelman DT. Watanabe M. Engelman RM. Rousou JA. Flack JE., 3rd Deaton DW. Gorbunov NV. Elsayed NM. Kagan VE. Das DK. Nitric oxide/carbon monoxide: a molecular switch for myocardial preservation during ischemia. Circulation. 1996;94:II398–II406. [PubMed] [Google Scholar]
- 85.Michel T. Feron O. Nitric oxide synthases: which, where, how, and why? J Clin Invest. 1997;100:2146–2152. doi: 10.1172/JCI119750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moldovan L. Mythreye K. Goldschmidt-Clermont PJ. Satterwhite LL. Reactive oxygen species in vascular endothelial cell motility: roles of NAD(P)H oxidase and Rac1. Cardiovasc Res. 2006;71:236–246. doi: 10.1016/j.cardiores.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 87.Morimoto A. Sakata Y. Watanabe T. Murakami N. Leucocytosis induced in rabbits by intravenous or central injection of granulocyte colony stimulating factor. J Physiol. 1990;426:117–126. doi: 10.1113/jphysiol.1990.sp018129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Muller WA. Leukocyte-endothelial cell interactions in the inflammatory response. Lab Invest. 2002;82:521–533. doi: 10.1038/labinvest.3780446. [DOI] [PubMed] [Google Scholar]
- 89.Nagano M. Yamashita T. Hamada H. Ohneda K. Kimura K. Nakagawa T. Shibuya M. Yoshikawa H. Ohneda O. Identification of functional endothelial progenitor cells suitable for the treatment of ischemic tissue using human umbilical cord blood. Blood. 2007;110:151–160. doi: 10.1182/blood-2006-10-047092. [DOI] [PubMed] [Google Scholar]
- 90.Nakamura Y. Tajima F. Ishiga K. Yamazaki H. Oshimura M. Shiota G. Murawaki Y. Soluble c-kit receptor mobilizes hematopoietic stem cells to peripheral blood in mice. Exp Hematol. 2004;32:390–396. doi: 10.1016/j.exphem.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 91.Ngo BT. Hayes KD. DiMiao DJ. Srinivasan SK. Huerter CJ. Rendell MS. Manifestations of cutaneous diabetic microangiopathy. Am J Clin Dermatol. 2005;6:225–237. doi: 10.2165/00128071-200506040-00003. [DOI] [PubMed] [Google Scholar]
- 92.Nishikawa SI. Nishikawa S. Hirashima M. Matsuyoshi N. Kodama H. Progressive lineage analysis by cell sorting and culture identifies FLK1+VE-cadherin+ cells at a diverging point of endothelial and hemopoietic lineages. Development. 1998;125:1747–1757. doi: 10.1242/dev.125.9.1747. [DOI] [PubMed] [Google Scholar]
- 93.Orlic D. Kajstura J. Chimenti S. Bodine DM. Leri A. Anversa P. Transplanted adult bone marrow cells repair myocardial infarcts in mice. Ann N Y Acad Sci. 2001;938:221–229. doi: 10.1111/j.1749-6632.2001.tb03592.x. discussion 229–230. [DOI] [PubMed] [Google Scholar]
- 94.Orlic D. Kajstura J. Chimenti S. Jakoniuk I. Anderson SM. Li B. Pickel J. McKay R. Nadal-Ginard B. Bodine DM. Leri A. Anversa P. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–705. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 95.Otani A. Kinder K. Ewalt K. Otero FJ. Schimmel P. Friedlander M. Bone marrow-derived stem cells target retinal astrocytes and can promote or inhibit retinal angiogenesis. Nat Med. 2002;8:1004–1010. doi: 10.1038/nm744. [DOI] [PubMed] [Google Scholar]
- 96.Palumbo R. Bianchi ME. High mobility group box 1 protein, a cue for stem cell recruitment. Biochem Pharmacol. 2004;68:1165–1170. doi: 10.1016/j.bcp.2004.03.048. [DOI] [PubMed] [Google Scholar]
- 97.Park IK. Qian D. Kiel M. Becker MW. Pihalja M. Weissman IL. Morrison SJ. Clarke MF. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 98.Phillips TJ. Chronic cutaneous ulcers: etiology and epidemiology. J Invest Dermatol. 1994;102:38S–41S. doi: 10.1111/1523-1747.ep12388556. [DOI] [PubMed] [Google Scholar]
- 99.Powell TM. Paul JD. Hill JM. Thompson M. Benjamin M. Rodrigo M. McCoy JP. Read EJ. Khuu HM. Leitman SF. Finkel T. Cannon RO., 3rd Granulocyte colony-stimulating factor mobilizes functional endothelial progenitor cells in patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2005;25:296–301. doi: 10.1161/01.ATV.0000151690.43777.e4. [DOI] [PubMed] [Google Scholar]
- 100.Rafii S. Avecilla S. Shmelkov S. Shido K. Tejada R. Moore MA. Heissig B. Hattori K. Angiogenic factors reconstitute hematopoiesis by recruiting stem cells from bone marrow microenvironment. Ann N Y Acad Sci. 2003;996:49–60. doi: 10.1111/j.1749-6632.2003.tb03232.x. [DOI] [PubMed] [Google Scholar]
- 101.Rafii S. Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med. 2003;9:702–712. doi: 10.1038/nm0603-702. [DOI] [PubMed] [Google Scholar]
- 102.Rafii S. Meeus S. Dias S. Hattori K. Heissig B. Shmelkov S. Rafii D. Lyden D. Contribution of marrow-derived progenitors to vascular and cardiac regeneration. Semin Cell Dev Biol. 2002;13:61–67. doi: 10.1006/scdb.2001.0285. [DOI] [PubMed] [Google Scholar]
- 103.Rathur HM. Boulton AJ. The diabetic foot. Clin Dermatol. 2007;25:109–120. doi: 10.1016/j.clindermatol.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 104.Rehman J. Li J. Parvathaneni L. Karlsson G. Panchal VR. Temm CJ. Mahenthiran J. March KL. Exercise acutely increases circulating endothelial progenitor cells and monocyte-/macrophage-derived angiogenic cells. J Am Coll Cardiol. 2004;43:2314–2318. doi: 10.1016/j.jacc.2004.02.049. [DOI] [PubMed] [Google Scholar]
- 105.Reiber GE. Vileikyte L. Boyko EJ. del Aguila M. Smith DG. Lavery LA. Boulton AJ. Causal pathways for incident lower-extremity ulcers in patients with diabetes from two settings. Diabetes Care. 1999;22:157–162. doi: 10.2337/diacare.22.1.157. [DOI] [PubMed] [Google Scholar]
- 106.Reyes M. Dudek A. Jahagirdar B. Koodie L. Marker PH. Verfaillie CM. Origin of endothelial progenitors in human postnatal bone marrow. J Clin Invest. 2002;109:337–346. doi: 10.1172/JCI14327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ruzinova MB. Schoer RA. Gerald W. Egan JE. Pandolfi PP. Rafii S. Manova K. Mittal V. Benezra R. Effect of angiogenesis inhibition by Id loss and the contribution of bone-marrow-derived endothelial cells in spontaneous murine tumors. Cancer Cell. 2003;4:277–289. doi: 10.1016/s1535-6108(03)00240-x. [DOI] [PubMed] [Google Scholar]
- 108.Salven P. Mustjoki S. Alitalo R. Alitalo K. Rafii S. VEGFR-3 and CD133 identify a population of CD34+ lymphatic/vascular endothelial precursor cells. Blood. 2003;101:168–172. doi: 10.1182/blood-2002-03-0755. [DOI] [PubMed] [Google Scholar]
- 109.Sata M. Saiura A. Kunisato A. Tojo A. Okada S. Tokuhisa T. Hirai H. Makuuchi M. Hirata Y. Nagai R. Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat Med. 2002;8:403–409. doi: 10.1038/nm0402-403. [DOI] [PubMed] [Google Scholar]
- 110.Schafer G. Cramer T. Suske G. Kemmner W. Wiedenmann B. Hocker M. Oxidative stress regulates vascular endothelial growth factor-A gene transcription through Sp1- and Sp3-dependent activation of two proximal GC-rich promoter elements. J Biol Chem. 2003;278:8190–8198. doi: 10.1074/jbc.M211999200. [DOI] [PubMed] [Google Scholar]
- 111.Sen CK. Khanna S. Babior BM. Hunt TK. Ellison EC. Roy S. Oxidant-induced vascular endothelial growth factor expression in human keratinocytes and cutaneous wound healing. J Biol Chem. 2002;277:33284–33290. doi: 10.1074/jbc.M203391200. [DOI] [PubMed] [Google Scholar]
- 112.Shandall A. Lowndes R. Young HL. Colonic anastomotic healing and oxygen tension. Br J Surg. 1985;72:606–609. doi: 10.1002/bjs.1800720808. [DOI] [PubMed] [Google Scholar]
- 113.Sheikh AY. Gibson JJ. Rollins MD. Hopf HW. Hussain Z. Hunt TK. Effect of hyperoxia on vascular endothelial growth factor levels in a wound model. Arch Surg. 2000;135:1293–1297. doi: 10.1001/archsurg.135.11.1293. [DOI] [PubMed] [Google Scholar]
- 114.Shen JT. Falanga V. Innovative therapies in wound healing. J Cutan Med Surg. 2003;7:217–224. doi: 10.1007/s10227-002-0106-5. [DOI] [PubMed] [Google Scholar]
- 115.Shi Q. Rafii S. Wu MH. Wijelath ES. Yu C. Ishida A. Fujita Y. Kothari S. Mohle R. Sauvage LR. Moore MA. Storb RF. Hammond WP. Evidence for circulating bone marrow-derived endothelial cells. Blood. 1998;92:362–367. [PubMed] [Google Scholar]
- 116.Shintani S. Murohara T. Ikeda H. Ueno T. Honma T. Katoh A. Sasaki K. Shimada T. Oike Y. Imaizumi T. Mobilization of endothelial progenitor cells in patients with acute myocardial infarction. Circulation. 2001;103:2776–2779. doi: 10.1161/hc2301.092122. [DOI] [PubMed] [Google Scholar]
- 117.Singer AJ. Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 118.Smiell JM. Clinical safety of becaplermin (rhPDGF-BB) gel: Becaplermin Studies Group. Am J Surg. 1998;176:68S–73S. doi: 10.1016/s0002-9610(98)00174-3. [DOI] [PubMed] [Google Scholar]
- 119.Stamm C. Westphal B. Kleine HD. Petzsch M. Kittner C. Klinge H. Schumichen C. Nienaber CA. Freund M. Steinhoff G. Autologous bone-marrow stem-cell transplantation for myocardial regeneration. Lancet. 2003;361:45–46. doi: 10.1016/S0140-6736(03)12110-1. [DOI] [PubMed] [Google Scholar]
- 120.Sumpio BE. Lee T. Blume PA. Vascular evaluation and arterial reconstruction of the diabetic foot. Clin Podiatr Med Surg. 2003;20:689–708. doi: 10.1016/S0891-8422(03)00088-0. [DOI] [PubMed] [Google Scholar]
- 121.Sweitzer SM. Fann SA. Borg TK. Baynes JW. Yost MJ. What is the future of diabetic wound care? Diabetes Educ. 2006;32:197–210. doi: 10.1177/0145721706286897. [DOI] [PubMed] [Google Scholar]
- 122.Takahashi T. Kalka C. Masuda H. Chen D. Silver M. Kearney M. Magner M. Isner JM. Asahara T. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5:434–438. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 123.Tam M. Moschella SL. Vascular skin ulcers of limbs. Cardiol Clin. 1991;9:555–563. [PubMed] [Google Scholar]
- 124.Tepper OM. Capla JM. Galiano RD. Ceradini DJ. Callaghan MJ. Kleinman ME. Gurtner GC. Adult vasculogenesis occurs through in situ recruitment, proliferation, and tubulization of circulating bone marrow-derived cells. Blood. 2005;105:1068–1077. doi: 10.1182/blood-2004-03-1051. [DOI] [PubMed] [Google Scholar]
- 125.Tepper OM. Galiano RD. Capla JM. Kalka C. Gagne PJ. Jacobowitz GR. Levine JP. Gurtner GC. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–2786. doi: 10.1161/01.cir.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- 126.Thom SR. Effects of hyperoxia on neutrophil adhesion. Undersea Hyperb Med. 2004;31:123–131. [PubMed] [Google Scholar]
- 127.Thom SR. Bhopale V. Fisher D. Manevich Y. Huang PL. Buerk DG. Stimulation of nitric oxide synthase in cerebral cortex due to elevated partial pressures of oxygen: an oxidative stress response. J Neurobiol. 2002;51:85–100. doi: 10.1002/neu.10044. [DOI] [PubMed] [Google Scholar]
- 128.Thom SR. Bhopale VM. Velazquez OC. Goldstein LJ. Thom LH. Buerk DG. Stem cell mobilization by hyperbaric oxygen. Am J Physiol Heart Circ Physiol. 2006;290:H1378–H1386. doi: 10.1152/ajpheart.00888.2005. [DOI] [PubMed] [Google Scholar]
- 129.Thom SR. Fisher D. Zhang J. Bhopale VM. Ohnishi ST. Kotake Y. Ohnishi T. Buerk DG. Stimulation of perivascular nitric oxide synthesis by oxygen. Am J Physiol Heart Circ Physiol. 2003;284:H1230–H1239. doi: 10.1152/ajpheart.01043.2002. [DOI] [PubMed] [Google Scholar]
- 130.Tolcher AW. Giusti RM. O'Shaughnessy JA. Cowan KH. Arterial thrombosis associated with granulocyte-macrophage colony-stimulating factor (GM-CSF) administration in breast cancer patients treated with dose-intensive chemotherapy: a report of two cases. Cancer Invest. 1995;13:188–192. doi: 10.3109/07357909509011689. [DOI] [PubMed] [Google Scholar]
- 131.Uhl E. Sirsjo A. Haapaniemi T. Nilsson G. Nylander G. Hyperbaric oxygen improves wound healing in normal and ischemic skin tissue. Plast Reconstr Surg. 1994;93:835–841. doi: 10.1097/00006534-199404000-00028. [DOI] [PubMed] [Google Scholar]
- 132.Vasa M. Fichtlscherer S. Aicher A. Adler K. Urbich C. Martin H. Zeiher AM. Dimmeler S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:E1–E7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- 133.Verma S. Kuliszewski MA. Li SH. Szmitko PE. Zucco L. Wang CH. Badiwala MV. Mickle DA. Weisel RD. Fedak PW. Stewart DJ. Kutryk MJ. C-reactive protein attenuates endothelial progenitor cell survival, differentiation, and function: further evidence of a mechanistic link between C-reactive protein and cardiovascular disease. Circulation. 2004;109:2058–2067. doi: 10.1161/01.CIR.0000127577.63323.24. [DOI] [PubMed] [Google Scholar]
- 134.Walter DH. Haendeler J. Reinhold J. Rochwalsky U. Seeger F. Honold J. Hoffmann J. Urbich C. Lehmann R. Arenzana-Seisdesdos F. Aicher A. Heeschen C. Fichtlscherer S. Zeiher AM. Dimmeler S. Impaired CXCR4 signaling contributes to the reduced neovascularization capacity of endothelial progenitor cells from patients with coronary artery disease. Circ Res. 2005;97:1142–1151. doi: 10.1161/01.RES.0000193596.94936.2c. [DOI] [PubMed] [Google Scholar]
- 135.Wild S. Roglic G. Green A. Sicree R. King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 136.Willert K. Brown JD. Danenberg E. Duncan AW. Weissman IL. Reya T. Yates JR., 3rd Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 137.Wu SC. Driver VR. Wrobel JS. Armstrong DG. Foot ulcers in the diabetic patient: prevention and treatment. Vasc Health Risk Manag. 2007;3:65–76. [PMC free article] [PubMed] [Google Scholar]
- 138.Wysocki AB. Wound fluids and the pathogenesis of chronic wounds. J Wound Ostomy Continence Nurs. 1996;23:283–290. doi: 10.1016/s1071-5754(96)90047-9. [DOI] [PubMed] [Google Scholar]
- 139.Yoder MC. Mead LE. Prater D. Krier TR. Mroueh KN. Li F. Krasich R. Temm CJ. Prchal JT. Ingram DA. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109:1801–1809. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Young PP. Hofling AA. Sands MS. VEGF increases engraftment of bone marrow-derived endothelial progenitor cells (EPCs) into vasculature of newborn murine recipients. Proc Natl Acad Sci U S A. 2002;99:11951–11956. doi: 10.1073/pnas.182215799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zamboni WA. Wong HP. Stephenson LL. Pfeifer MA. Evaluation of hyperbaric oxygen for diabetic wounds: a prospective study. Undersea Hyperb Med. 1997;24:175–179. [PubMed] [Google Scholar]