

Abstract
Vascular progenitor cells derived from stem cells could potentially lead to a variety of clinically relevant applications including cell-based therapies and tissue engineering. Here we describe methods for isolating purified proliferating populations of vascular endothelial cells from mouse embryonic stem cells (mESC) using Flk-1 positive sorted cells, VEGF supplementation, and a rigorous manual selection technique required for endothelial cell purification and expansion. Using this in vitro derivation procedure, it is possible to obtained millions of cells at various stages of differentiation, with the potential for up to 25 population doublings.
Keywords: embryonic stem cells, endothelial cells, endothelial progenitor cells, vascular progenitor cells, Flk-1, VEGF
INTRODUCTION
Vascular endothelial cells or endothelial progenitor cells derived from stem cells could potentially lead to a variety of clinically relevant applications (Dzau et al., 2005). These cells could be used in therapeutic strategies for the repair and revascularization of ischemic tissue in patients exhibiting vascular defects (Kalka et al., 2000; Soker et al., 2000). Endothelial progenitor cell transplantation has been shown to induce new vessel formation in ischemic myocardium and hind limb (Kalka et al., 2000; Kawamoto et al., 2001; Kocher et al., 2001). Since it is well known that endothelial cells inhibit platelet adhesion and clotting, they are needed for lining the lumen of a synthetic or tissue-engineered vascular graft or for re-endothelization of injured vessels (Kaushal et al., 2001; Griese et al., 2003). Moreover, because endothelial cells line the lumen of blood vessels and can release proteins directly into the blood stream, they are ideal candidates to be used as vehicles of gene therapy. Endothelial cells may also be used for vascularizing tissue-engineered materials prior to implantation and for investigating mechanisms of angiogenesis and vasculogenesis.
One potential source for these therapeutic endothelial cells is the embryonic stem cell (ESC). The ESC possesses some advantages over adult stem cells in that the ESC provide an excellent in vitro culture system for studying cellular differentiation events, and because the ESC is thought to have the capacity for an unlimited number of cell divisions, it may retain greater potential for in vitro expansion of large numbers of tissue-specific cells.
The methodology presented in this unit expands on the work of Nishikawa's group (Yamashita et al. 2000; Nishikawa et al., 1998, 2001a) for the in vitro differentiation and purification (>96% pure) of EC populations from mouse ESC (McCloskey et al., 2003). These ESC-derived endothelial cells display characteristics of the vascular endothelial cell in that they express several endothelial markers (McCloskey et al., 2003), and they form two-dimensional tube-like structures, as well as complex vessel-like structures in three-dimensional collagen type-I gels (McCloskey et al., 2005).
In this unit, we will describe detailed protocols for derivation of EC from mouse ESC (for both R1 and D3 cell lines). We will also provide a second protocol for the 2) maintenance and initial differentiation of EC from mouse ESC under serum-free conditions. Lastly, we will 3) review current methods of EC differentiation from human ESC.
BASIC PROTOCOL
ENDOTHELIAL CELL DIFFERENTIATION FROM MOUSE ESC
This first section will present detailed methods for isolating purified proliferating populations of endothelial cells from mouse embryonic stem cells using a 2-D induction on collagen IV, followed by sorting of the Flk-1+ cells that are generated, VEGF supplementation, and a second, more rigorous manual selection technique for isolation of highly purified populations of EC. Using this in vitro derivation procedure, large numbers of endothelial cells can be expanded for up to 25 population doublings.
The ESC culturing methods described here provide approximately 106 ESC per 35-mm dish at confluence. These small dishes are maintained due to the expense of reagents however, if larger numbers of cells are desired, this protocol may be scaled up proportionally keeping constant the cell seeding density (the number of cells per cm2). Generalized protocols for freezing, thawing, and of cells used in these experiments—feeder cells, ESC, EC—are provided in Support Protocols 1, 2, and 3.
Materialst
ESC Cell Culture
ES-D3 or ES-R1 cells (American Type Culture Collection; cat. No. CRL-1934 or SCRC-1036)
Primary mouse embryonic fibroblasts or STO cells (American Type Culture Collection, Manassas, VA; cat. no. CRL-1503).
Dulbecco's Phosphate Buffer Solution (PBS) (500-ml; Gibco, Carlsbad, CA; cat. no. 14190−144).
Gelatin (Sigma, Milwaukee, WI, cat. no. G-1890).
35-mm polystyrene cell culture dishes (Corning, Acton, MA; cat. no. 430165).
Trypsin/EDTA (100-ml; Gibco, cat. no. 25300−054).
15-ml centrifuge tubes (VWR, West Chester, PA, cat. no. 21008−103).
T-175 cell culture flasks (tissue culture treated with 0.2-mm vent cap; Corning; cat. no. 431080).
Nunc Cryotube vials (VWR; cat. no. 66021−986).
Nalgene Cryo 1C freezing containers (Research Products International, Mt. Prospect, IL; cat. no. 5100−0001).
Mitomycin-C (2-mg, Sigma, cat. no. M4287). Mytomycin C solution: dissolve 2.0mg of mytomycin C powder in 200ml of Fibroblast Feeder Cell Medium (10μg/ml). Stock may be stored in the dark at 4°C for up to 6 weeks, or −20°C for long term storage.
Dulbecco's Modified Eagle Medium (DMEM)-High Glucose (Gibco, cat. no. 119650−092).
Fetal Bovine Serum (FBS)-Heat Inactivated (500-ml; Cellgro, Herndon, VA; cat. no. 35−001-CV).
Penicillin/Streptomycin (from 100 stock) (100-ml; Gibco, cat. no. 15070−063).
L-Glutamine (from 100 stock) (100-ml; Gibco, cat. no. 25030−081)
Knockout Dulbecco's Modified Eagle Medium (KO-DMEM) (500-ml; Gibco, cat. no. 10829−018).
Embryonic Stem (ES) Cell Qualified Fetal Bovine Serum (500-ml; Gibco, cat. no. 16141−079).
Knockout Serum Replacement (500-ml; Gibco, cat. no. 10828−028).
Leukemia inhibitory factor (106 units; Chemicon International, Temecula, CA; cat. no. ESG1106).
β-mercaptoethanol (250-ml; Sigma, cat. no. M-7522).
MEM Nonessential amino acids (100-ml; Gibco, cat. no. 11140−050).
Dimethyl Sulphoxide (DMSO) Hybri-Max (100-ml, Sigma, cat. no. D2650).
ESC-to-EC cell differentiation
Collagen IV Biocoat Cellware 35-mm dishes (Becton-Dickinson, Franklin Lakes, NJ; cat. no. 354459).
Alpha-Minimal Essential Medium (500-ml; Gibco, cat. no.12561−056).
Fetal Bovine Serum (FBS)-Heat Inactivated (500-ml; Cellgro, cat. no. 35−001-CV).
β-mercaptoethanol (250-ml; Sigma, cat. no. M-7522).
Penicillin/Streptomycin (from 100 stock) (100-ml; Gibco, cat. no. 15070−063).
L-Glutamine (from 100 stock) (100-ml bottle; Gibco, cat. no. 25030−081).
Purification of vascular progenitor cells
Cell Dissociation Solution (100-ml; Sigma, cat. no. C-5914).
Fetal Bovine Serum (FBS)-Heat Inactivated (500-ml; Cellgro, cat. no. 35−001-CV).
Dulbecco's Phosphate Buffer Solution (PBS) (500-ml; Gibco, cat. no. 14190−144).
Bovine Albumin Cohn-Analog (25-g, Sigma, cat. no. A-1470). Add 0.6g of Bovine albumin to 200ml PBS to make 0.3% BSA buffer solution. Place this mixture in the water bath until the albumin is dissolved (sterile filter the solution if needed).
Rabbit anti-mouse Flk-1 (Alpha Diagnostic International, San Antonio, TX; cat. no. FLK11-A).
Donkey anti-rabbit Phycoerythrin (PE)-conjugated (Research Diagnostics Inc, Flanders, NJ; cat. no. RDI-711116152).
Normal donkey serum (10-ml, Research Diagnostics Inc, cat. no. RDI-NSDNKY).
5-ml polystyrene round bottom tubes (sterile, Becton-Dickinson, cat. no. 352058).
Collagen IV Biocoat Cellware 35-mm dishes (Becton-Dickinson, cat. no. 354459).
Recombinant human vascular endothelial growth factor (VEGF165) (R&D Systems, Minneapolis, MN; cat. no. 293-VE).
Purification and expansion of ECs from vascular progenitor cells
Aspirator assembly (5-pk; Sigma; cat. no. A5177).
Acrodisc syringe filter (0.2 μm membrane; Pall, East Hills, NY; cat. no. 4192).
Glass pastuer pipettes (9”, VWR; cat. no. 53283−915).
Pippette tips (200−1000 μl, autoclavable, VWR; cat. no. 53508−819).
Cell Dissociation Solution (100-ml bottle; Sigma; cat. no. C-5914).
EGM-2 medium Bullet Kit (500-ml bottle plus growth factors; Clonetics, East Rutherford, NJ; cat. no. CC-3162).
Recombinant human vascular endothelial growth factor (VEGF165) (R&D Systems, cat. no. 293-VE).
Collagen IV (1mg; Becton-Dickinson; cat. no. 354233).
Collagen I (100mg; Becton-Dickinson; cat. no. 354236).
Fibronectin (2mg; Sigma; cat. no. F-1141).
Gelatin (Sigma; cat. no. G-1890).
General equipment
Water bath at 37°C.
Coulter counter or hemacytometer for cell counting.
Inverted microscope with 5× and 10× objectives.
Humidified incubator at 37°C and 5% CO2.
Laminar flow cabinet.
Liquid nitrogen storage tank.
Micropipettes and tips (10-, 20-, 100-, 200-, and 1000μl).
Clean hood.
Stereomicroscope.
Disposable pipettes (1-, 5-, 10-, 25-, and 50ml).
Refrigerator (4°C) and freezer (−20°C and −80°C).
Tabletop centrifuge.
Culture ESC
Thaw D3-ES cells (Support Protocol 2) making sure to use Mouse Embryonic Stem Cell Medium (ESC medium).
Plate 1×105 to 5×105 D3-ES cells per fibroblast feeder cell-coated 35-mm dish with 2.5 ml of ESC Medium.
Replace culture medium daily.
Subculture ESC
4. Subculture the cells before colonies begin to touch.
If 2 ×105 cells per 35-mm dish are plated, they will need to be subcultured in 3 days.
ES cells maintain their undifferentiated state best when the colonies are subcultured before the colonies contact with other colonies (Fig. 1F.5.1)
Figure 1F.5.1.
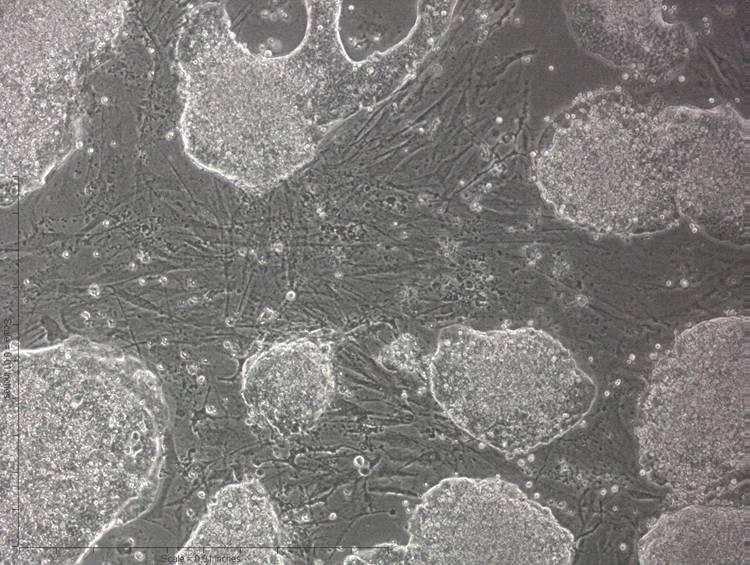
Mouse ESC colonies on embryonic fibroblast feeder cells. Note that, in general, the ESC colonies are not in contact with one another and should be subcultured well before colonies begin to contact one another. This figure shows what is considered a “confluent” dish. These cells should be subcultured within 24 hr.
5. Gently remove culture medium and rinse cells twice with 3 ml of PBS per 35-mm dish.
6. Add 1ml of trypsin/EDTA per 35-mm dish and place cells in the incubator for 1 to 2 min.
7. Gently pipet cells up and down 10 times to disaggregate cells.
8. Add 3 ml of mouse ESC medium and transfer all of the cell suspension to a 15-ml centrifuge tube.
9. Add 3−5 ml more of ESC medium to completely neutralize the trypsin.
10. Again, gently pipet cells up and down 10−30 more times to disaggregate cells.
The goal is to obtain a single-cell suspension and fibroblasts tend to stick together. Therefore, allow 2 min for the large cell clumps to sink to the bottom of the centrifuge tube and transfer the top ¾ of the cell suspension to another centrifuge tube and discard the fibroblast cell clumps in the first tube. This technique also ensures that fewer fibroblasts are subcultured in the next dish.
11. Count cells (UNIT 1C.3).
12. Centrifuge at 200 × g for 4−5 min at room temperature.
13. Remove supernatant.
14. Resuspend pellet in appropriate quantities of mouse ESC medium and replate at 1×105 to 5×105 D3-ES cells per fibroblast feeder cell-coated 35-mm dish with 2.5 ml ESC medium per dish.
Collect ESC
15. Gently remove culture medium and rinse cells twice with 3 ml of PBS per 35-mm dish.
16. Add 1 ml of trypsin/EDTA per 35-mm dish and place cells in the incubator for 1 to 2 min.
17. Gently pipet cells up and down 10 times to disaggregate cells.
18. Add 3 ml of ESC-to-EC differentiating medium and transfer all of the cell suspension to a 15-ml centrifuge tube.
19. Add 3−5 ml more of ESC-to-EC differentiating medium to completely neutralize the trypsin.
20. Again, gently pipet cells up and down 10−30 more times to disaggregate cells.
21. Centrifuge at 200 × g for 4−5 min at room temperature.
22. Remove supernatant.
Replate cells for differentiation
The cells are subcultured on 0.1% gelatin (no feeders) for 3−6 days before switching to differentiation conditions. This allows expansion of the embryonic stem cells, while minimizing the number of feeder cells in the culture.
23. Resuspend pellet in 1 ml of ESC-to-EC differentiating medium and gently pipet cells up and down 10−30 more times to disaggregate cells.
24. Count cells.
25. Add 2.5 ml of ESC-to-EC differentiating medium each to 2−4 Collagen IV Biocoat Cellware 35-mm dishes.
26. Add 30,000 cells (calculated volume) to each 35-mm collagen IV-coated dish.
27. Incubate at 37°C and 5% CO2 for 4 days. Do not change culture medium during these 3 to 4 days.
Collect Flk-1+ vascular progenitor cells
After 3 to 4 days of differentiation, the ESC will consist of a heterogeous mixture of progenitor cells. When ESC begin to differentiate, they will lose typical 3-D colony appearance and begin to grow more like monolayer cell cultures (Fig.1F.5.2). Included in the mixture will be a population of Flk-1 expressing cells that are vascular progenitor cells and blood precursor cells (for discussion see refs. (Hirashima et al., 1999; Nishikawa et al., 1998, 2001a; Yamashita et al., 2000; McCloskey et al., 2003). Using flow cytometry, the brightest Flk-1 expressing cells can be isolated from the heterogeneous mixture of cells.
Figure 1F.5.2.
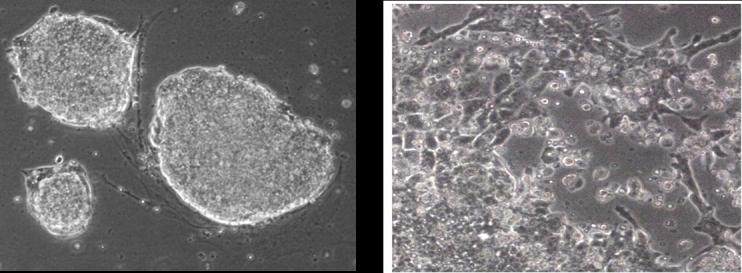
Mouse ESC colonies on gelatin (left). Mouse ES cells after 3−4 days of differentiation on collagen type-IV (right). Note the distinct changes in morphology between undifferentiated ES cells and differentiated ES cells.
28. Remove culture medium and wash cells twice with 3 ml of PBS per 35-mm dish.
29. Add 3 ml of cell dissociation solution to each dish and allow cells to incubate at 37°C for 20 to 30 min.
When staining cells for extracellular surface markers, it is very important to use a non-enzymatic method for removing the cells from the culture dishes, therefore, do not use trypsin when staining cells. Trypsin will degrade the surface markers that you are attempting to stain.
30. Pipet up and down 10 times while washing solution over the bottom of the dish to remove all the cells. If some cells are still adhering to the bottom of the dish, then use a cell scraper to remove the remaining cells.
31. Transfer cells to a 15-ml centrifuge tube and add 3 ml of FBS.
32. At this stage, pool up to five 35-mm dishes of cells for staining and sorting.
33. Centrifuge cells at 200 ×g for 4−5 min at room temperature.
The cells are now ready for immunostaining.
Take care to keep the cells sterile during the entire staining and sorting procedure. All solutions for staining will be kept at 4°C or on ice.
Stain cells for sorting
34. Chill the buffer solution at 4°C or keep buffer solution on ice.
35. Remove supernatant from the cell pellet.
36. Resuspend the entire cell pellet in 1 ml of BSA buffer solution/10% donkey serum. Vortex gently.
37. Incubate on ice or at 4°C for 1 hr.
38. Add 4 ml of BSA buffer solution and centrifuge at 200 × g for 4−5 min. Vortex gently.
39. Remove supernatant and resuspend the cell pellet in 400 μl of BSA buffer solution. Pipet up and down to evenly distribute the cells in the solution.
Expose cells to Flk-1 antibody
40. Place 50 μl of cell suspension in another 15-ml centrifuge tube and label it “cells only”.
41. Place 50 μl of cell suspension in another 15-ml centrifuge tube and label it “PE only” (or use an IgG PE isotype control).
42. Label the original cell suspension “Flk-1 PE”.
43. Add 250 μl of BSA buffer to the two new centrifuge tubes.
All tubes should now be at 300 μl.
44. Add 8 μl of Flk-1 antibody to the tube labeled “Flk-1 PE”.
45. Incubate all tubes on ice or at 4°C for 30 min.
46. Add 4 ml of BSA buffer solution to all tubes and centrifuge at 200 × g for 4−5 min at 4°C.
47. Remove supernatant and resuspend the cell pellets in 300 μl of BSA buffer solution each. Pipet up and down to evenly distribute the cells in the solution.
Expose cells to secondary antibody
48.Add 8 μl of donkey anti-rabbit PE to the tube labeled “Flk-1 PE” and the tube labeled “PE only”. Alternatively, use an IgG PE isotype control.
Fluorescent antibodies should be kept in the dark during storage and when labeling cells. Exposure to too much light may cause the fluorescent molecules to emit light prematurely.
49. Incubate all tubes on ice or at 4°C for 30 min.
50. Add 4 ml of BSA buffer solution to all tubes and centrifuge at 200 × g for 4−5 min at 4°C.
51. Remove supernatant and repeat step 60.
Sort the cells
52. Resuspend the cells in “Flk-1 PE” tube in 1 ml of BSA buffer and transfer the cell solution to a labeled 5-ml round bottomed polystyrene FACS tube.
53. Resuspend the cells in “cells only” and “PE only” tubes in 300 μl of BSA buffer and transfer the cell solutions to labeled 5-ml round bottomed polystyrene FACS tubes.
54. Fill a 4th 5-ml round bottomed polystyrene FACS tube with 1.5 ml of ESC-to-EC differentiating medium.
This tube will serve as your “collection” tube for flow activated cell sorting (FACS).
55. Sort the “brightest” population of Flk-1 expressing cells into the “collection” tube. Usually there will be a subpopulation of cells that is expressing a very high number of Flk-1 surface molecules. This population will be the “brightest” population of cells falling in the highest channels of your FACS histogram. This population of cells typically ranges from 10−30% of your total cell population (Fig. 1F.5.3).
Figure 1F.5.3.
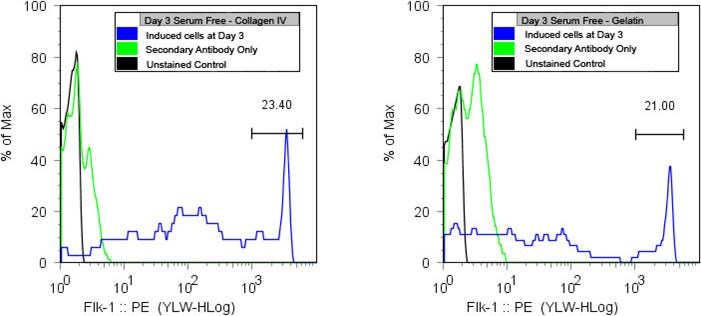
R1 ESC exhibit high Flk-1 expression after 3 days on collagen type-IV or gelatin. These are the vascular progenitor cells that will be isolated from the heterogeneous mixture of cells.
Plate and culture the Flk-1+ cells
56. Centrifuge the “collection” tube containing Flk-1 positive cells at 200 × g for 3−4 min at room temperature.
57. Remove supernatant and resuspend cells in 1 ml of ESC-to-EC differentiating medium. Based on number of cell sorting events, calculate the volume of cell suspension to add to each 35-mm collagen IV coated dish.
You will want approximately 50,000−100,000 cells per dish.
58. Fill each dish to a total volume of 2.5 ml of ESC-to-EC differentiating medium.
59. Add 125 μl of VEGF (50 ng/ml) to each dish.
60. Put the cells in a 37°C incubator and do not move dishes for 4 days.
61. On day 4, aspirate off old medium and add 2.5 ml fresh ESC-to-EC differentiating medium plus 125 μl of VEGF per 35-mm dish.
Most of these cells will die due to staining and FACS sorting procedures. Do not move the dishes or change medium for 4 days and then allow at least 1 week before expecting to see any cell growth.
Purify ECs from vascular progenitor cells
After culturing for approximately 1 week, the Flk-1 positive cell outgrowths exhibit predominantly two different morphologies (see Fig. 1F.5.4). These include endothelial-like cells with a cobblestone morphology, and elongated smooth muscle-like cell populations. Since these two populations are distinctly different in appearance, it is possible to manually isolate the endothelial cells and re-plate them in clean dishes for further purification.
Figure 1F.5.4.
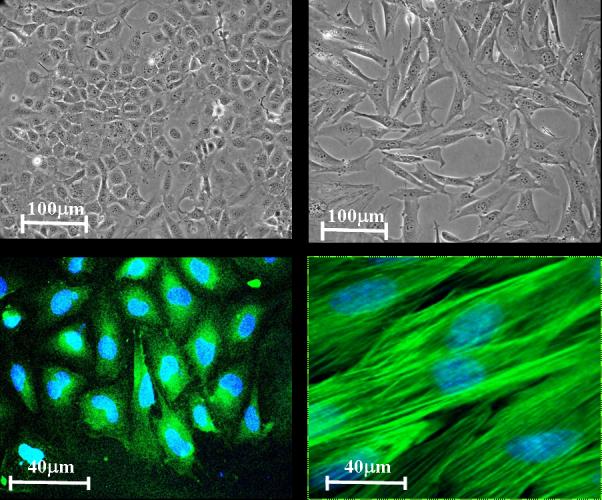
Outgrowths of Flk-1 positive cells consist of primarily two cell populations: endothelial-like cells exhibiting a cobblestone-like morphology (A) stained with endothelial marker PE-CAM1 (B), and elongated smooth muscle-like cells (C) stained with alpha-smooth muscle actin (D).
62. Prepare dissecting pipets (Support Protocol 4) and mouth aspirator (Support Protocol 5).
63. Aspirate culture medium and wash cells twice with 3 ml of PBS per 35-mm dish.
64. Incubate cells with cell dissociation solution for 5 min.
As cells begin to detach from the culture dish, their distinct cell morphologies may become vague. It is helpful to mark the bottom of the dish with the appropriate location of the desired cells and work quickly.
65. Meanwhile fill 6−10 collagen IV coated 35-mm dishes with 2 ml of EC medium.
66. Carve around a 5−10 cell cluster with the edge of the mouth-pasteur pipet assembly (see Fig. 1F.5.5).
Figure 1F.5.5.

Endothelial-like cells exhibiting a cobblestone-like morphology are manually picked based on proper morphology and replated on a second dish coated with collagen IV (top) and a photograph of the aspiration device used for the manual picking (bottom). Note that several batches of endothelial cells may be isolated from one dish. These batches may vary slightly, so it is a good idea to expand the batches separately.
67. Aspirate the cells into the pipet and transfer to the new 35-mm dishes containing 2 ml of ESC-EC differentiation medium.
68. Repeat carving out another 5− 10 cluster of cells and plate in a new 35-mm dish.
69. Repeat for 6−10 dishes, using a separate dish for each cluster.
The number of clusters obtained depends on the quality of the EC sheets, 6−10 clusters per 35-mm dish of Flk-1+ outgrowths is normal.
70. Add 50 ng/ml of VEGF to each dish.
71. Incubate cells at 37°C and 5% CO2 for 7 to 10 day. Change medium every 4 days.
Expand ECs in vitro
72 . After allowing the cells 7 to 10 days of uninterrupted growth, observe the dishes carefully for EC colonies.
Once the cell colonies are well established, you will see 50 to 100 cells in a circular sheet. These cells will be highly confluent in the center and appear to grow outward at the edges of the colony.
73. To encourage further cell proliferation, the cells should be subcultured in EC medium to allow cells to grow easily on the entire surface of the collagen IV coated 35-mm dish.
74. Continually expand the cells in larger dishes (35-mm dish, then 25-cm2 flask, then 75-cm2 flask, then 175-cm2 flask, and then multiple 175-cm2 flasks).
Make sure to coat the surface of each flask with collagen IV, collagen I, fibronectin, or gelatin for 2 hr prior to cell seeding and wash off the extra substrate with PBS. The ESC derived EC can be frozen and thawed normally (Support Protocols 1 and 2).
SUPPORT PROTOCOL 1
FREEZING CULTURED CELLS
Feeder cells, ESCs, and ECs can be frozen to maintain stocks of cells until they are needed. This is a generalized procedure for freezing cells. The cells are removed from the dish, resuspended in freezing medium and frozen.
Materials
Cultures to be frozen
Trypsin/EDTA
Phosphate-buffered saline
35-mm tissue culture dishes
Appropriate medium for cells containing serum
Freezing medium (see recipe)
Cryovials
Cryo 1°C freezing container
1. Trypsinize cells in the exponential phase of growth (varies for each cell type, but typically is after 3 days of growth). First aspirate the medium and wash the culture twice with 3 mL of PBS per 35-mm dish and then add 1 mL of trypsin/EDTA. Incubate under the phase contrast microscope.
After about 3 min cells begin to round with clearly defined edges.
2. Once cell rounding is observed, add 3 mL of mediim with serum and pipet several times to disaggregate cells from the dish and from each other until a single cell suspension is achieved.
This is a general trypsinization procedure. The medium added after trypsinization should be the same as the cells are currently cultured in.
3. Pellet the cells by centrifugation (200 × g, 4−5 min, at room temperature) and resuspend in an appropriate amount of cell culture medium.
For convenience, cells are frozen in 1-ml aliquots at cell numbers that correspond to the appropriate numbers that will be needed upon thawing. The upper limit would be 5−10 × 106 cells/ml.
4. Slowly add an equal volume of the freezing medium drop-wise over 2 min. Continuously shake the cell suspension for even distribution of the DMSO/FBS freezing medium.
5. Aliquot 1-ml of cell suspension into cryovials.
ESC are typically frozen between 5×105 and 1×106 cells/mL.
6. Immediately transfer cryovials to a Cryo 1°C Freezing Container and place the container in a −70°C or a −80°C freezer for 24 hr.
7. Transfer the vials to liquid nitrogen.
SUPPORT PROTOCOL 2
THAWING CULTURED CELLS
Frozen stocks of cultured cells need to be carefully thawed to ensure viability. This is a generalized method applicable to feeder cells, ESCs, and ECs.
Materials
Frozen stocks of cells
Appropriate cell medium
15-ml centrifuge tube
Thawing cells
1. Thaw the cells in a 37°C water bath until only a small ice droplet remains (approximately 1 min, the last drop will thaw as you carry the vial to the laminar flow cabinet).
2. While the vial is thawing, fill a 15-ml centrifuge tube with 10-ml of the appropriate cell culture medium for the cell types.
You will use embryonic fibroblast feeder cell medium for fibroblasts, ESC medium for mouse ESC, and EC medium for fully differentiated and purified EC. Cells at intermediate stages of differentiation are not usually frozen.
3. Transfer the thawed cells to the centrifuge tube and collect the cells by centrifuging at 200 × g for 4−5 min at room temperature.
4. Remove the supernatant and gently resuspend the cells in 4−5 ml fresh growth medium.
5. Transfer cells to the prepared culture dish and place in a 37°C incubator.
ESC should be plated at 1×105 cells per 35-mm dish. Fibroblasts feeder cells should be plated at 4×105 cells per 35-mm dish.
Both ESC and EC cells are maintained on dishes or flasks coated with the appropriate substrate, therefore, when thawing or passing cells, make sure to have allowed time (1−2 hr) for the substrate to adhere to the culture dish and wash off excess substrate with PBS. For ESCs that will be cultured on fibroblasts, make sure to prepare those dishes with a layer of fibroblast cells at least 4 hr prior to ESC seeding.
6. Replace the medium with fresh ESC medium the next day.
SUPPORT PROTOCOL 3
MITOTIC INACTIVATION OF FIBROBLAST FEEDER CELLS
Typically, ES cells are cultured on fibroblast feeder cells that are inactivated with mitomycin C or irradiation. The inactivation of the fibroblast cells allows the ES cells to benefit from the co-culture feeder conditions without fibroblast proliferation Mouse embryonic fibroblast feeder cells are typically used, however, the isolation of these cells requires several animals to be sacrificed and labor intensive dissection of the fetal tissue. If mouse embryonic feeders are unavailable, or undesirable, STO cells may also be used (available from ATCC). Before disposing, mytomycin C must be neutralized with Clorox bleach for at least 15 min.
Materials
Feeder cells to be inactivated: mouse fibroblasts or STO cells (ATCC)
Fibroblast feeder cell medium (see recipe)
Mitomycin C solution (see recipe)
175-cm2 tissue culture flasks
Phosphate-buffered saline (PBS)
Trypsin/EDTA
1. After thawing (Support Protocol 2), allow mouse fibroblasts to grow to 90% to 95% confluency in T-175 tissue-culture treated flasks in fibroblast feeder cell medium.
2. Aspirate culture medium from flask and replace with 16 ml of mitomycin C solution.
3. Incubate the treated flasks for 2 hr at 37°C, 5%CO2.
4. After 2 hr, aspirate mitomycin C solution and wash each 175-cm2 flask five times with 20 ml PBS.
5. Add 3 ml of trypsin/EDTA per flask and monitor cell detachment.
After approximately 1 min, cells should detach from the flask surface (gently rock flask side-to-side).
6. After cells have detached, add 5−10 ml of fibroblast feeder cell medium.
7. Transfer the cell suspension from each flask to 15-ml centrifuge tubes.
8. Centrifuge at 200 × g for 4−5 min at room temperature.
9. Remove supernatant and wash again with 10-ml fibroblast feeder cell medium per tube.
10. Centrifuge at 200 × g for 4−5 min at room temperature.
11. Repeat washing one more time. Resuspend the cells 1 ml fibroblast feeder cell medium.
12. Count an aliquot of cells.
13. Plate between 3×105 and 4×105 cells per 35-mm dish that will be needed for ESC culture. Fill dishes to a total of 3 ml fibroblast feeder cell medium each, allow at least 4 hr, preferably overnight, for the cells to adhere to dishes before adding embryonic stem cells.
Excess inactivated fibroblasts may also be frozen at this point for future use. Inactivated fibroblasts may be used for up to 1 week.
SUPPORT PROTOCOL 4
PREPARATION OF DISSECTING PIPETS
Pipets must be modified for manual dissection of EC progenitor cells for passaging.
Materials
Pasteur pipets
Bunsen burner
1. Hold the narrow tip of a Pasteur pipet in your left hand and larger end in your right hand. Pass the center of the narrow portion through a low flame of a Bunsen burner until the pipet is hot.
2. Quickly pull on the tip of the pipet while lifting the pipet out of the flame to generate a pipet region with a smaller diameter just above the tip of the pipet.
3. Loop back the pulled glass and rub glass to glass to create a point of friction. Tap the glass to break the tip off at the point of friction.
The technique for pulling Pasteur pipettes will take some practice.
4. Polish the new end of the pipet by passing the new tip gently over a low flame.
The Pasteur pipettes should remain sterile, so use immediately after pulling, or pull several pipettes and sterilize them ahead of time.
SUPPORT PROTOCOL 5
PREPARING A MOUTH ASPIRATOR
A mouth aspirator is used with the dissecting pipet when passaging EC progenitor cells.
Materials
1000-μl micropipette tip
Aspirator assembly with rubber tubing
0.2-μm syringe filter
Dissecting pipet (Support Protocol 4)
1. Fit the narrow end of a 1000-μl micropipette tip into the rubbing tubing of an aspirator assembly fitted with a 0.2-μm syringe filter.
2. Insert the modified Pasteur pipet into the wide end of the 1000-μl micropipette tip. This aspirator assembly allows for simultaneous microscope viewing and cell colony manipulations.
ALTERNATE PROTOCOL 1
EC differentiation from mouse ESC culture under Serum-Free Conditions
The methods described above employ methods of cell culture and differentiation where the ESC are grown in medium containing fetal bovine serum (FBS). However, the reproducibility of some aspects of these experiments can vary since FBS composition can vary significantly from batch-to-batch. This leads to tiresome batch testing and buying up entire lots of screened batches of FBS at once time. This process must then be repeated when the desired lot is exhausted. By using an induction system that does not require serum, the conditions under which the cells are grown are chemically defined, and more reproducible.
Based on the formulas previously developed (Adelman et al. 2002; Tanaka et al. 2006), it is possible to maintained murine ESC in culture on gelatin in a chemically-defined serum-free medium The cells retain their morphology well and replicate quickly with a doubling time of approximately 3 days (This is a slower growth rate than achieved with serum).
Efforts to develop a chemically defined medium for differentiation have been more difficult. In the absence of serum and LIF, the cells differentiate, but proliferate much more slowly in comparison to the induction medium with serum. However, the percentage of Flk-1+ cells in the serum-free induction is comparable to that obtained from inductions with serum-containing medium, and therefore; can be scaled-up to achieve the desired number of Flk-1+ cells.
EC differentiation in a two-dimensional system has been traditionally performed on collagen IV-coated dishes on the premise that collagen IV induces the greatest number of mesodermal cells (Nishikawa et al., 2001b, 2007). However, our lab has succeeded in inducing equally sufficient expression of Flk-1 on gelatin-coated dishes. When compared to a serum-containing differentiation medium, our serum-free mixture yielded a comparable percentage of Flk-1+ cells, 20%.
Addition Materials (also see Basic Protocol)
1. Bone Morphogenic Protein 4 [BMP-4] (R&D Systems)
Serum-free ESC culture medium (see recipe)
Serum-free ESC differentiating medium (see recipe)
The basic steps for serum-free culture and EC induction follow those of Basic Protocol. The serum-free ESC culture medium is substituted in the steps for ESC culture (steps 1 to 22). The serum-free ESC differentiating medium is substituted in the steps for differentiating ESC and purification of ECs (steps 23 to 71). Serum-free subculture for mature EC (steps 72 to 74) is currently under investigation.
REAGENTS AND SOLUTIONS
Embryonic fibroblast feeder cell medium
88% (v/v) high-glucose DMEM
10% (v/v) heat inactivated fetal bovine serum
1% penicillin/streptomycin
1% (v/v) L-glutamine
Store for up to 1 month at 4°C
Mouse ESC cell medium
78% (v/v) Knockout Dulbecco's modified Eagle medium
15% (v/v) ES cell-qualified fetal bovine serum
5% (v/v) Knockout serum replacement
1% (v/v) penicillin/streptomycin
1% (v/v) L-glutamine
1,000 U/ml leukemia inhibitory factor
5×10−5 M β-mercaptoethanol
Store for up to 2 weeks at 4°C.
Freezing medium
80% (v/v) fetal bovine serum
20% (v/v) DMSO
Prepare fresh for each use
To freeze cells, mix equal volumes of the appropriate cell culture medium and freezing medium.
ESC-to-EC differentiating medium
93% (v/v) α-minimal essential medium
5% (v/v) fetal bovine serum
1% (v/v) penicillin/streptomycin
1% (v/v) L-glutamine
5×10−5 M β-mercaptoethanol
Store for up to 1 month at 4°C
EC medium
This is a commercially available EC medium kit; EGM-2 medium Bullet Kit (500-ml bottle plus growth factors; Clonetics, East Rutherford, NJ; cat. no. CC-3162).
Serum-Free ESC Culture Medium
15% (v/v) KnockOut Serum Replacement [KSR] (Gibco)
1× penicillin-streptomycin (Invitrogen)
2 mM L-glutamine (Invitrogen)
1× non-essential amino acids (Invitrogen)
0.1mM β-mercaptoethanol (Calbiochem)
2000 U/ml leukemia inhibitory factor [LIF] (Chemicon)
10 ng/ml bone morphogenic protein 4 [BMP-4] (R&D Systems)
Knock-Out DMEM [KO DMEM] (Gibco).
Serum-free ESC differentiating medium
20% (v/v) KnockOut Serum Replacement [KSR] (Gibco)
1× penicillin/streptomycin (Invitrogen)
2mM L-glutamine (Invitrogen)
1× non-essential amino acids (Invitrogen)
5×10−5M β-mercaptoethanol (Calbiochem)
5 ng/ml bone morphogenic protein 4 [BMP-4] (R&D Systems)
30 ng/ml vascular endothelial growth factor [VEGF] (R&D Systems)
α-minimum essential medium [αMEM] (Cellgro)
A Chemically Defined Medium (CDM) has also been used for serum-free induction (Johansson and Wiles, 1995; Wiles and Johansson, 1999; Ng et al., 2005). In the cited study, the addition of BMP-4 or Activin A was found to enhance mesoderm differentiation (Johansson and Wiles, 1995).
COMMENTARY
Background Information
Endothelial cells have been derived from mouse and human ESC by isolating the differentiating endothelium from an embryoid body (Levenberg et al., 2002). Although the embryoid body system enables investigation of vasculogenesis virtually as it occurs in the embryo ( Risau et al., 1988; Wang et al., 1992; Vittet et al., 1996; Choi et al., 1998), the multiple cell-cell contacts and cell lineages make it difficult to study and control the behavior of the maturing endothelial cell in detail.
Endothelial, hematopoietic, and smooth muscle cells have also been derived from Flk-1+ outgrowths from murine ESCs grown on type-IV collagen coated surfaces (Nishikawa et al., 1998; Yamashita et al., 2000; McCloskey et al., 2003), showing that the three-dimensional structure is not necessary for endothelial maturation from ESC (Nishikawa et al., 1998). The two-dimensional monolayer technique of endothelial differentiation not only allows closer study and control of the in vitro maturation, molecular events, and growth factor requirements of endothelial cell derivation (Nishikawa et al., 1998; Hirashima et al., 1999; Yamashita et al., 2000), but also uses an induction method that is devoid of the three-dimensional embryo-like self-programmed machinery for vascular differentiation. Although the two-dimensional monolayer derivation methods have been very successful in isolating and studying the maturation of endothelial cells from murine ESCs, the long-term maintenance of these murine ESC-derived endothelial cells has been limited. Without genetic manipulation, the longest these ESC-derived EC were maintained in culture was 7 days, increasing to two or three passages by culturing cells on OP9 stromal cells (Nishikawa et al., 2001a).
In addition to the limitations in the proliferative capabilities of the endothelial cells from murine ESCs (Nishikawa et al., 1998; Hirashima et al., 1999; Yamashita et al., 2000), the reported studies did not isolate uniform populations of endothelial cells from the contaminating smooth muscle cell, or other cell populations. Based on our studies (techniques presented in this unit), the isolation of pure populations of EC is critical for further expansion of these cells (McCloskey et al., 2003). Pure cell populations are also essential for studying the effectiveness of these cells for cell-based therapies and should alleviate the problem of teratomas that form when ESC are implanted in vivo.
Recent discoveries of molecular markers for arterial, venous, and lymphatic endothelial cells allow a more sophisticated characterization of endothelial diversity (Aranguren et al., 2007; Yamashita, 2007). Arterial specification, promoted by Notch signaling, is characterized by ephrinB2, Delta-like (Dll)-4, Notch-1 and 4, Jagged-1, and connexin-40 expression. Venous endothelium, potentially a default pathway of EC differentiation, is characterized by EphB4 and COUP-TFII. Committed lymphatic EC, differentiated from venous EC, express Prox-1 as the most specific lymphatic EC marker.
EC differentiation from human ESC (hESC)
Although both mouse and hESC exhibit similar expression of key transcription factors, including Oct-3/4, Nanog, and Sox2, there are some fundamental differences between mouse and human ESC. For example, the population doubling time of hESC is 36 hr compared with 12 hr for mouse ESC. The hESC grow in relatively flat compact colonies compared with mouse ESC. Expression of stage-specific embryonic antigens (SSEA) also varies. Undifferentiated human ESC express SSEA-3 and -4, and do not express SSEA-1, while mouse ESC express SSEA-1 and do not express SSEA-3 or -4. Most importantly, for hESC culture, the presence of LIF does not support undifferentiated feeder-free growth, while LIF is sufficient in mouse ESC cultures.
EC differentiation and isolation from hESC was first published by the Langer laboratory in 2002 (Levenberg et al., 2002). In this study, embryoid bodies (EBs) were employed for the initial induction of EC. Endothelial markers CD31, CD34, and VE-cadherin peaked between days 13 and 15 of induction. Sorting CD31+ cells on day 13, allowed for expansion of EC progenitors. After several passages in culture, 78% of the cells still expressed CD31. More recently, the same laboratory used similar protocols for generation of EBs, but sorted CD34+ cells on day 10 to generate vascular progenitor cells retaining the potential to generate both endothelial and smooth muscle cells (Ferreira et al., 2007).
Because the formation of EBs from ESC triggers spontaneous differentiation of all cell types, it is an inefficient method for the generation of specific cell types because the microenvironment within the EB is difficult to control. Methods for two-dimensional induction of hESC to EC have also been published (Wang et al., 2007). In this study, hESC were placed on mouse embryonic feeders in differentiation medium containing 15% fetal bovine serum (FBS) for 10 days. By day 10, 5−10% of these cells expressed CD34, a common hematopoietic and endothelial progenitor marker. Two rounds of magnetic bead sorting enriched the cells to 80% to 95% purity. When these cells were cultured in endothelial growth medium, the majority of the cells expressed endothelial markers CD31 and VE-cadherin. These researchers were also able to remove FBS by substitution with BIT 9500, VEGF, and BMP-4 growth factors for a serum-free induction, and reported a similar number of CD34+ cells.
Critical Parameters
The optimal day of initial induction of Flk-1+ vascular progenitor cells is a very small window (less than 1 day) and will depend on the cell line and culture conditions. ESC-D3 cells will take approximately 4 days for optimal induction, while ESC-R1 cells will take only 3 days to generate a significant number of very bright (high expressing) Flk-1+ vascular progenitors. Past this optimal window, the Flk-1+ expression is reduced and most of the vascular progenitor cells will be lost. This stage is very important and should be optimized (induce using the ESC induction medium for 2, 2.5, 3, 3.5, 4, and 4.5 days) for the cells before attempting to move forward with the cell sorting of Flk-1+ cells.
Anticipated Results
The methods presented here can generate up to 1−3 billion EC from each initial induction of 30,000 mouse ESC.
Time Considerations
The differentiation, isolation, and expansion procedures (Fig. 1F.5.6) for the generation of EC from mouse ESC will take approximately 1 month, but new practitioners should allow another few months for optimization of the methods described. Allow at least 2 weeks, or more, for optimization of the timing for the generation of Flk-1+ cells and sorting these cells. Add another month for practicing the manual selection technique described. Fluorescence activated cell sorting or magnetic cell sorting can also be used for selection of EC progenitors from smooth muscle cells, but will not yield the high purity needed for a significant expansion of EC. If the two cell types are not purified away from each other, smooth muscle cells will eventually outgrow EC and the cell cultures will need to be discarded.
Figure 1F.5.6.
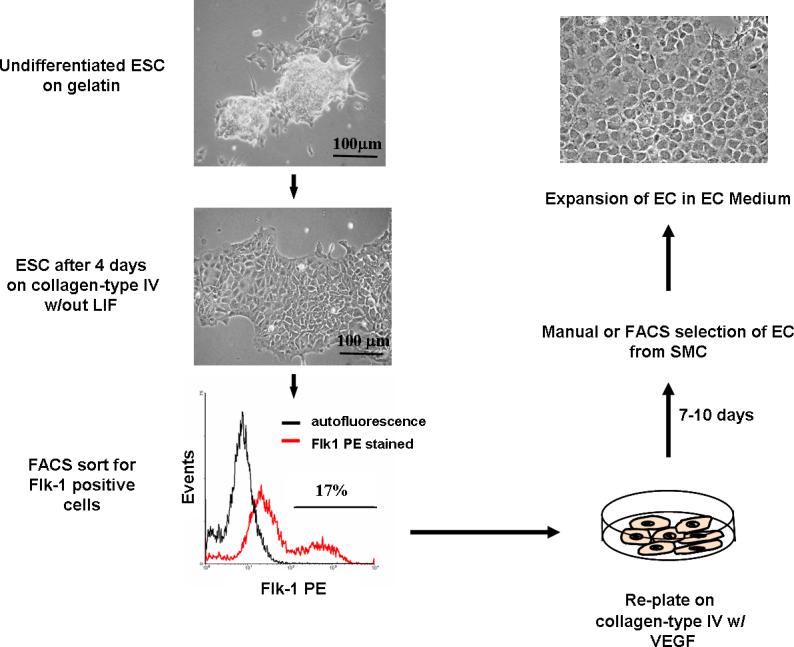
Flow diagram of the steps for EC differentiation from mouse ESC.
Literature Cited
- Adelman CA, Chattopadhyay S, Bieker JJ. The BMP/BMPR/Smad pathway directs expression of the erythroid-specific EKLF and GATA1 transcription factors during embryoid body differentiation in serum-free media. Development. 2002;129:539–549. doi: 10.1242/dev.129.2.539. [DOI] [PubMed] [Google Scholar]
- Aranguren XL, Luttun A, Clavel C, Moreno C, Abizanda G, Barajas MA, Pelacho B, Uriz M, Arana M, Echavarri A, Soriano M, Andreu EJ, m Merino J, Garcia-Verdugo JM, Verfaillie CM, Prosper E. In vitro and in vivo arterial differentiation of human multipotent adult progenitor cells. Blood. 2007;109:2634–2642. doi: 10.1182/blood-2006-06-030411. [DOI] [PubMed] [Google Scholar]
- Choi K, Kennedy M, Kazarov A, Papadimitriou JC, Keller G. A common precursor for hematopoietic and endothelial cells. Development. 1998;125:725–32. doi: 10.1242/dev.125.4.725. [DOI] [PubMed] [Google Scholar]
- Dzau VJ, Gnecchi M, Pachori AS, Morello F, Melo LG. Therapeutic potential of endothelial progenitor cells in cardiovascular diseases. Hypertension. 2005;46:7–18. doi: 10.1161/01.HYP.0000168923.92885.f7. [DOI] [PubMed] [Google Scholar]
- Ferreira LS, Gerecht S, Shieh HF, Watson N, Rupnick MA, Dallabrida SM, Vunjak-Novakovic G, Langer R. Vascular progenitor cells isolated from human embryonic stem cells give rise to endothelial and smooth muscle like cells and form vascular networks in vivo. Circ. Res. 2007;101:286–294. doi: 10.1161/CIRCRESAHA.107.150201. [DOI] [PubMed] [Google Scholar]
- Griese DP, Ehsan A, Melo LG, Kong D, Zhang L, Mann MJ, Pratt RE, Mulligan RC, Dzau VJ. Isolation and transplantation of autologous circulating endothelial cells into denuded vessels and prosthetic grafts: implications for cell-based vascular therapy. Circulation. 2003;108:2710–2715. doi: 10.1161/01.CIR.0000096490.16596.A6. [DOI] [PubMed] [Google Scholar]
- Hirashima M, Kataoka H, Nishikawa S, Matsuyoshi N. Maturation of embryonic stem cells into endothelial cells in an in vitro model of vasculogenesis. Blood. 1999;93:1253–1263. [PubMed] [Google Scholar]
- Johansson BM, Wiles MV. Evidence for involvement of activin A and bone morphogenetic protein 4 in mammalian mesoderm and hematopoietic development. Mol. Cell Biol. 1995;15:141–151. doi: 10.1128/mcb.15.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalka C, Masuda H, Takahashi T, Kalka-Moll WM, Silver M, Kearney M, Li T, Isner JM, Asahara T. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc. Natl. Acad. Sci. U.S.A. 2000;97:3422–3427. doi: 10.1073/pnas.070046397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal S, Amiel GE, Guleserian KJ, Shapira OM, Perry T, Sutherland FW, Rabkin E, Moran AM, Schoen FJ, Atala A, Soker S, Bischoff J, Mayer JE., Jr. Functional small-diameter neovessels created using endothelial progenitor cells expanded ex vivo. Nat. Med. 2001;7:1035–1040. doi: 10.1038/nm0901-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto A, Gwon HC, Iwaguro H, Yamaguchi JI, Uchida S, Masuda H, Silver M, Ma H, Kearney M, Isner JM, Asahara T. Therapeutic potential of ex vivo expanded endothelial progenitor cells for myocardial ischemia. Circulation. 2001;103:634–637. doi: 10.1161/01.cir.103.5.634. [DOI] [PubMed] [Google Scholar]
- Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, Wang J, Homma S, Edwards NM, Itescu. S. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat. Med. 2001;7:430–436. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- Levenberg S, Golub JS, Amit M, Itskovitz-Eldor J, Langer R. Endothelial cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 2002;99:4391–4396. doi: 10.1073/pnas.032074999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCloskey KE, Gilroy ME, Nerem RM. Use of embryonic stem cell-derived endothelial cells as a cell source to generate vessel structures in vitro. Tissue Eng. 2005;11:497–505. doi: 10.1089/ten.2005.11.497. [DOI] [PubMed] [Google Scholar]
- McCloskey KE, Lyons I, Rao RR, Stice SL, Nerem RM. Purified and proliferating endothelial cells derived and expanded in vitro from embryonic stem cells. Endothelium. 2003;10:329–336. doi: 10.1080/10623320390272325. [DOI] [PubMed] [Google Scholar]
- Ng ES, Azzola L, Sourris K, Robb L, Stanley EG, Elefanty AG. The primitive streak gene Mixl1 is required for efficient haematopoiesis and BMP4-induced ventral mesoderm patterning in differentiating ES cells. Development. 2005;132:873–884. doi: 10.1242/dev.01657. [DOI] [PubMed] [Google Scholar]
- Nishikawa SI, Hirashima M, Nishikawa S, Ogawa M. Cell biology of vascular endothelial cells. Ann. N.Y. Acad. Sci. 2001a;947:35–40. doi: 10.1111/j.1749-6632.2001.tb03928.x. discussion 41. [DOI] [PubMed] [Google Scholar]
- Nishikawa SI, Hirashima M, Nishikawa S, Ogawa M. Cell Biology of Vascular Endothelial Cells. Ann. N.Y. Acad. Sci. 2001b;947:35. doi: 10.1111/j.1749-6632.2001.tb03928.x. [DOI] [PubMed] [Google Scholar]
- Nishikawa SI, Jakt LM, Era T. Opinion: Embryonic stem-cell culture as a tool for developmental cell biology. Nat. Rev. Molec. Cell Biol. 2007;8:502–507. doi: 10.1038/nrm2189. [DOI] [PubMed] [Google Scholar]
- Nishikawa SI, Nishikawa S, Hirashima M, Matsuyoshi N, Kodama H. Progressive lineage analysis by cell sorting and culture identifies FLK1+VE-cadherin+ cells at a diverging point of endothelial and hemopoietic lineages. Development. 1998;125:1747–1757. doi: 10.1242/dev.125.9.1747. [DOI] [PubMed] [Google Scholar]
- Risau W, Sariola H, Zerwes HG, Sasse J, Ekblom P, Kemler R, Doetschman T. Vasculogenesis and angiogenesis in embryonic-stem-cell-derived embryoid bodies. Development. 1988;102:471–478. doi: 10.1242/dev.102.3.471. [DOI] [PubMed] [Google Scholar]
- Soker S, Machado M, Atala A. Systems for therapeutic angiogenesis in tissue engineering. World J. Urol. 2000;18:10–18. doi: 10.1007/pl00007070. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Takeuchi T, Neri QV, Sills ES, Palermo GD. Laser-assisted blastocyst dissection and subsequent cultivation of embryonic stem cells in a serum/cell free culture system: applications and preliminary results in a murine model. J. Translat. Med. 2006;4:20. doi: 10.1186/1479-5876-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vittet D, Prandini MH, Berthier R, Schweitzer A, Martin-Sisteron H, Uzan G, Dejana E. Embryonic stem cells differentiate in vitro to endothelial cells through successive maturation steps. Blood. 1996;88:3424–3431. [PubMed] [Google Scholar]
- Wang R, Clark R, Bautch VL. Embryonic stem cell-derived cystic embryoid bodies form vascular channels: an in vitro model of blood vessel development. Development. 1992;114:303–316. doi: 10.1242/dev.114.2.303. [DOI] [PubMed] [Google Scholar]
- Wang ZZ, Au P, Chen T, Shao Y, Daheron LM, Bai H, Arzigian M, Fukumura D, Jain RK, Scadden DT. Endothelial cells derived from human embryonic stem cells form durable blood vessels in vivo. Nat. Biotechnol. 2007;25:317–318. doi: 10.1038/nbt1287. [DOI] [PubMed] [Google Scholar]
- Wiles MV, Johansson BM. Embryonic stem cell development in a chemically defined medium. Exp. Cell Res. 1999;247:241–248. doi: 10.1006/excr.1998.4353. [DOI] [PubMed] [Google Scholar]
- Yamashita J, Itoh H, Hirashima M, Ogawa M, Nishikawa S, Yurugi T, Naito M, Nakao K. Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature. 2000;408:92–96. doi: 10.1038/35040568. [DOI] [PubMed] [Google Scholar]
- Yamashita JK. Differentiation of arterial, venous, and lymphatic endothelial cells from vascular progenitors. Trends Cardiovasc. Med. 2007;17:59–63. doi: 10.1016/j.tcm.2007.01.001. [DOI] [PubMed] [Google Scholar]