

Abstract
The Bio-Dictionary-based Gene Finder was used to reassess the coding potential of the AD169 laboratory strain of human cytomegalovirus and sequences in the Toledo strain that are missing in the laboratory strain of the virus. The gene-finder algorithm assesses the potential of an ORF to encode a protein based on matches to a database of amino acid patterns derived from a large collection of proteins. The algorithm was used to score all human cytomegalovirus ORFs with the potential to encode polypeptides ≥50 aa in length. As a further test for functionality, the genomes of the chimpanzee, rhesus, and murine cytomegaloviruses were searched for orthologues of the predicted human cytomegalovirus ORFs. The analysis indicates that 37 previously annotated ORFs ought to be discarded, and at least nine previously unrecognized ORFs with relatively strong coding potential should be added. Thus, the human cytomegalovirus genome appears to contain ≈192 unique ORFs with the potential to encode a protein. Support for several of the predictions of our in silico analysis was obtained by sequencing several domains within a clinical isolate of human cytomegalovirus.
Human cytomegalovirus (HCMV) is a ubiquitous β-herpes-virus. Infection is usually asymptomatic in healthy individuals, but the virus is not cleared and persists in a latent state. Primary infection or reactivation of latent virus in immunocompromised individuals can lead to severe disease, including pneumonitis (1). The double-stranded DNA genome of HCMV is composed of so-called unique long (UL) and unique short (US) domains, which are flanked on one end by terminal repeated sequences (TRL and TRS) and on the other end by internal repeats (IRL and IRS). In a population of HCMV DNA, four isoforms are attributable to recombination events that result in the US and UL regions being present in both orientations with respect to each other (2).
The ≈230,000-bp sequence and annotation of the AD169 laboratory strain of HCMV was published in 1990 (3). This first annotation predicted that AD169 has the potential to encode 208 ORFs, of which 14 are duplicated within the TRL/IRL repeats. The criteria that were used for ORF prediction required that a presumptive ORF encode a polypeptide at least 100 aa in length and that it not overlap a longer ORF by 60% or more. In regions where more than one overlapping ORF was identified, the longer of the two ORFs was generally accepted as the valid ORF. In accordance with the predicted genomic layout of Epstein-Barr virus (4) and herpes simplex virus 1 (5), the AD169 ORFs generally lay in an end-to-end configuration with little noncoding sequence. In a few regions lacking ORFs that fulfilled the above-mentioned criteria, ORFs were predicted by identifying transcriptional regulatory signals upstream of coding sequences, by considering codon usage bias, and by searching for protein motifs. The authors noted that the annotation criteria could miss ORFs that are small or that represent a single exon within a gene that is spliced. Since the original annotation, several additional AD169 ORFs have been identified, including UL21.5, UL111a, UL48/49, and UL80.5 (6-9). In related work, HCMV ORFs were processed by using two different computational methods to predict their putative functions and other properties (10, 11).
AD169 has a restricted cellular tropism as compared with clinical isolates (12), because it is tissue culture-adapted and has undergone duplications, deletions, and numerous more subtle sequence changes in comparison with clinical isolates of HCMV. The Toledo strain, which has been passaged to a more limited extent in cell culture, contains a block of additional ORFs that are absent from AD169 (13).
Recently, the AD169 genome was compared with that of chimpanzee cytomegalovirus, which has not been extensively passaged in the laboratory (14). It was assumed that protein-coding ORFs would be conserved between the two viruses, but noncoding ORFs would diverge. The comparison revealed that the original annotation of AD169 contained 51 ORFs not found in chimpanzee cytomegalovirus. In addition, the comparison identified 10 ORFs present in both viruses that had not been considered in earlier studies. The search for orthologues has made a major contribution to our understanding of the viral genome, but HCMV ORFs with human virus-specific functions would be missed.
To reassess the coding potential of HCMV, we used the Bio-Dictionary-based Gene Finder (BDGF) algorithm (15), which was originally designed to predict functional ORFs in archaeal and bacterial genomes. The BDGF program evaluates the coding potential of a given region of DNA by interrogating a collection of amino acid patterns that were derived by carrying out pattern discovery on the Swiss-Prot/TrEMBL database of amino acid sequences. Each pattern appears twice or more in the processed input, and the collection as a whole, which is termed the Bio-Dictionary (15), nearly completely accounts for the sequence space of identified proteins. The BGDF program gauges whether a DNA region is likely to code for a gene by determining the number and composition of matches it contains to entries in the Bio-Dictionary.
HCMV ORFs were processed by using the BDGF algorithm, and they also were used to interrogate the sequences of chimpanzee, rhesus, and murine cytomegaloviruses to search for orthologues. The analysis indicates that 37 of the HCMV ORFs predicted in earlier studies are not likely to encode proteins, and it identifies additional ORFs that were not considered in earlier studies. Several of the coding regions predicted by the BDGF were validated by sequence analysis of a HCMV clinical isolate.
Methods
BDGF Analysis. The sequence of the HCMV AD169 strain (GenBank accession no. X17403) was analyzed by using the macvector program (Accelrys, San Diego) to identify ORFs with a coding capacity ≥50 aa. A total of 2,434 ORFs were identified and then analyzed with the BDGF algorithm (15). The algorithm used a Bio-Dictionary that was derived from the January 2002 release of the Swiss-Prot/TrEMBL database, and contains ≈55 million amino acid patterns. These patterns account for essentially all the sequence space of identified proteins. In the original version of the BDGF algorithm (15), weights for the Bio-Dictionary patterns were derived by training the program with a collection of archaeal and bacterial sequences. To make the BDGF algorithm “sensitive” to sequences that appear in a virus that has evolved in human cells, a new version of BDGF was trained with all Homo sapiens sequences available within the SwissProt database (16) and all of the reported genes of the eukaryote Ciona intestinalis (17). Moreover, we allowed each the 61 possible non-stop codons to act as the start of an ORF. We normalized the scores that BDGF assigned to the reported ORFs by dividing the score by the amino acid length of the corresponding ORF. The C. intestinalis/H. sapiens-trained BDGF algorithm can be accessed at http://cbcsrv.watson.ibm.com/Tgi.html.
Orthologue Identification. The sequences of chimpanzee (accession no. NC_003521), rhesus (accession no. AY186194), and murine (accession no. NC_004065) cytomegaloviruses were used to identify all possible ORFs ≥50 aa long. Each ORF was translated and used as a query sequence in a BLASTP analysis against a database generated from the translations of all predicted ORFs present within the AD169 HCMV genome and the ≈15 kB of additional sequence found in the Toledo HCMV strain (accession no. U33331). A chimpanzee, rhesus, or murine cytomegalovirus query sequence with a BLASTP alignment to HCMV at a statistical significance ≤10-5 was scored as an orthologue. A sequence was determined to contain a Kozak motif if it had six of the nine nucleotides within the consensus sequence CC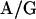 CCATGG with the ATG required (18).
CCATGG with the ATG required (18).
Viral DNA Isolation and Sequence Analysis. The Coz clinical strain of HCMV (a kind gift of S. Spector, University of California, San Diego), isolated from the blood of an AIDS patient with cytomegalovirus retinitis, was minimally passaged in human foreskin fibroblasts that were maintained in medium containing 10% FCS. Viral DNA was prepared from partially purified virions (19) by phenol extraction. Virion DNA was used as template for PCR amplification by using the Expand High Fidelity PCR system (Roche Applied Science). PCR products were gel-purified by isolation into GF/A glass microfiber filters (Whatman) backed by Spectra/Por cellulose dialysis membranes with a molecular weight cutoff of 12,000-14,000 Da (Spectrum Laboratories, Houston), and used as a template for dye-terminator sequencing. Sequences were analyzed by using the macvector program.
Results
BDGF Analysis of HCMV ORFs Identified in Previous Annotations. To reevaluate the coding potential of HCMV, we used the BDGF algorithm to process the sequence of HCMV. We gauged the coding potential of each ORF that was ≥50 aa in length by taking into account the number and nature of Bio-Dictionary patterns that could be located in the amino acid translation of the ORF under consideration. The higher the normalized BDGF score, the greater the likelihood that the ORF encodes a protein. Fig. 1 displays the results of the BDGF analysis of the original annotated AD169 ORFs (3), the additional Toledo ORFs (13), and several AD169 ORFs identified by their correspondence to chimpanzee cytomegalovirus orthologues (14). The ORFs in are listed from the highest to the lowest normalized BDGF score.
Fig. 1.

BDGF analysis of HCMV ORFs. All previously annotated ORFs of HCMV are listed from high (upper left) to low (lower right) normalized BDGF (N-BDGF) scores. Expression of the ORF was scored as positive if a report exists in the published literature directly demonstrating the expression of a protein or showing that a mutation within the ORF generates a viral growth phenotype. NR indicates that no report was found. The presence of orthologues corresponding to HCMV ORFs within the genomes of chimpanzee cytomegalovirus (CCMV), rhesus cytomegalovirus (RhCMV), and murine cytomegalovirus (MCMV) are indicated. Green boxes designate characteristics favoring the conclusion that an ORF encodes a protein, and red boxes mark characteristics arguing that an ORF does not encode a protein. The length of the polypeptide potentially encoded by each ORF and the presence of a Kozak translational initiation motif (Kozak AUG) are provided for informational purposes. The UL65 ORF was reported to be expressed (26), but the reported polypeptide does not match the UL65 sequence (3).
The scores range from 131.3 for US19 to 0.9 for UL143. The average score is 19.2 and the median is 15.2. To determine how well the normalized BDGF scores capture the likelihood that an ORF encodes an expressed protein, we searched for published reports that described the expression of proteins encoded by ORFs or demonstrated a phenotype when an ORF is mutated (6, 19-39). A report of expression or function for an ORF unambiguously identifies it as a genuine ORF, although the lack of a report does not guarantee that the ORF lacks coding potential. We also tested for the presence of orthologous ORFs in the chimpanzee, rhesus, and murine cytomegaloviruses. Of the ORFs receiving a normalized BDGF score above the median, 93% express a functional protein or have an orthologue in one of the other cytomegalovirus genomes. In contrast, only 66% of the ORFs scoring below the median are known to express a protein or have a chimpanzee, rhesus, or murine virus orthologue. Thus, a correlation exists between high normalized BDGF scores and other characteristics indicative of functional ORFs.
It is not possible to set an absolute score threshold that can reliably discriminate between ORFs encoding proteins and those that do not, because poorly scoring HCMV ORFs might encode proteins with characteristics unique to cytomegaloviruses. For example, RL12 has a normalized BDGF score of 2.0, but the chimpanzee and rhesus viruses contain orthologues of the gene, arguing that it is likely a functional ORF. Consequently, to call into question the functionality of a previously annotated ORF, we set three criteria: (i) a normalized BDGF score below the median, i.e., <15.2; (ii) no published evidence for its expression; and (iii) no orthologue within the chimpanzee, rhesus, or murine viruses. These criteria identified 37 ORFs that are not likely to encode a polypeptide (Fig. 1, red-background entries). Among the questionable ORFs, UL129 had the highest BDGF score at 12.7. When the questionable ORFs are set aside, 183 ORFs with reasonable potential to encode a protein remain (Fig. 1, green-background entries).
Fig. 2 displays a map of the HCMV genome. Green arrows represent previously predicted ORFs that are likely to be coding, as determined by BDGF analysis in combination with the above-described additional filters. Red arrows mark previously annotated ORFs that our analysis suggests are not likely to be genuine coding ORFs. Many of the ORFs that are not likely to code for proteins are relatively small and form groups on the viral genome, i.e., RL5-RL7, UL58-UL59, UL62-UL68, UL106-UL111, UL125-UL126, and US35-US36.
Fig. 2.

Map of the HCMV genome. The green arrows represent previously annotated ORFs of HCMV likely to encode a bona fide polypeptide, red arrows designate ORFs unlikely to encode a polypeptide, and blue arrows indicate previously unrecognized ORFs that the present analysis predicts have high potential to encode proteins. The gray box marks the additional sequence found in the HCMV Toledo strain, locating it with respect to the AD169 genome. Rectangles superimposed on the line represent the sequence-identify terminal repeats. Each mark on the sequence line represents 1,000 bp.
It is possible that some regions of the HCMV AD169 laboratory strain were substantially altered during its extensive passage in fibroblasts. Alternatively, these regions of the viral genome might serve a purpose other than encoding translated ORFs. To distinguish between these possibilities, the genome of the HCMV Coz clinical isolate was sequenced in the regions corresponding to UL57-UL61, UL61-UL69, and US34-TRS1 (deposited in GenBank under accession nos. AY372064, AY372065, and AY372066, respectively). No ORFs were identified in the clinical isolate that were not present in the AD169, Towne, or Toledo strains, and the clinical isolate clearly lacked several of the poor-scoring ORFs in these regions, including UL58, UL63-UL66, UL68, US35, and US36, further validating our computational predictions. The UL106-UL111 region was sequenced in several clinical isolates for another purpose (C. Kulesza and T.S., unpublished data), and again the poor-scoring ORFs were not conserved.
Newly Recognized HCMV ORFs Likely to Encode Proteins. We searched for additional ORFs in AD169 with the potential to encode proteins by using the following criteria: (i) a normalized BDGF score >8.0, chosen because >95% of previously annotated ORFs with a score in this range were classified as genuine; (ii) the ability to encode a polypeptide with an N-terminal methionine that is ≥80 aa in length, chosen because >95% of previously annotated ORFs that we have classified as genuine start with an AUG codon and meet this size requirement; and (iii) a location on the genome that does not completely overlap another ORF with a higher normalized BDGF score. Twelve ORFs fulfilled all criteria and are listed in Table 1, along with their genomic coordinates and BDGF scores. The previously uncharacterized candidate ORFs have average (36.4) and median (23.8) normalized BDGF scores that are considerably higher than the corresponding scores for the set of previously annotated ORFs. At three locations on the genome, two candidate ORFs (ORFs 3 and 4; ORFs 7 and 8, and ORFs 9 and 10) occupy the same region. Chimpanzee cytomegalovirus contains orthologous ORFs corresponding to ORFs 7 and 9. This finding argues that these ORFs, rather than ORFs 8 and 10, which overlap them, are true protein-coding ORFs. Further experimentation will be required to confirm the functionality of the newly predicted ORFs and to unambiguously discriminate between substantially overlapping ORFs.
Table 1. Candidate HCMV ORFs.
| ORF | Position | Strand | Kozak | AA | N-BDGF | CCMV | RhCMV | MCMV |
|---|---|---|---|---|---|---|---|---|
| 1 | 19107-19358 | − | − | 83 | 15.2 | − | − | − |
| 2 | 36827-37144 | − | − | 105 | 84.8 | − | − | − |
| 3 | 94103-94417 | + | + | 104 | 50.3 | − | − | − |
| 4 | 94156-94464 | + | + | 102 | 25.2 | − | − | − |
| 5 | 95528-95788 | + | − | 86 | 17.3 | − | − | − |
| 6 | 133412-133783 | + | + | 123 | 8.4 | + | + | + |
| 7 | 145007-145588 | + | − | 193 | 22.0 | + | − | − |
| 8 | 144976-145251 | − | + | 91 | 12.7 | − | − | − |
| 9 | 170171-170854 | + | + | 227 | 79.8 | + | − | − |
| 10 | 170547-170825 | + | − | 92 | 54.6 | − | − | − |
| 11 | 3333-3599 | + | − | 88 | 22.3 | + | − | − |
| 12 | 228504-228773 | − | + | 89 | 44.5 | − | − | − |
Nucleotide position corresponds to accession no. X17403, except ORF 11, which corresponds to X33331. Kozak, presence or absence of a Kozak translational initiation motif; AA, length in amino acids of the protein that could be encoded by an ORF; N-BDGF, normalized BDGF score; CCMV, RhCMV, and MCMV, presence or absence of orthologues to the predicted HCMV ORF in the genome of the chimpanzee, rhesus, and murine cytomegalovirus, respectively.
These newly predicted ORFs are designated by blue arrows in Fig. 2. Two regions containing newly predicted ORFs were sequenced in the Coz clinical isolate (deposited in GenBank under accession nos. AY372067 and AY372068), and the analysis demonstrated that ORFs 6, 9, and 10 are present, demonstrating that these ORFs are not unique to the AD169 laboratory strain.
Discussion
The original annotation of the AD169 sequence designated functional ORFs primarily by choosing a set of the longest nonoverlapping ORFs. A problem with this approach is that the virus is G/C-rich (57.2%) with some regions of very high G/C content (3). Because the three stop codons are A/T-rich, the high G/C content allows for the generation of spurious ORFs because of the reduced number of randomly occurring stop codons (40, 41). This occurrence could cause larger nongenuine ORFs to be accepted at the expense of smaller genuine ORFs or it could identify nongenuine ORFs in noncoding regions. We have reevaluated the coding potential of HCMV by using the BDGF algorithm (15), together with supporting evidence provided by reports describing the expression of a polypeptide from an ORF or a functional consequence to mutating an ORF and by screening for orthologues of HCMV ORFs in chimpanzee, rhesus, and murine cytomegaloviruses. The BDGF predictions correlated reasonably well with the supporting evidence (Fig. 1), and our analysis indicated that 37 previously described ORFs are not likely to encode proteins. In general, the questionable ORFs were relatively small and were located in groups on the viral genome (Fig. 2). Sequence analysis of several regions within the genome of the HCMV clinical isolate did not find homologues for eight members of this group of ORFs that were tested, providing further support for the validity of these in silico predictions. Additional regions within clinical isolates must be sequenced to confirm the designations of the remaining questionable ORFs.
Several limitations characterize both statistics- and similarity-based gene-finding methods (15). In general, statistical methods will identify coding regions whose statistical behavior is similar to that of the set used to train the underlying model; they will thus have difficulty in identifying coding regions that are unlike the genome's average and will generally work best if a different model is built for each target genome. Heuristics-based sequence similarity programs have limitations when the length of the query is small and the size of the target database is large; most importantly, they make the implicit assumption that a gene such as the one coded by a candidate region already exists in the searched database, which is not always the case. The BDGF algorithm is built on a method that borrows the best attributes of the similarity searches and, at the same time, relies on implicit sequence statistics as in the Markov models. The reader is referred to ref. 15 for a detailed description of this gene-finding method.
The results of our analysis are in general agreement with the conclusions of Davison et al. (14), who searched for conserved orthologues of AD169 ORFs in the closely related chimpanzee cytomegalovirus. Indeed, the presence of a chimpanzee virus orthologue was a consideration in our analysis. However, use of the chimpanzee cytomegalovirus as the sole criterion for defining the genes of HCMV will eliminate ORFs that may have evolved after the divergence of the related viral hosts and are therefore specific to the human virus. For example, chimpanzee cytomegalovirus lacks a UL111a or UL21.5 orthologue, and, as a result, orthologue analysis would predict that UL111a and UL21.5 are not real ORFs. However, both UL111a and UL21.5 have been reported to be expressed (7, 8) and both score well by BDGF analysis (Fig. 1, scores of 42.8 and 21.9, respectively). Additional ORFs with BDGF scores above the median (RL2, RL8, UL41, UL60, UL101, and UL137) would be removed from the HCMV annotation if one used only chimpanzee orthologue analysis. Several of the new ORFs that were predicted by the chimpanzee virus orthologue comparison (UL15a, UL41a, and UL148d) scored poorly by BDGF analysis (Fig. 1, scores of 3.4, 1.1, and 2.0, respectively). The poor BDGF scores imply that the proteins encoded by these ORFs must have amino acid sequence organizations relatively unique to cytomegaloviruses. Our analysis also identified several ORFs with the potential to encode proteins (Table 1). Orthologues corresponding to four of these ORFs were identified in the chimpanzee virus, and sequence analysis of two of the ORFs revealed their presence in a clinical isolate of HCMV.
Our analysis predicts that regions of the HCMV strain AD169 genome exist that are devoid of bona fide gene-coding ORFs (R L5-R L7, UL58-UL59, UL62-UL68, UL106-UL111, UL125-UL127, and US35-US36). This prediction is supported by the fact that sequence analysis of a clinical isolate did not find conserved ORFs in the regions occupied by UL58-UL59, UL62-UL68, UL106-UL111, and US35-US36. Viruses are constrained in the amount of genetic information that they can encode in their genomes, given limits to the amount of nucleic acid that can be packaged into virions. Consequently, as a rule, they do not carry nonfunctional DNA sequences. Some of these gene-less regions likely encode cis-acting functions, as does the UL58-UL62 region, which includes the lytic origin of DNA replication (42). Several of these regions are highly transcribed, e.g., RL7 and UL106-UL111 (43, 44), and it is possible that the transcripts do not encode proteins, but rather function directly as RNAs. To delineate the coding potential of HCMV more definitively, it will be necessary to sequence several clinical isolates of the virus.
Acknowledgments
We thank S. Spector for the generous gift of the HCMV Coz clinical isolate. This work was supported in part by National Institutes of Health Grants CA82396, CA85786, and CA87661 (to T.E.S.).
Abbreviations: HCMV, human cytomegalovirus; BDGF, Bio-Dictionary-based Gene Finder; UL, unique long; US, unique short.
Data deposition: Sequences of selected regions in the Coz HCMV isolate have been deposited in the GenBank database (accession nos. AY372064-AY372068).
References
- 1.Britt, W. J. & Alford, C. A. (1996) in Fields Virology, eds. Fields, B. N., Knipe, D. M. & Howley, P. M. (Lippincott-Raven, Philadelphia), Vol. 2, pp. 2493-2524. [Google Scholar]
- 2.Bankier, A. T., Beck, S., Bohni, R., Brown, C. M., Cerny, R., Chee, M. S., Hutchison, C. A., III, Kouzarides, T., Martignetti, J. A., Preddie, E., et al. (1991) DNA Sequences 2, 1-12. [DOI] [PubMed] [Google Scholar]
- 3.Chee, M. S., Bankier, S., Beck, S., Bohni, R., Brown, C. R., Horsnell, T., Hutchisno, C. A., III, Kouzarides, T., Martignetti, J. A., Preddie, E., et al. (1990) Curr. Top. Microbiol. Immunol. 154, 125-169. [DOI] [PubMed] [Google Scholar]
- 4.Baer, R., Bankier, A. T., Biggin, M. D., Deininger, P. L., Farrell, P. J., Gibson, T. J., Hatfull, G., Hudson, G. S., Satchwell, S. C., Seguin, C., et al. (1984) Nature 310, 207-211. [DOI] [PubMed] [Google Scholar]
- 5.McGeoch, D. J., Dalrymple, M. A., Davison, A. J., Dolan, A., Frame, M. C., McNab, D., Perry, L. J., Scott, J. E. & Taylor, P. (1988) J. Gen. Virol. 69, 1531-1574. [DOI] [PubMed] [Google Scholar]
- 6.Gibson, W., Clopper, K. S., Britt, W. J. & Baxter, M. K. (1996) J. Virol. 70, 5680-5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kotenko, S. V., Saccani, S., Izotova, L. S., Mirochnitchenko, O. V. & Pestka, S. (2000) Proc. Natl. Acad. Sci. USA 97, 1695-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mullberg, J., Hsu, M. L., Rauch, C. T., Gerhart, M. J., Kaykas, A. & Cosman, D. (1999) J. Gen. Virol. 80, Part 2, 437-440. [DOI] [PubMed] [Google Scholar]
- 9.Wood, L. J., Baxter, M. K., Plafker, S. M. & Gibson, W. (1997) J. Virol. 71, 179-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rigoutsos, I., Novotny, J., Huynh, T., Chin-Bow, S. T., Parida, L., Platt, D., Coleman, D. & Shenk, T. (2003) J. Virol. 77, 4326-4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Novotny, J., Rigoutsos, I., Coleman, D. & Shenk, T. (2001) J. Mol. Biol. 310, 1151-1166. [DOI] [PubMed] [Google Scholar]
- 12.Prichard, M. N., Penfold, M. E., Duke, G. M., Spaete, R. R. & Kemble, G. W. (2001) Rev. Med. Virol. 11, 191-200. [DOI] [PubMed] [Google Scholar]
- 13.Cha, T. A., Tom, E., Kemble, G. W., Duke, G. M., Mocarski, E. S. & Spaete, R. R. (1996) J. Virol. 70, 78-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davison, A. J., Dolan, A., Akter, P., Addison, C., Dargan, D. J., Alcendor, D. J., McGeoch, D. J. & Hayward, G. S. (2003) J. Gen. Virol. 84, 17-28. [DOI] [PubMed] [Google Scholar]
- 15.Shibuya, T. & Rigoutsos, I. (2002) Nucleic Acids Res. 30, 2710-2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bairoch, A. & Apweiler, R. (2000) Nucleic Acids Res. 28, 45-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dehal, P., Satou, Y., Campbell, R. K., Chapman, J., Degnan, B., De Tomaso, A., Davidson, B., Di Gregorio, A., Gelpke, M., Goodstein, D. M., et al. (2002) Science 298, 2157-2167. [DOI] [PubMed] [Google Scholar]
- 18.Kozak, M. (1984) Nucleic Acids Res. 12, 857-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alderete, J. P., Child, S. J. & Geballe, A. P. (2001) J. Virol. 75, 7188-7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hitomi, S., Kozuka-Hata, H., Chen, Z., Sugano, S., Yamaguchi, N. & Watanabe, S. (1997) Arch. Virol. 142, 1407-1427. [DOI] [PubMed] [Google Scholar]
- 21.Adair, R., Douglas, E. R., Maclean, J. B., Graham, S. Y., Aitken, J. D., Jamieson, F. E. & Dargan, D. J. (2002) J. Gen. Virol. 83, 1315-1324. [DOI] [PubMed] [Google Scholar]
- 22.Fraile-Ramos, A., Pelchen-Matthews, A., Kledal, T. N., Browne, H., Schwartz, T. W. & Marsh, M. (2002) Traffic 3, 218-232. [DOI] [PubMed] [Google Scholar]
- 23.Huber, M. T., Tomazin, R., Wisner, T., Boname, J. & Johnson, D. C. (2002) J. Virol. 76, 5748-5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huber, M. T. & Compton, T. (1998) J. Virol. 72, 8191-8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lilley, B. N., Ploegh, H. L. & Tirabassi, R. S. (2001) J. Virol. 75, 11218-11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Atalay, R., Zimmermann, A., Wagner, M., Borst, E., Benz, C., Messerle, M. & Hengel, H. (2002) J. Virol. 76, 8596-8608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis, M. G. & Huang, E. S. (1985) J. Virol. 56, 7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wing, B. A., Lee, G. C. & Huang, E. S. (1996) J. Virol. 70, 3339-3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang, S. K., Duh, C. Y. & Chang, T. T. (2000) J. Gen. Virol. 81, 2407-2416. [DOI] [PubMed] [Google Scholar]
- 30.Krosky, P. M., Underwood, M. R., Turk, S. R., Feng, K. W., Jain, R. K., Ptak, R. G., Westerman, A. C., Biron, K. K., Townsend, L. B. & Drach, J. C. (1998) J. Virol. 72, 4721-4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu, Y. & Biegalke, B. J. (2002) J. Virol. 76, 2460-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Furman, M. H., Dey, N., Tortorella, D. & Ploegh, H. L. (2002) J. Virol. 76, 11753-11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dal Monte, P., Pignatelli, S., Zini, N., Maraldi, N. M., Perret, E., Prevost, M. C. & Landini, M. P. (2002) J. Gen. Virol. 83, 1005-1012. [DOI] [PubMed] [Google Scholar]
- 34.Battista, M. C., Bergamini, G., Boccuni, M. C., Campanini, F., Ripalti, A. & Landini, M. P. (1999) J. Virol. 73, 3800-3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LaPierre, L. A. & Biegalke, B. J. (2001) J. Virol. 75, 6062-6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stamminger, T., Gstaiger, M., Weinzierl, K., Lorz, K., Winkler, M. & Schaffner, W. (2002) J. Virol. 76, 4836-4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spaderna, S., Blessing, H., Bogner, E., Britt, W. & Mach, M. (2002) J. Virol. 76, 1450-1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benedict, C. A., Butrovich, K. D., Lurain, N. S., Corbeil, J., Rooney, I., Schneider, P., Tschopp, J. & Ware, C. F. (1999) J. Immunol. 162, 6967-6970. [PubMed] [Google Scholar]
- 39.Mocarski, E. S. (1996) in Fields Virology, eds. Fields, B. N., Knipe, D. M. & Howley, P. M. (Lippincott-Raven, Philadelphia), Vol. 2, pp. 2447-2492. [Google Scholar]
- 40.Ikehara, K. & Okazawa, E. (1993) Nucleic Acids Res. 21, 2193-2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silke, J. (1997) Gene 194, 143-155. [DOI] [PubMed] [Google Scholar]
- 42.Masse, M. J., Karlin, S., Schachtel, G. A. & Mocarski, E. S. (1992) Proc. Natl. Acad. Sci. USA 89, 5246-5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDonough, S. H., Staprans, S. I. & Spector, D. H. (1985) J. Virol. 53, 711-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hutchinson, N. I., Sondermeyer, R. T. & Tocci, M. J. (1986) Virology 155, 160-171. [DOI] [PubMed] [Google Scholar]