

Abstract
The biological basis or mechanism whereby folate supplementation protects against heart and neural tube defects is unknown. It has been hypothesized that the amino acid homocysteine may be the teratogenic agent, since serum homocysteine increases in folate depletion; however, this hypothesis has not been tested. In this study, avian embryos were treated directly with d,l-homocysteine or with l-homocysteine thiolactone, and a dose response was established. Of embryos treated with 50 μl of the teratogenic dose (200 mM d,l-homocysteine or 100 mM l-homocysteine thiolactone) on incubation days 0, 1, and 2 and harvested at 53 h (stage 14), 27% showed neural tube defects. To determine the effect of the teratogenic dose on the process of heart septation, embryos were treated during incubation days 2, 3, and 4; then they were harvested at day 9 following the completion of septation. Of surviving embryos, 23% showed ventricular septal defects, and 11% showed neural tube defects. A high percentage of the day 9 embryos also showed a ventral closure defect. The teratogenic dose was shown to raise serum homocysteine to over 150 nmol/ml, compared with a normal level of about 10 nmol/ml. Folate supplementation kept the rise in serum homocysteine to ≈45 nmol/ml, and prevented the teratogenic effect. These results support the hypothesis that homocysteine per se causes dysmorphogenesis of the heart and neural tube, as well as of the ventral wall.
Although a number of congenital defects are known to be the result of chromosomal aberrations [e.g., “CATCH22” (1)], a major proportion of congenital malformation appears to be the result of environmental factors including nutritional deficiency or toxicity. The association between folic acid deficiency and congenital malformation is especially well-known: both animal experimentation and epidemiologic studies have shown that a deficiency of folic acid may be associated with defects of neural tube closure, as well as defects of aorticopulmonary (or conotruncal) septation (2–10). Because the morphogenetic events that produce normal neural tube closure and normal heart septation are quite separate in embryonic time and space, it is not obvious that the two processes are intimately related and thus potentially susceptible to the same teratogens. This intimate relationship is in the common origin of the cells involved: the neural crest cells that form the vascular smooth muscle cells of the aorticopulmonary septum originate in a neural ectoderm site that is conterminus with the site of neural tube closure (11, 12). Undifferentiated cells from these two adjacent sites are interchangeable early in development, with each able to differentiate into the ultimate phenotype of the other (13). In some way, the teratogenic process that results from folate insufficiency appears to select for these particular lateral neuroepithelial cells from the entire neural ectoderm and produces congenital defects of both the neural tube per se and the neural crest-derived aorticopulmonary septum.
A number of recent studies have demonstrated that the incidence of congenital defects related to interruption of both neurulation and conotruncal septation could be reduced by the use of supplementary folate during pregnancy (e.g., refs. 3–5). However, according to a 1994 Ciba Symposium (6), the biological basis of this effect is unknown. One result of folate deficiency that has been cited as a potential cause of these congenital defects is based upon the central role of folate in maintaining one-carbon groups as well as S-adenosylmethionine, the primary methyl donor in transmethylation reactions (6). According to this hypothesis, the teratogenic effect of folate deficiency is a result of an insufficient supply of nucleic acid precursors in the rapidly dividing embryonic cells. However, the highly specific and predictable nature of the congenital defects caused by folate deficiency argues against a generalized lesion: although derivatives of the neural ectoderm are effected more than other tissues by folate deficiency, all of the embryonic tissues are dividing rapidly during the susceptible developmental period (6, 7). Furthermore, it appears that the degree and duration of folate deficiency necessary to inhibit DNA synthesis may be greater than the conditions that are necessary to produce congenitally disordered organ sytems (6), and folate deficiency is not by itself sufficient to produce neural tube defects in mice (14). These data argue that folate deficiency does not induce congenital neural tube and heart defects by generally limiting the availability of nucleic acids but by producing some more specific effect upon a narrow region of the neural ectoderm. Therefore, a mechanism other than disrupted transmethylation must be at work during development of the septal and neural tube defects that are associated with folate insufficiency. This mechanism may relate to elevated homocysteine (Hcys).
A recent overview of the issue stated that Hcys levels were always high in cases of folate-specific defects (15). Because of its central role in methionine metabolism, Hcys inevitably increases during folate deficiency (16, 17). Hcys may be increased in some individuals in spite of their being replete with folate, because of abnormal enzymes in the Hcys/methionine cycle (6). Thus Hcys, which is now recognized as an independent risk factor for atherosclerosis (18), also has been given attention as it relates to the occurrence of congenital defects, especially of the neural tube. Recently Eskes et al. (3) and Steegers-Theunissen et al. (19) hypothesized that elevated Hcys per se may be teratogenic based upon epidemiologic evidence, but they were unable to cite any direct experimental evidence to support a role for Hcys in the induction of neural tube or heart defects. To provide a test of the hypothesis that Hcys per se was teratogenic, we treated avian embryos with doses of 0.5–20 μM exogenous Hcys per embryo. This treatment resulted in physiologic increases of Hcys in the embryonic serum and reliably produced heart and neural tube defects that were typical of folate deficiency, in a dose- and time-dependent fashion.
METHODS
Effect of Exogenous Hcys During Gastrulation/Neurulation.
The objective of the present study was to test the hypothesis that exogenous Hcys would induce congenital defects in avian embryos in the absence of folate depletion. The serum Hcys concentration in mature vertebrates typically is reported as 10 ± 5 μmol/liter (nmol/ml; refs. 1, 2, and 5) and was found to be similar in the serum of the avian embryo (see Fig. 8). To effect an increase of serum Hcys in the absence of folate depletion, exogenous Hcys is given to mature vertebrates as a dietary supplement or in water ad libitum (e.g., ref. 20). To get a similar effect in the present study, a solution of Hcys was placed upon the inner embryonic membrane; the approximate equivalent of the absorptive membrane of the alimentary system. A dose size of 50 μl was selected, which appears to be a typical dose size for experiments wherein micronutrients are delivered to avian embryos (e.g., refs. 21 and 22).
Figure 8.
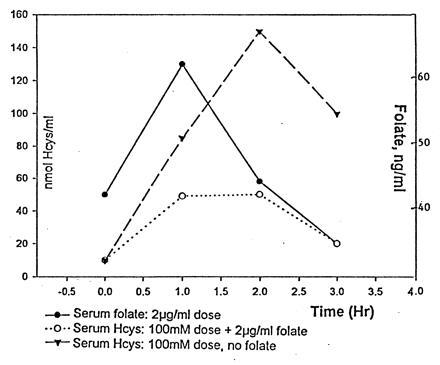
Embryos were treated with a 5-μmol dose of l-Hcys thiolactone or with 5 μmol of l-Hcys thiolactone and 0.1 μg (100.0 ng) of folic acid. Embryonic blood samples were withdrawn, and the levels of Hcys and folate were measured.
Albumen (2 cc) was withdrawn from each egg to enlarge the airspace slightly over the inner membrane. To titrate for a dysmorphogenic effect, doses of d,l-Hcys (Sigma) of 0.5–20 μmol were applied through the enlarged airspace at each of the following developmental time points: the prestreak stage (3 h after the initiation of incubation); the one-somite stage (24 h); and the 19-somite stage (48 h). l-Hcys is the metabolically available form, and these doses were equivalent to doses of 0.25–10 μmol of l-Hcys. Control embryos were treated identically with comparable doses of leucine, methionine, or cysteine. The embryos were harvested after ≈53 h of incubation (the 22-somite stage, or Hamburger–Hamilton stage 14 (23). This schedule was designed to expose the embryos to Hcys during the processes of gastrulation and neurulation; they were harvested upon the completion of neurulation to maximize survival. Embryos collected at 53 h were stained with the vital dye neutral red so that they could be photographed and evaluated before fixation; then they were fixed with Bouin’s solution, which we had found provided the best fixation of early embryos. The best teratogenic dose was found to be 10 μmol of d,l-Hcys (5 μmol of l-Hcys; see Results). Because the thiolactone form of Hcys is more stable, simulates the physiologic effects of Hcys per se, and is available in the l-only form (Sigma), we treated another set of embryos with 5 μmol of l-Hcys thiolactone and found that it reproduced the teratogenic effect of 10 μmol of d,l-Hcys. Hereafter, therefore, the term HcysTD will be used as an abbreviated reference to the term “teratogenic dose of l-Hcys” and should be taken to refer to treatment based upon a dose of either 10 μmol of d,l-Hcys or 5 μmol of l-Hcys thiolactone, either of which may be used as the HcysTD. There was no detectable effect of d-Hcys (see Results).
After the HcysTD was established, a larger series of embryos was treated with HcysTD at time 0 and each 24 h thereafter. These embryos were harvested after three treatments (0 h, 24 h, and 48 h) as described above.
Effect of Exogenous Hcys During Conotruncal Septation.
A second regimen was devised where HcysTD treatment would be limited to three points around the window of time when the principal features of aorticopulmonary septation are underway: (i) 24 h, corresponding with the initial formation of the cranial neural fold; (ii) 48 h, corresponding with the emergence of the nascent aortic arch ectomesenchyme (24); and (iii) 72–75 h, corresponding with the arrival of the neural crest-derived ectomesenchyme in the truncus arteriosus and the initial expression of the elastogenic phenotype (25, 26). This is approximately equivalent to days 18–24 in human development, when the ectomesenchymal cells are migrating through the aortic arches; and treatment with agents effecting these cells may result in congenital cardiovascular malformations. This window of time also includes the latter part of the process of neurulation. By using the same series of dilutions as described above, the HcysTD again was found to be 10 μmol of d,l-Hcys or 5 μmol of l-Hcys thiolactone (see below, Results). Embryos thereafter were treated with the HcysTD once, at either 24, 48, or 72 h; twice, at 24 and 48 or 48 and 72 h; or three times, at 24, 48, and 72 h. These embryos were harvested at day 9 following the completion of the process of conotruncal septation.
Continuous Treatment.
A small series of embryos was treated with the HcysTD at each 24 h interval, from time 0, day 9 of incubation, and the embryos were harvested at day 9.
Effect of Exogenous Hcys on Serum Hcys.
The HcysTD was determined for this study empirically, by titrating for the best rate of survival/teratogenic effect; that is, the concentration of Hcys that maintained a survival near that of control while producing more defects than lower concentrations. However, it was essential to learn the dynamics of l-Hcys passage into the serum and to compare these results with other experimental and epidemiologic results, to determine actual serum levels of l-Hcys. Therefore, 5 μmol of l-Hcys as l-Hcys thiolactone or saline control was placed in the enlarged airspace, on the highly vascularized membrane of embryos aged 9–12 days, and a 100-μl serum sample was drawn at times 0 min, 15 min, 1 h, and 2 h. This procedure was repeated, with the addition of folate in the concentrations shown above. Serum samples were frozen immediately and thawed at the time of analysis. l-Hcys and folate concentrations were measured according to our published methods (16, 17).
Circulation does not begin in the avian embryo until 44 h, and the vessels of the 53 h embryo are too small for reliable acquisition of serum samples of adequate size for Hcys analysis. However, it was possible to gain some information about l-Hcys passage into the early embryos by the following procedure: 5 μmol of l-Hcys thiolactone or saline control was placed on the chorioallantoic membrane of a series of 53 h embryos, which were harvested and frozen after intervals of 0 min, 15 min, 1 h, and 2 h. Then the l-Hcys content per whole embryo was analyzed by our published methods (16, 17).
Effect of Supplementary Folate.
To determine if supplementary folate would effect the results of exogenous Hcys treatment, the above experiments with the HcysTD were repeated, with the simultaneous addition of a supplementary daily dose of 0.1 μg (100.0 ng) of folic acid. To determine the dynamics of folate crossing the membranes into the embryonic serum, serum folate was measured in embryos of 9–12 days following the administration of 0.1 μg of folate, given in the same solution as the HcysTD.
Histologic Methods.
Embryos harvested at 53 h and fixed in Bouin’s solution were embedded and sectioned serially by our published methods (25, 26). Embryos harvested at day 9 were fixed in methanol-based Carnoy fluid; those that showed grossly visible defects were photographed whole. Control and treated embryos were embedded and sectioned serially. Consecutive serial sections were mounted and observed without stain. Even-numbered sections were stained either with the method of Spicer (27) or with periodic acid-Schiff. Selected odd-numbered methacarn fixed sections from day 9 embryos were used for immunofluorescence identification of smooth muscle α-actin according to our published methods (2).
RESULTS
Result of Hcys Treatment During Neurulation.
No congenital defects were found with any of the control amino acids, and few defects were observed with 0.5–7.5 μmol of d,l-Hcys. At the opposite pole, 20 μmol of d,l-Hcys was lethal. Grossly obvious neural tube defects were observed in embryos treated with 5–15 μmol of d,l-Hcys (Table 1). The highest ratio of survival/neural tube defects was with 10 μmol of d,l-Hcys. In general, we found that the preparation and use of d,l-Hcys was made complex by the rapid decline of its effectiveness in solution; therefore, we turned to the more stable thiolactone form. Unlike Hcys per se, Hcys thiolactone is available commercially in the l-only form (Sigma). When embryos were treated with 5 μmol of l-Hcys thiolactone, the result was dysmorphogenesis that was equal to that induced by a fresh solution of d,l-Hcys. From this result it was concluded that the d-isomer in the presence of the l-isomer had not contributed to the dysmorphogenic effect. Since 10 μmol of d,l-Hcys was equivalent to 5 μmol of l-Hcys thiolactone, data that was derived from these can be pooled and referred to as the HcysTD (Table 2).
Table 1.
Hcys treatment at time 0, stage 14 (53 h)
| Control | Hcys, 0.5–2.5 μmol | Hcys, 5–7.5 μmol | Hcys, 10 μmol | Hcys, 15 μmol | Hcys, 20 μmol | |
|---|---|---|---|---|---|---|
| Survival | ≥85% | ≥85% | ≥85% | ≥85% | ≤25% | 0.0% |
| NTD | 0% | 0–5% | ≤18% | ≥30% | ≥75% | — |
Embryos were treated with d,l-Hcys in the amounts indicated, at incubation time 0, 24, and 48 h. They were harvested at 53 h (stage 14) and evaluated for the occurrence of grossly or histologically visible defects in neural tube closure. d,l-Hcys (10 μmol) gave the best rate of (survival) × (% defects). NTD, neural tube defects; control, pooled embryos with no treatment, saline treatment, and treatment with amino acids other than Hcys.
Table 2.
Fifty-three-hour embryos
| Treatment | Survivors/treated (%) | Delay (%) | NTD
|
Total NTD (%) | ||
|---|---|---|---|---|---|---|
| Spinal (%) | Cranial (%) | Multiple (%) | ||||
| HcysTD | 167/187 (89%) | 29/167 (17%) | 5 (3%) | 18* (11%) | 23 (14%) | 46/167 (27%) |
| HcysTD plus FA | 15/22 (68%) | 6 (40%) | 4 (27%) | 1 (7%) | 3 (20%) | 8/15 (53%) |
Embryos were treated with 5 μmol of l-Hcys thiolactone (HcysTD) as in Fig. 1, and analyzed for the occurrence of neural tube closure defects. Embryos in the Delay column were at stages 10–12 rather than at stage 14, as predicted by the time of incubation (53 h). A second set of embryos was treated simultaneously with 5 μmol of l-homocysteine thiolactone to which was added 0.1 μg (100.0 ng) folic acid. The folic acid offered no protection from the dysmorphogenic effects of the Hcys. Some members of the HcysTD-treated group that showed cranial defects also showed a duplication or branching of the notochord just caudal to the cranial closure defect. FA, folic acid; NTD, neural tube defects.
Four of 18 showed duplication of the notochord.
About half of the embryos treated with HcysTD were indistinguishable from control embryos. Some HcysTD-treated embryos showed a delay in development of 6–12 h that was independent of any obvious anatomic defect. Although it was not possible to stage the most severely defective embryos (i.e., most of those with cranial defects and all of those with multiple neural tube defects), the embryos with spina bifida did not appear to have suffered any consistent delay in development compared with controls (Fig. 1). Therefore, the significance of the observed delays is not obvious.
Figure 1.

Embryos stained with neutral red after 53 h of incubation (stage 14). The embryo treated with leucine (a) has experienced normal neural tube closure including the lumbar region (arrowheads), whereas the embryo treated with HcysTD (b) shows a failure of closure (nascent spina bifida) in the lumbar region (arrowheads).
Some of the d,l-Hcys-treated embryos showed a pronounced “torsion defect,” with a twist of up to 270° in the thoracic or lumbar. This phenomenon was not necessarily associated with any detectable gross anatomic or histologic defect, and its significance is not known.
Singular spina bifida (Fig. 1) was the least common defect of neural tube closure; the most common neural tube defect included both cranial and spinal defects (“MULTIPLE,” Table 2). An interesting feature in embryos with cranial defects was the occurrence of notochord duplication (Fig. 2).
Figure 2.

Nonconsecutive serial sections from rostral to caudal through a 53-h embryo that showed a failure of neural tube closure in the cranial region (a). Sections caudal to the head (b–d) show a paired or branched notochord (arrowheads) that fuses caudally (d). Sections b–d are at a slightly higher magnification than a to show notochord details. NO, open neural tube; NC, closed neural tube.
Gross anatomic and histologic examinations both indicated that the defects resulting from HcysTD treatment were limited to the neural tube and face, including the branchial arches. The heart was of generally normal size and shape (e.g., Fig. 1b) and appeared to be beating normally even in severely defective embryos. Histologic examination of hearts from 53 h embryos revealed no obvious anatomic difference between HcysTD-treated and untreated embryos. In no case did any somites appear to be abnormal independent of a neural tube defect.
Result of Nine Daily Hcys Treatments: Time 0, Day 9.
The most prominent feature of continual Hcys treatment was its lethality. Nine consecutive daily treatments with HcysTD were lethal to nearly 90% (21/24) of the treated embryos. One survivor was normal; one showed a ventral midline closure defect with ventricular septal defect (VSD) similar to Fig. 3; and the third had VSD, ventral midline closure defect, and anencephaly.
Figure 3.
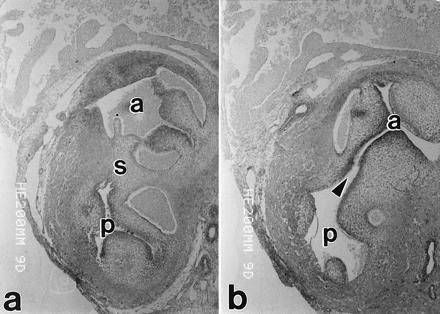
Nonconsecutive serial sections through the cardiac outflow tract from a 9-day embryo that had been treated with HcysTD at three time points during the formation of the aorticopulmonary septum (incubation days 2–4). The septum has separated the aorta (a) from the pulmonary trunk (p) down to the level shown on the left; but a few sections upstream (toward the heart), there is an obvious subaortic VSD (arrowhead).
Result of Three Daily Hcys Treatments During Conotruncal Septation.
No defects were induced by HcysTD-treatment at only one stage or at two consecutive stages, or with leucine, methionine, or cysteine. Three consecutive treatments led to heart, neural tube, and ventral closure defects (Table 3). Details of these results are discussed below.
Table 3.
Nine-day embryos
| Treatment | Survivors/treated (%) | Ventral closure defects (%) | Heart defects (%) | Neural tube defect (%) |
|---|---|---|---|---|
| Control | 45/54 (83%) | 0 | 0 | 0 |
| FA | 16/18 (89%) | 0 | 0 | 0 |
| HcysTD plus FA | 27/36 (75%) | 1/27 (4%) | 0 | 0 |
| HcysTD | 84/108 (78%) | 75/84 (79%) | 19/84 (23%) | 9/84 (11%) |
Embryos were treated with 5 μmol l-Hcys (HcysTD) on days 2, 3, and 4; or with HcysTD to which was added 0.1 μg (100.0 ng)/day folic acid. Embryos were harvested at day 9 and evaluated for the occurrence of grossly and histologically identifiable developmental defects. The addition of folic acid eliminated the high rate of developmental defects that was found with HcysTD alone. FA, folic acid.
Heart defects.
Histologic analysis showed VSDs in the hearts of 4/18 of the surviving 9-day embryos treated with HcysTD (Fig. 3). One potential cause of VSD that could be identified histologically was the failure of the neural crest-derived ectomesenchyme to aggregate at the appropriate site. As we have reported previously, ectomesenchyme aggregates between the aorta and the pulmonary artery during normal conotruncal septation and the cells express vascular smooth muscle α-actin during the process of septation (11). In the present study in embryos with VSD, there either was no sign of such an aggregation of cells or there was an ectopic aggregation of α-actin-positive cells (Fig. 4). The coronary arteries appeared to be normal.
Figure 4.
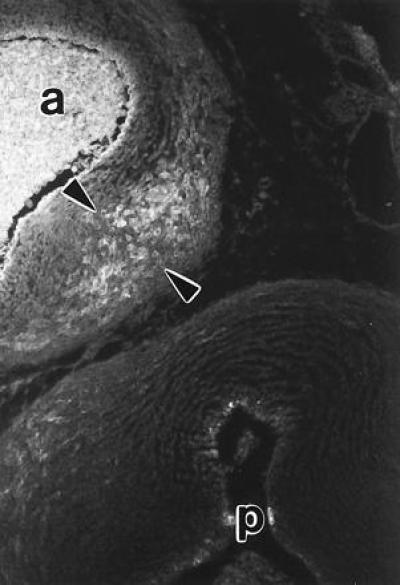
This 9-day embryo was treated with HcysTD and showed a VSD as in Fig. 6. The normal condensation of differentiated vascular smooth muscle cells that forms the aorticopulmonary septum at the level of the valves was absent. However, an ectopic condensation of cells that was positive for vascular smooth muscle α-actin (arrowheads) was located downstream from the heart between the aorta (a) and the point of bifurcation of the pulmonary trunk (p). This finding indicates that the HcysTD had interfered with the timing of migration or differentiation of the neural crest precursors of the septal smooth muscle cells. The smooth muscle cells of the aorta and pulmonary arteries per se do not express α-actin until about day 14.
Neural tube defects.
Spina bifida (Fig. 5) and anencephaly (Fig. 6) were observed, but there was no occurrence of multisite defects or of the extensive failure of closure that was most common at stage 14. The spinal lesion showed anatomic features that indicated a duplication of portions of the spinal cord (Fig. 7).
Figure 5.
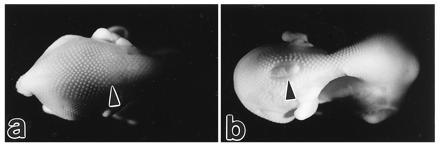
The control 9-day embryo (a) shows a normal closure of the thoraco-lumbar neural tube (arrowhead), while the embryo treated with HcysTD shows an obvious spina bifida (arrowhead).
Figure 6.
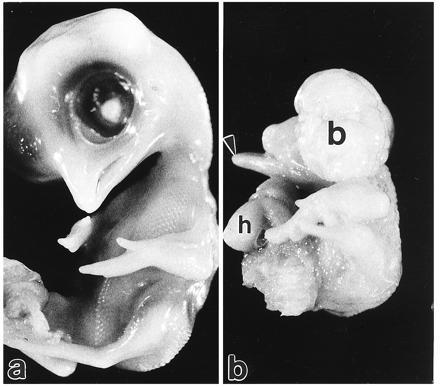
The control 9-day embryo (a) demonstrates the normal appearance of the head and ventral body wall. An embryo of the same age treated with HcysTD (b) is grossly abnormal. There is a lower beak (arrowhead) but no upper beak, indicating a failure of closure of the face. The cranium is completely open and brain tissue is exposed (b), as with anencephaly. The ventral wall of the thorax is open and the heart is extruded as in ectopia cordis (h).
Figure 7.
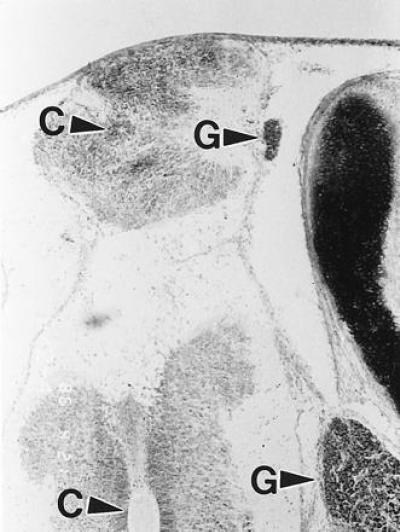
A 9-day embryo treated with HcysTD showed a spina bifida grossly similar to that shown in Fig. 5. Histologic sections of this lesion stained with the Spicer method showed a small, abnormal duplication of a fairly normal cord; the redundant branch was separated from the outside by a thin membrane. C, central canal; G, spinal ganglion.
Ventral closure defects.
A ventral closure syndrome occurred in about three/quarters of the embryos treated with HcysTD during conotruncal septation (Table 3). This anomaly appeared to be an avian equivalent to the mammalian “pentalogy of Cantrell” defect (28) and included schisis defects over the heart and abdominal viscera; as well as the ectopia cordis and lack of a visceral pericardium that is illustrated in Fig. 6b. In about half of these pentalogy-like cases, there was an associated VSD, as predicted by the literature (28).
Effect of Exogenous Hcys on Serum Hcys.
The normal level of serum Hcys (i.e., in the absence of exogenous Hcys) was 9.24 ± 1.4 nmol/ml in the untreated embryos (Fig. 8). When 5 μmol of l-Hcys thiolactone was placed on the embryonic membrane, there was a rapid rise in serum Hcys with a peak in 1 h at ≈150 nmol/ml, followed by a decline.
In the 53 h embryos, Hcys was present in low levels (<3 nmol per whole embryo). In 15 min following treatment with 5 μmol of l-Hcys thiolactone, Hcys per whole embryo rose to 37 μmol per embryo, and in 1 h posttreatment it rose slightly more, to ≈39 μmol. By 2 h posttreatment, Hcys had declined to 14 μmol per embryo.
Effect of Supplementary Folate.
Day 9 embryos that received the 5 μmol of l-Hcys thiolactone on days 2, 3, and 4 (the conotruncal septation interval) were protected from dysmorphogenesis by a daily folate supplement of 100.0 ng given simultaneously with the Hcys (Table 3). This is consistent with the finding that folate given simultaneously with HcysTD sharply curtails the rise in serum Hcys (Fig. 8). On the other hand, supplementary folate offered no protection to 53 h embryos that had been given the solution containing folate and the HcysTD at times 0, 24, and 48 h (Table 2).
DISCUSSION
These results show that Hcys per se is teratogenic when avian embryos are exposed to concentrations that are known to occur in humans. This direct teratogenic effect has been hypothesized previously based upon epidemiologic studies (19) but has not been demonstrated experimentally. Based upon the present results, it is probable that the congenital defects that accompany experimental folate deficiency are a result of elevated maternal Hcys; and conversely, that the primary useful effect of folate supplementation for protection against dysmorphogenesis is in the reduction of maternal serum Hcys. The importance of regulation of maternal Hcys is implied by the results of the present study, where supplementary folate provided directly to early (up to 53 h) embryos was not protective—i.e., the embryos did not use the folate to protect themselves. This situation is analogous to the teratogenic effect of folate deficiency in humans, which must effect the embryos at the same early stage of development. This scenario would provide one explanation about the observation that elevated Hcys may be associated with dysmorphogenesis in offspring of hyperhomocysteinemic women with normal serum and erythrocyte folate (29).
Folate supplementation in the older embryos limited the rise in the first hour in exogenous Hcys to an average level of ≈45 nmol/ml, compared with 150 nmol/ml in the absence of supplementary folate, and in the closed sytem of the avian egg this was sufficient to prevent most of the dysmorphogenesis induced by Hcys. There is no data available regarding the concentration of Hcys that may be teratogenic to the human embryo; data regarding maternal Hcys may or may not mimic the level that reaches the embryo in man. Therefore it is not shown by this study that the same levels will be teratogenic in man.
The ability of the older embryos by themselves to use the folate for reduction of ambient Hcys concentration may relate to the presence of a liver. Liver cells are rich in the enzyme complex that rapidly converts Hcys to methionine, and the liver is the major site of Hcys conversion. Visible liver anlagen are not present in these embryos until the roots of the omphalomesenteric vessels have fused, after ≈50 h. Thus for the early embryos that were not protected by folate, there was no liver for virtually the entire treatment period, 0–53 h, and presumably therefore they had limited ability to metabolize the exogenous Hcys. On the other hand, the embryos that were treated during days 2, 3, and 4 had a liver undergoing rapid growth for the entire treatment period and were afforded significant protection.
The validity of these results is supported by the fact that the Hcys concentrations at the level of the embryo per se were found to be in the physiological range and thus should escape the criticism that applies when extremely high, cytotoxic levels of Hcys are used to produce an effect. Our ability to regulate both the Hcys concentration and the folate concentration at the level of the embryo was improved significantly through our use of the avian embryo, which is highly accessible compared with the mammalian embryo. As Bradley Keller has stated (personal communication), the use of the avian embryo probably removes an order of magnitude of experimental complexity, compared with the use of mammalian embryos in similar experiments. To overcome the complexity and potential ambiguity introduced with vaso-placental interactions, numerous investigators have used mammalian embryos in vitro for analyses such as these. However, mammalian embryos only are viable in vitro for ≈48 h, and during this brief interval they are maintained in conditions that are profoundly different from the conditions that prevail during normal embryogenesis. For these reasons, perturbations of the development of mammalian embryos in vitro may be difficult to interpret.
The avian embryo also has unique value for these studies on the basis of retrospective and prospective scientific considerations. A substantial amount of our basic knowledge about both neural crest and neural tube development has been derived from studies of the avian embryo. Furthermore, as these studies advance to the molecular level, the avian genome presents special features that give it a high priority among the vertebrates for the study of regulatory genes during development (30), including the comparative lack of untranslated DNA in the avian genome.
In spite of the scientific and technical value of the avian embryo for developmental studies, the caveat must be borne in mind that no experimental model system reproduces exactly the conditions that exist for the human embryo in utero. The results of this study of avian embryos in ovo therefore should be interpreted broadly: Hcys is dysmorphogenic to the neural tube and heart, but neither the chronology of events nor the exact Hcys concentrations shown here may be extrapolated directly to the human embryo.
This study has duplicated the widely reported “schisis association” (31). In this association, ventral and dorsal midline closure defects including cleft palate are associated statistically; the closure defects in turn are associated with cardiac septal and other neural crest defects. A phenotype that shows many of the associated defects or relatively few may be included in this statistical association, for which the biological basis is unknown. It has been suggested that the members of this association may represent a specific developmental field (32), but the the developmental field in question has not been defined.
The data given here are not especially informative about any action of Hcys at the molecular level that may result in the observed dysmorphogenesis. However, some of these results seem at odds with the popular idea that Hcys is simply a cytotoxic agent and that it may exert a teratogenic role by killing cells: in the case of the notochord in the 53-h embryos and the spinal cord in the 9-day embryos, there was duplication rather than reduction of tissues. Similar duplication of the embryonic spinal cord results from a growth-promoting effect of retinoic acid (33), indicating that Hcys also may be a growth promoter. Indeed, Hcys has a growth factor-like effect upon vascular smooth muscle cells (34). We hypothesize that the teratogenic effect of Hcys is based on this growth factor-like effect, altering with this effect the expression of genes critical to the process of neural tube closure/neural crest migration. This hypothesis is consistent with the possibility that Hcys may have growth factor-like activity and at the same time be cytotoxic under some circumstances: Hcys at low concentrations appears to be essential for the normal growth of rat embryos, but is lethal at higher concentrations (35).
Acknowledgments
This study was supported by National Institutes of Health Grant HL55940-01.
Footnotes
Abbreviations: Hcys, homocysteine; HcysTD, teratogenic dose of d,l-Hcys or l-Hcys thiolactone; VSD, ventricular septal defect.
References
- 1.Seaver L H, Pierpont J W, Erickson R P, Donuerstein R L, Cassidy S B. J Med Genet. 1994;31:830–834. doi: 10.1136/jmg.31.11.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baird C D, Nelson M M, Monie I W, Evans H M. Circ Res. 1954;11:544–554. doi: 10.1161/01.res.2.6.544. [DOI] [PubMed] [Google Scholar]
- 3.Eskes T K, Steegers-Theunissen R P. Eur J Obstet Gynecol Reprod Biol. 1994;53:147–152. doi: 10.1016/0028-2243(94)90225-9. [DOI] [PubMed] [Google Scholar]
- 4.Lucock M D, Wild J, Schorah C J, Levene M I, Hartley R. Biochem Med Metab Biol. 1994;52:101–114. doi: 10.1006/bmmb.1994.1040. [DOI] [PubMed] [Google Scholar]
- 5.Milunsky A, Jick H, Jick S S, Bruell C L, MacLaughlin D S, Rothman K J, Willett W. J Am Med Assoc. 1989;262:2847–2852. doi: 10.1001/jama.262.20.2847. [DOI] [PubMed] [Google Scholar]
- 6.Scott J M, Weir D G, Molloy A, McPartlin J, Daly L, Kirke P. Ciba Found Symp. 1994;181:180–191. doi: 10.1002/9780470514559.ch11. [DOI] [PubMed] [Google Scholar]
- 7.Seller M J. Ciba Found Symp. 1994;181:161–179. doi: 10.1002/9780470514559.ch10. [DOI] [PubMed] [Google Scholar]
- 8.Gunther T, Struwe M, Aguzzi A, Shughart K. Development (Cambridge, UK) 1994;120:3119–3130. doi: 10.1242/dev.120.11.3119. [DOI] [PubMed] [Google Scholar]
- 9.Simpson J L, Mills J L, Rhoads G G, Cunningham G C, Hoffman H J, Conley M R. Prenatal Diagn. 1991;11:641–648. doi: 10.1002/pd.1970110823. [DOI] [PubMed] [Google Scholar]
- 10.Wald N J. Ciba Found Symp. 1994;181:192–211. doi: 10.1002/9780470514559.ch12. [DOI] [PubMed] [Google Scholar]
- 11.Beall A C, Rosenquist T H. Anat Rec. 1990;226:360–366. doi: 10.1002/ar.1092260313. [DOI] [PubMed] [Google Scholar]
- 12.Rosenquist T H, Fray-Gavalas C A, Waldo K, Beall A C. Am J Anat. 1990;188:339–356. doi: 10.1002/aja.1001890406. [DOI] [PubMed] [Google Scholar]
- 13.Serbedzija G N, Bronner-Fraser M, Fraser S E. Development (Cambridge, UK) 1994;120:1709–1718. doi: 10.1242/dev.120.7.1709. [DOI] [PubMed] [Google Scholar]
- 14.Heid M K, Bills N D, Hinrichs S H, Clifford A J. J Nutr. 1992;122:888–894. doi: 10.1093/jn/122.4.888. [DOI] [PubMed] [Google Scholar]
- 15.Kingman S. J NIH Res. 1995;7:44–46. [Google Scholar]
- 16.Miller J W, Nadeau M R, Smith D, Selhub J. Am J Clin Nutr. 1994;59:1033–1039. doi: 10.1093/ajcn/59.5.1033. [DOI] [PubMed] [Google Scholar]
- 17.Miller J W, Nadeau M R, Smith J, Smith D, Selhub J. Biochem J. 1994;298:415–419. doi: 10.1042/bj2980415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCully K S. Nat Med. 1996;2:386–389. doi: 10.1038/nm0496-386. [DOI] [PubMed] [Google Scholar]
- 19.Steegers-Theunissen R P, Boers G H J, Trijbels F J M, Finkelstein J D, Blom H J, Thomas C M G, Borm G F, Wouters M G A J, Eskes T K A B. Metabolism. 1994;43:1475–1480. doi: 10.1016/0026-0495(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 20.Brown J C, Strain J J. J Nutr. 1990;120:1068–1074. doi: 10.1093/jn/120.9.1068. [DOI] [PubMed] [Google Scholar]
- 21.Hart R C, McCue P A, Ragland W L, Winn K J, Unger E R. Teratology. 1990;41:463–472. doi: 10.1002/tera.1420410411. [DOI] [PubMed] [Google Scholar]
- 22.Hart R C, Winn K J, Unger E R. Tetratology. 1992;46:533–539. doi: 10.1002/tera.1420460602. [DOI] [PubMed] [Google Scholar]
- 23.Hamilton H L, Hamburger V. J Morphol. 1951;38:49–92. [PubMed] [Google Scholar]
- 24.Kirby M L, Kumiski D H, Myers T, Cerjan C, Mishima N. Dev Dyn. 1993;198:296–311. doi: 10.1002/aja.1001980407. [DOI] [PubMed] [Google Scholar]
- 25.Rosenquist T H, McCoy J R, Waldo K, Kirby M L. Anat Rec. 1988;221:860–871. doi: 10.1002/ar.1092210411. [DOI] [PubMed] [Google Scholar]
- 26.Rosenquist T H, Modis L. Anat Rec. 1991;229:116–124. doi: 10.1002/ar.1092290113. [DOI] [PubMed] [Google Scholar]
- 27.Spicer S S. Stain Technol. 1961;36:337–340. doi: 10.3109/10520296109113306. [DOI] [PubMed] [Google Scholar]
- 28.Carmi R, Boughman J A. Am J Med Genet. 1992;42:90–95. doi: 10.1002/ajmg.1320420118. [DOI] [PubMed] [Google Scholar]
- 29.Mills J L, McPartlin J M, Kirke P N, Lee Y J, Conley M R, Weir D G, Scott J M. Lancet. 1995;345:149–151. doi: 10.1016/s0140-6736(95)90165-5. [DOI] [PubMed] [Google Scholar]
- 30.Fainsod A, Gruenbaum Y. Poultry Avian Biol Rev. 1995;6:19–34. [Google Scholar]
- 31.Czeizel A. Am J Med Genet. 1981;10:25–35. doi: 10.1002/ajmg.1320100105. [DOI] [PubMed] [Google Scholar]
- 32.Opitz J M, Gilbert E F. Am J Med Genet. 1982;12:443–455. doi: 10.1002/ajmg.1320120408. [DOI] [PubMed] [Google Scholar]
- 33.Kohga H, Obata K. Neurosci Res. 1992;13:175–187. doi: 10.1016/0168-0102(92)90057-j. [DOI] [PubMed] [Google Scholar]
- 34.Tsai J-C, Perrella M A, Youshizumi M, Hsich C-M, Haber E, Sclegel R, Lee M-E. Proc Natl Acad Sci USA. 1994;91:6369–6373. doi: 10.1073/pnas.91.14.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Aerts L A, Blom H J, Deaubreu R A, Trijbels F J, Eskes T K, Copius J H, Noordhoek J. Teratology. 1994;50:348–360. doi: 10.1002/tera.1420500506. [DOI] [PubMed] [Google Scholar]