

Abstract
To determine the role of PTHrP in fetal calcium metabolism, blood calcium was measured in mice homozygous (HOM) for deletion of the PTHrP gene. On day 18.5 of gestation, ionized calcium and the maternal–fetal calcium gradient were significantly reduced in HOM PTHrP-ablated fetuses compared with that of their littermates. To assess the placental contribution to the effect of PTHrP, 45Ca and 51Cr-EDTA (as a blood diffusional marker) were administered by intracardiac injection to pregnant, heterozygous dams on day 17.5 of gestation. Five minutes after the injection, whole fetal 45Ca accumulation was significantly decreased in HOM PTHrP-ablated fetuses compared with that of their littermates. Next, two fetuses from each litter were injected in utero with fragments of PTHrP, PTH, or diluent 1 h before administering 45Ca and 51Cr to the dam. PTHrP-(1–86) and PTHrP-(67–86) significantly increased relative 45Ca accumulation in HOM PTHrP-ablated fetuses, but PTHrP(1–34), PTH-(1–84), and the diluent had no effect. Finally, similar studies were performed on fetal mice that lacked the PTH/PTHrP receptor gene. Ionized calcium was significantly reduced in HOM PTH/PTHrP receptor-ablated fetuses. However, 5 min after maternal injection of 45Ca and 51Cr, relative accumulation of 45Ca was significantly increased in these fetuses. It was concluded that PTHrP is an important regulator of fetal blood calcium and placental calcium transport. In addition, the bioactivity of PTHrP for placental calcium transport is specified by a mid-molecular region that does not use the PTH/PTHrP receptor.
In the latter part of gestation in humans, rodents, and probably all mammals, calcium is actively transported across the placenta, presumably to meet the needs of the rapidly mineralizing fetal skeleton. This active transport probably occurs at the fetus-facing plasma membranes of the syncytiotrophoblast in the human, at the trophoblast in the rodent, and at the basal surface of the intraplacental yolk sac in both (1, 2). Relative fetal hypercalcemia (total and ionized), representing a positive maternal–fetal calcium gradient, also has been found in most fetal mammals and is believed to result from this active transport of calcium (3, 4). It is also likely that the fetal blood calcium level is affected by calcium fluxes in and out of the mineralizing skeleton and by excretion of calcium by the fetal kidney, but the exact relative contributions of placenta, bone, and kidney to the fetal blood calcium level have not been determined.
The traditional calcitropic hormones [parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D (1,25-D)] are likely to have limited roles in regulating the fetal blood calcium level or the placental transport of calcium. Although maternally derived 1,25-D can cross the placenta, fetal blood levels of 1,25-D are low (5, 6). Furthermore, although 1,25-D (given at pharmacologic doses to the dam or fetus) can increase the fetal calcium content and blood level of calcium (7, 8), fetuses of severely vitamin D-deficient, hypocalcemic rats have normal blood calcium levels and fully mineralized skeletons (9, 10). Maternally derived PTH cannot cross the placenta (11), and fetal immunoreactive PTH levels are low throughout gestation in mammals (3, 12).
The regulation of placental calcium transport and the fetal blood calcium level has been investigated through the use of the ovine placental perfusion model described by Weatherley et al. (13). Fetal thyroparathyroidectomy with thyroid hormone replacement lowered fetal blood calcium and abolished the maternal–fetal calcium gradient (14). Seven to 10 days after thyroparathyroidectomy, the fetus was removed from the uterus, and the placentas were artificially perfused in situ. Calcium transfer across perfused placentas from previously thyroparathyroidectomized fetuses was slower than calcium transfer across perfused placentas from previously intact fetuses (14). Administration of PTH could not reverse the effect of previous fetal parathyroidectomy on calcium transport in perfused placentas (15). However, PTHrP, originally discovered as the mediator of humoral hypercalcemia of malignancy (16), increased the rate of calcium transport in perfused placentas from these previously parathyroidectomized fetal lambs (15, 17). In particular, fragments of PTHrP that encompassed the mid-region of the molecule stimulated placental calcium transport, especially PTHrP-(1–86) and, to some extent, PTHrP-(67–86) (17, 18).
However, other investigators who studied intact fetal lambs (19, 20) and artificially perfused placentas from decapitated (to mimic thyroparathyroidectomy) fetal rats (21, 22) did not find an effect of the PTHrP mid-molecule on calcium transport. Instead, PTHrP-(1–86), PTHrP-(1–34), PTH-(1–34), and 1,25-D were found to have a small stimulatory effect on calcium transport.
In this study, fetal mice that were either homozygous (HOM) for deletion of the PTHrP gene (23) or HOM for deletion of the PTH/PTHrP receptor gene (24) were studied to define the role of PTHrP in determining the fetal blood calcium level and the rate of placental calcium transfer in intact animals. The results showed that, without a functional PTHrP gene, the maternal–fetal calcium gradient was abolished and transplacental transport of calcium ions was slowed. In contrast, without a functional PTH/PTHrP receptor gene, the maternal–fetal calcium gradient was abolished, but placental transport of calcium was increased. PTHrP-(1–86) and PTHrP-(67–86) can stimulate calcium transport in mice lacking a functional PTHrP gene but PTHrP-(1–34) and PTH-(1–84) cannot.†
MATERIALS AND METHODS
Materials.
Murine and human PTHrP proteins differ by 1 amino acid through the first 100 amino acids (25), so, in these experiments, the commercially available synthetic human PTHrP protein fragments were used. PTHrP-(1–86) and human PTH-(1–84) were obtained from Bachem. PTHrP-(67–86) and PTHrP-(1–34) were obtained from Peninsula Laboratories. 45Ca and 51Cr-EDTA were obtained from DuPont/NEN. Scintigest, Scintiverse, Taq DNA polymerase, and proteinase K were obtained from Fisher.
PTHrP and PTH/PTHrP Receptor Knockout Mice.
PTHrP and PTH/PTHrP receptor knockout mice were obtained by targeted disruption of the murine genes in embryonic stem cells as described (23, 24). The data presented below on PTHrP knockout mice were completed using mice in the original hybrid C57BL/6–129/SvJ background. The PTH/PTHrP receptor knockout mice were originally created in the same C57BL/6–129/SvJ background, but few HOM fetuses survived after day 14 of gestation. After crossing the original hybrid strain into Black Swiss for one or two generations, 20% of PTH/PTHrP receptor knockout fetuses were HOM at day 18.5 of gestation; this hybrid background was used for the experiments described herein. It was subsequently discovered that normal or mutant adult and fetal mice in the Black Swiss hybrid had ionized calcium levels that were 0.10–0.20 mmol/liter higher than their respective counterparts in the original C57BL/6 strain. Therefore, to allow side-by-side comparison of the results obtained from the PTHrP knockout mice and the PTH/PTHrP receptor knockout mice, the PTHrP knockout mice were also back-crossed into Black Swiss for several generations to produce a similar strain. This new hybrid Black Swiss strain of PTHrP knockout mice was then used to verify the findings obtained with the original C57BL/6–129/SvJ strain.
Heterozygous (HET) males and HET females were mated overnight; the presence of a vaginal mucus plug on the morning after mating marked gestational day 0.5. Normal gestation in these mice is 19 days. All mice were allowed a standard chow diet and ad lib access to water. All studies described were performed with the prior approval of the Institutional Animal Care and Use Committee of Massachusetts General Hospital.
Ionized Calcium Measurements.
Two samples of whole blood were collected into heparinized, 50-μl capillary tubes from the tail vein of each dam on day 18 ± 0.5 of pregnancy and placed on ice. The dam was then killed by cervical dislocation, and a cesarean section was quickly performed to remove the uterus intact. In turn, each individual fetus was removed from its amniotic sac, the neck was incised to transect the left carotid and jugular vessels, and whole blood was collected into heparinized capillary tubes. The volume of fetal blood collected measured 30–50 μl on average from each fetus—sufficient for one calcium measurement. All capillary tubes were capped and immediately immersed in ice. The order that each fetus was collected in the litter was recorded, and the tail of each fetus was clipped and placed in lysis buffer (100 mM Tris·HCl, pH 8.0/500 mM EDTA, pH 8.0/0.2% SDS/200 mM NaCl/100 μg/ml proteinase K) for later DNA extraction and genotyping. Each blood sample was then analyzed within 15 min of collection on a CIBA/Corning 634 Ca2+/pH analyzer to obtain the ionized calcium and pH. The raw, ionized calcium data were used; “normalized” ionized calcium values (i.e., adjusted by the machine to the expected equivalent value for a normal pH of 7.4) were not used.
Placental Calcium Transport.
Pregnant dams on day 17 ± 0.5 of gestation were lightly anesthetized by isoflurane inhalation and then were given an intracardiac injection of 50 μCi of 45Ca and 50 of μCi 51Cr-EDTA in 0.9% saline (total injection volume, 100 μl) (1 Ci = 3 GBg). These doses of isotopes were chosen after preliminary experiments showed that they would be detectable at levels 500- to 10,000-fold above background in the recovered fetuses. At specific time points after the injection of the isotopes (5, 15, or 30 min), the dam was killed by cervical dislocation. A cesarean section was quickly performed, and the uterus was removed intact. Each fetus was removed from its gestational sac, and a tail clipping was obtained for subsequent genetic analysis by PCR. Each fetus was asphyxiated, weighed, and placed into a capped test tube, and the 51Cr activity was determined using a Packard γ-counter. The fetuses were subsequently solubilized with Scintigest at 60°C for 24–48 h and vortexed twice. Once the fetuses were fully solubilized, scintillation fluid (Scintiverse I, Fisher) was added. To minimize chemoluminescence, 5 drops of glacial acetic acid was added to each vial, and the vials were wiped with an antistatic cloth, shielded from light, and equilibrated to room temperature over 12–24 h. 45Ca activity was then counted on a Beckman liquid scintillation counter, and the ratio of 45Ca/51Cr activity was calculated for each fetus. In the subsequent data analysis, the mean ratio for the heterozygotes of each litter was set at 100% so that the results from different litters could be compared.
In preliminary experiments, it was established that the amount of 51Cr activity in the vials registered on the liquid scintillation counter at a level that was <1% of the activity of the 45Ca. In turn, the 45Ca activity did not register above background on the γ-counter. Thus, it was not necessary to adjust the windows on either instrument in an attempt to exclude the activity of the other isotope.
Backflux of Calcium.
In separate experiments designed to measure relative fetal–maternal calcium flow (backflux), a pregnant dam on day 18 ± 0.5 of gestation was anesthetized with isoflurane. A cesarean section was quickly performed, and the individual gestational sacs were partly opened to identify a HOM fetus. Two apparently normal fetuses were removed, and their placental vessels were cauterized to prevent maternal blood loss. A HOM fetus (left in situ with umbilical vessels patent) was given an intraabdominal injection of 2 μCi of 45Ca in 2 μl of 0.9% saline by microinjection pipette. The maternal incision was closed, and the dam recovered from the anesthesia. The two removed (control) fetuses were asphyxiated and given an intraabdominal injection of 1/100 or 1/1000 the activity of 45Ca. After 30 min, the dam was killed by cervical dislocation, and the injected, HOM fetus was removed. The dam, HOM fetus, and two control fetuses were ashed at 500°C for 24 h. The ashes were dissolved in 10 M HCl, scintillation cocktail was added, and the samples were counted on a Beckman liquid scintillation counter.
PTHrP Fragment Injections.
All lyophilized stock peptides were dissolved in 0.9% saline at a concentration of 0.5 nmol/μl and kept at −20°C until used. Anesthesia in a HET PTHrP-deleted dam was induced by inhalation of isoflurane and was maintained by a single i.p. injection of 200 μl of a 1% solution of methohexital (Brevital, Lilly Research Laboratories, Indianapolis). This combination of anesthetics rapidly induced 10 min of surgical anesthesia but allowed the dam to be fully awake within 20–30 min of the initial injection. A transverse incision was used to open the right lower quadrant and to allow access to two fetuses. After grasping with forceps, the abdomen of each selected fetus was identified and pierced through the uterine wall and amniotic sac from the left side (to avoid puncturing the liver) with a microinjection pipette, and 1.0 nmol of peptide in 2 μl of 0.9% saline was injected under positive pressure. A suture was placed in the uterine wall overlying each injected fetus to facilitate its later identification. After injection of two fetuses in each litter, the pelvic incision was closed, and the dam was allowed to recover.
One hour after fetal injection of the peptides or vehicle, the HET PTHrP-deleted dam was again lightly anesthetized with isoflurane alone to allow an intracardiac injection of radioisotopes, as described above. The dam was subsequently killed 30 min after injection of the isotopes (90 min after fetal injection of peptides or vehicle), and, after identifying the two injected fetuses, all fetuses were harvested and processed as described above.
PCR.
Tail clippings were digested at 55°C for 12–24 h in the lysis buffer containing proteinase K. Genomic DNA was extracted and purified using phenol/chloroform, precipitated with a 25:1 solution of ethanol and 3 M sodium acetate (pH 5.2), and resuspended in TE buffer (10 mM Tris·HCl, pH 8.0/1 mM EDTA, pH 8.0). Using pairs of oligonucleotide primers specific for the native PTHrP gene sequence and the inserted neomycin gene sequence (23) or the native PTH/PTHrP receptor gene sequence and the inserted neomycin gene sequence (24), the genomic DNA was amplified in a single-tube, 36-cycle PCR using a PTC-200 Peltier Thermal Cycler (MJ Research, Cambridge, MA). The validity of the PCR genotyping was confirmed in preliminary experiments by Southern blotting of the genomic DNA with independent probes (23, 24).
Statistical Analysis.
Data were analyzed using systat 5.2.1 for Macintosh (Systat, Evanston, IL). Two-tailed probabilities are reported, and all data are presented as mean ± SE.
RESULTS
Ionized Calcium in PTHrP Knockout Mice.
In preliminary experiments in normal fetuses (obtained from mating normal litter mate males to normal litter mate females), the fetal pH was found to fall quickly from an initial mean pH of 7.2 during the harvesting of fetuses (Fig. 1A). This value is consistent with published data of late term, fetal pH in rats (26). However, the ionized calcium values were only modestly affected by the fall in pH. As shown in Fig. 1B, there was a slight rise in ionized calcium from the first to last pup of each litter. However, because the pups were sampled in random order (so genotypes were not known), these modest effects of pH on the ionized calcium did not affect importantly the analysis of the results that followed. This finding of a small effect of pH on the ionized calcium is consistent with published data from fetal rats, which showed that ionized calcium comprises >80% of the total calcium value and, therefore, is little affected by the action of H+ to release calcium bound to albumin (27).
Figure 1.
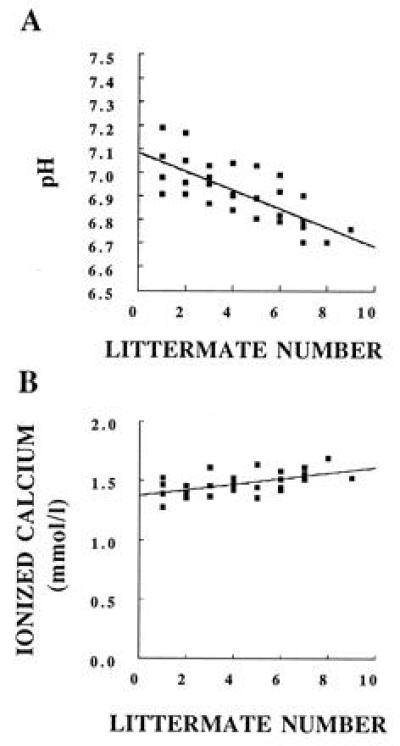
Normal fetal pH and ionized calcium. Data from offspring of normal litter mate dams are presented. The x axis indicates the order in which the fetal blood was collected in each litter.
In the offspring resulting from mating HET, PTHrP-deleted dams to HET, PTHrP-deleted males (all obtained from the original C57BL/6–129/SvJ strain), HOM, PTHrP-deleted fetuses had an ionized calcium of 1.22 ± 0.02 mmol/liter. This was significantly lower than the values obtained from their HET and wild-type littermates (1.48 ± 0.02 mmol/liter and 1.52 ± 0.03 mmol/liter, respectively; P ≪ 0.001) (Fig. 2A). Although the mean ionized calcium was 0.04 mmol/liter lower in HET, PTHrP-deleted fetuses than in their wild-type littermates, this difference was not statistically significant.
Figure 2.
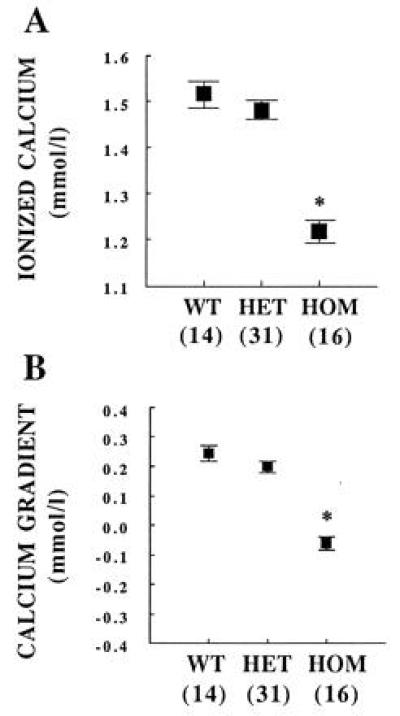
Ionized calcium and maternal–fetal calcium gradient in PTHrP knockout fetuses. WT, wild-type fetus. ∗, P ≪ 0.001 vs. WT or HET. The number of observations is indicated in parentheses.
The maternal–fetal ionized calcium gradient was determined by subtracting the corresponding maternal calcium value from each individual fetal calcium value. HET and wild-type fetuses had a positive calcium gradient of 0.24 ± 0.03 mmol/liter and 0.20 ± 0.02 mmol/liter, respectively (P = not significant). The gradient in HOM fetuses was −0.06 ± 0.02 mmol/liter, significantly lower than that of the wild-type and HET littermates (P ≪ 0.001) (Fig. 2B).
Calcium Transport in PTHrP Knockout Mice.
To determine if placental calcium transport was also affected in the HOM, PTHrP-deleted fetuses, pregnant HET dams from the original C57BL/6–129/SvJ strain were given an intracardiac injection of 45Ca and 51Cr-EDTA. 51Cr-EDTA remains in the extracellular fluid and has been used in placental perfusion studies in rodents to serve as a marker for blood flow differences between placentas (28). Therefore, the ratio of 45Ca/51Cr activities accumulated in each fetus was determined as an index of relative calcium transport differences among fetuses of a given litter.
Repeated sets of experiments were terminated at one of three time points: 5, 15, or 30 min after the injection of the isotopes to the dam. At all three time points, HOM, PTHrP-deleted fetuses had an accumulated 45Ca/51Cr activity ratio that was significantly lower than that of their wild-type or HET littermates (Fig. 3). For example, at 15 min, the value of the HOM, PTHrP-deleted fetuses was 82.3 ± 3.8% compared with 104.5 ± 5.4% (wild-type littermates) and 101.7 ± 3.4% (HET littermates) (P < 0.005). The relative placental transport of 45Ca was significantly lower in HET fetuses compared with wild-type fetuses at the 30-min time point only. This was an inconstant finding in additional experiments (data not shown), so the true difference in placental calcium transport between wild-type and HET fetuses is likely to be modest.
Figure 3.
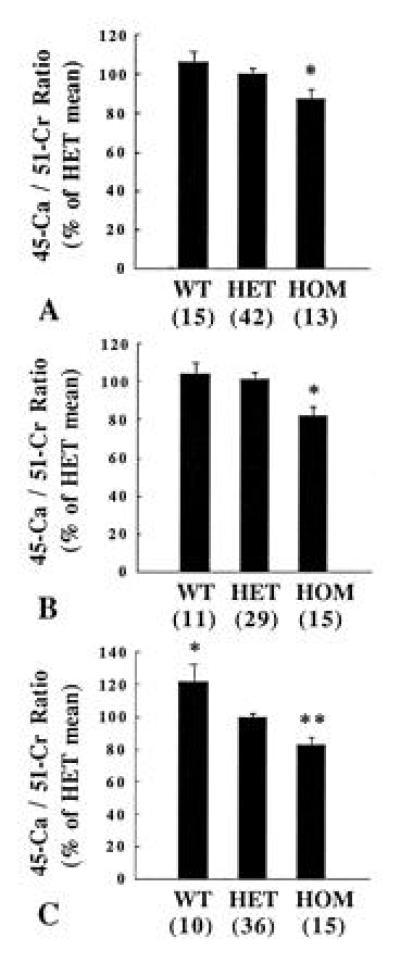
Placental transport of 45Ca and 51Cr-EDTA in PTHrP knockout fetuses. The ratio of 45Ca/51Cr activity accumulated in fetuses was determined (A) 5, (B) 15, and (C) 30 min after maternal administration of the isotopes. The mean heterozygote 45Ca/51Cr ratio of each litter was set at 100% to allow the results of multiple litters to be compared. The number of observations is indicated in parentheses. WT, wild-type fetus. (A) ∗, P < 0.02 vs. HET; (B) ∗, P < 0.001 vs. HET; (C) ∗, P < 0.005 and ∗∗, P < 0.001 vs. HET.
Backflux of 45Ca and 51Cr-EDTA across the placenta from the fetus to the dam could have affected these results if the amount of backflux was comparable to the forward flow. In control experiments, a single HOM, PTHrP-deleted fetus was injected with 45Ca, and, after 30 min, the amount of activity remaining in the fetus and the amount of activity present in the dam were determined. Under these conditions, <1% of the activity injected into the fetus was lost from the fetus and recovered in the dam (data not shown). These observations agree with those of others who have found backflux of calcium in rats and sheep to be <10% and probably <1% of the forward flow (29, 30). Furthermore, because the small volume of 45Ca crossing the placenta from dam to fetus would be expected to mix with the nonradioactive calcium within the total fetal blood volume before returning to the dam, it is unlikely that backflux of 45Ca could have accounted for the consistent 20% decrease in the 45Ca/51Cr ratio observed in the HOM fetuses at all time points.
Acute Effects of PTHrP Administration to PTHrP Knockout Mice.
In the following experiments, three questions were addressed: (i) Are the effects of gene deletion on calcium transport acutely reversible? (ii) Can systemically delivered PTHrP accomplish this reversal? (iii) What portions of the PTHrP molecule control placental calcium transport? Two fetuses of each litter were blindly (i.e., without determining the phenotype or genotype) given an intraabdominal injection of specific fragments of PTHrP, PTH, or diluent (0.9% saline) alone as described in Materials and Methods, and the radioisotopes were administered 1 h later.
In the first series of these experiments, HOM, PTHrP-deleted fetuses that did not receive an injection had a 45Ca/51Cr ratio of 75.3 ± 3.3%; 1 nmol of PTHrP-(1–86) increased the 45Ca/51Cr ratio of injected HOM fetuses to 98.9 ± 5.2% (P < 0.001) (Table 1). 45Ca/51Cr ratios of wild-type and HET fetuses were not significantly increased by PTHrP-(1–86) (not shown).
Table 1.
Acute effects on 45Ca transport by HOM fetuses treated with PTHrP, PTH, or diluent alone
| Treatment |
45Ca/51Cr
activity ratios
|
||
|---|---|---|---|
| Homozygote (n) | Treated homozygote (n) | P value | |
| PTHrP-(1-86) | 75.3 ± 3.3 (20) | 98.9 ± 5.2 (8) | <0.001 |
| PTHrP-(67-86) | 78.1 ± 2.9 (25) | 92.6 ± 5.8 (6) | <0.04 |
| PTHrP-(1-34) | 79.0 ± 4.5 (11) | 69.9 ± 5.6 (7) | NS |
| PTH-(1-84) | 75.0 ± 3.6 (18) | 80.9 ± 4.8 (10) | NS |
| 0.9% saline | 75.1 ± 3.9 (14) | 76.1 ± 2.7 (7) | NS |
All peptides were administered in 1-nmol amounts in an injection volume of 2 μl. The number of observations is indicated in parentheses. For the purpose of comparing data from different litters, the mean 45Ca/51Cr ratios of the untreated heterozygotes of each litter from which these basal and treated homozygote values were obtained have been set at 100%. For further details, see Materials and Methods. NS, not significant.
Similarly, PTHrP-(67–86) also increased the baseline 45Ca/51Cr ratio of HOM fetuses from 78.1 ± 2.6% to 92.6 ± 5.8% (P < 0.04) (Table 1). 45Ca/51Cr ratios of wild-type and HET fetuses also were unaffected by PTHrP-(67–86) (not shown).
Our study further determined that PTHrP-(1–34), PTH-(1–84), and diluent (0.9% saline) alone had no significant effect on the 45Ca/51Cr ratios of HOM, HET, and wild-type fetuses (Table 1). To independently verify that the aliquots of PTHrP-(1–34) and PTH-(1–84) were biologically active, we found that these same stocks of PTHrP-(1–34) and PTH-(1–84) were as active as PTHrP-(1–86) in stimulating cAMP production in a cultured chondrocyte cell line (data not shown). Therefore, the lack of effect of PTHrP-(1–34) and PTH-(1–84) on placental calcium transport in these experiments cannot be attributed to biologically ineffective stocks of these peptides.
Ionized Calcium in PTH/PTHrP Receptor Knockout Mice.
The requirement for the mid-molecular region of PTHrP for stimulation of transplacental calcium transport suggests that this activity is mediated by a receptor distinct from the cloned PTH/PTHrP receptor (which can respond to either amino-terminal PTH or PTHrP). Nevertheless, this receptor, which is expressed in fetal bone, kidney, yolk sac, and other tissues (31), could well contribute to fetal calcium homeostasis. To evaluate the role of the PTH/PTHrP receptor in fetal calcium metabolism, blood calcium was first measured in fetuses HOM for deletion of the PTH/PTHrP receptor gene.
In the offspring resulting from mating HET, PTH/PTHrP receptor gene-deleted dams and males (in a Black Swiss hybrid strain; see Materials and Methods), HOM fetuses had an ionized calcium of 1.00 ± 0.04 mmol/liter. This was significantly lower than the values obtained from their HET and wild-type littermates (1.65 ± 0.04 mmol/liter and 1.66 ± 0.05 mmol/liter, respectively; P ≪ 0.001). The maternal–fetal gradient of the HOM fetuses was −0.31 ± 0.05 mmol/liter compared with the maternal–fetal gradients of their HET and wild-type littermates (0.35 ± 0.04 mmol/liter and 0.36 ± 0.04 mmol/liter, respectively). By comparison, the ionized calcium of HOM, PTHrP-deleted fetuses in a Black Swiss hybrid background was 1.34 ± 0.05 mmol/liter with a maternal–fetal calcium gradient of 0.04 ± 0.03 mmol/liter (HET and wild-type fetuses had ionized calcium values of 1.70 ± 0.04 mmol/liter and 1.73 ± 0.06 mmol/liter and calcium gradient values of 0.34 ± 0.03 mmol/liter and 0.36 ± 0.03 mmol/liter, respectively). Thus, like the HOM PTHrP-deleted fetuses, the PTH/PTHrP receptor-deleted fetuses had lower than normal levels of ionized calcium.
Calcium Transport in PTH/PTHrP Receptor Knockout Mice.
To independently confirm that the action of PTHrP on placental calcium transport is a function of a mid-molecular region of PTHrP and is not mediated by the PTH/PTHrP receptor, relative placental calcium transport in HOM PTH/PTHrP receptor gene-deleted fetuses was determined. Pregnant, HET dams for deletion of the PTH/PTHrP receptor gene were given an intracardiac injection of 45Ca and 51Cr-EDTA, and the activities of 45Ca and 51Cr accumulated in each fetus after 5 min were determined. The relative accumulation of 45Ca was significantly increased in HOM fetuses (150.5 ± 12.1%) compared with their wild-type and HET littermates (97.9 ± 8.8% and 100.1 ± 9.3%, respectively; P < 0.005) (Fig. 4). In contrast, the placental transfer of 45Ca into HOM PTHrP-deleted fetuses was reduced in the Black Swiss hybrid strain (data not shown), just as in the original C57BL/6–129/SvJ strain.
Figure 4.
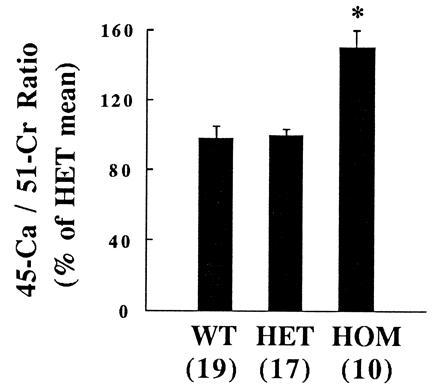
Placental transport of 45Ca and 51Cr-EDTA in PTH/PTHrP receptor knockout fetuses. The ratio of 45Ca/51Cr activity accumulated in fetuses was determined 5 min after maternal administration of the isotopes. The mean heterozygote 45Ca/51Cr ratio of each litter was set at 100% to allow the results of multiple litters to be compared. The number of observations is indicated in parentheses. WT, wild-type fetus. ∗, P < 0.005 vs. HET.
DISCUSSION
We have shown that absence of the murine PTHrP gene results in relative fetal hypocalcemia, obliteration of the normal maternal/fetal calcium gradient, and reduced placental transport of calcium. These observations reflect direct control of placental calcium transport by PTHrP but do not reflect complicated effects of the genetic ablation on placental development; injections of PTHrP-(1–86) were able to rapidly reverse the abnormalities in 45Ca transport. The fact that the wild-type and HET fetuses did not respond to PTHrP-(1–86) or PTHrP-(67–86) injections suggests that, late in pregnancy, calcium transport cannot be further up-regulated by additional exogenous PTHrP.
These results partly confirmed those of others in the ovine, perfused placental preparation (17, 18); PTHrP-(1–86) and, to some extent, PTHrP-(67–86) could increase placental calcium transport, but PTH could not. Thus, the structurally different epitheliochorial (ovine) and hemochorial placentas (rodent and human) share similar responses to PTHrP. However, the results of this study differ from the data obtained from the ovine placental perfusion model in some respects. PTHrP-(67–86) stimulated calcium transport in perfused placentas obtained from some intact ovine fetuses (18) but did not do so in our studies with intact wild-type or HET mice. This difference may be accounted for by the experimental method used in the ovine placental perfusion experiments, which disrupted the normal fetal-placental unit. Fetuses were acutely removed from the uterus, and, after 2 to 3 h of baseline perfusion, the experiments were performed. This time period was probably sufficient to eliminate all residual PTHrP in the perfusate that was derived from fetal sources.
In contrast, in the PTHrP gene knockout mice, placental calcium transport was studied in vivo with intact fetuses and placentas, which permitted normal fetal-placental interaction, with the exception that fetally derived PTHrP was absent in all tissues of the HOM, PTHrP gene-ablated mice. PTHrP apparently is produced not only in the fetal parathyroid gland (32) but also in the umbilical cord (33), amnion (34), and trophoblast cells of the placenta (35), and all of these sources (and perhaps others) might be relevant for placental calcium transport. Thus, in this model, addition of PTHrP to the endogenously produced PTHrP in normal (wild-type) fetuses did not further increase placental calcium transport.
The sole characterized receptor for PTHrP is the PTH/PTHrP receptor (31). PTHrP-(1–34) and PTH-(1–34) bind to and stimulate this receptor in virtually identical fashion. Another receptor specific for PTH has recently been identified and cloned (36), but, as yet, no specific PTHrP receptors have been identified. The PTH/PTHrP receptor has been detected as early as day 7.5 of gestation in mouse parietal and visceral extraembryonic endoderm (37) and, therefore, might be well situated for PTHrP to act on placental calcium transport. However, PTHrP-(1–34) and PTH-(1–84), which activate the PTH/PTHrP receptor, did not increase placental calcium transport in HOM, PTHrP-deleted fetuses. Furthermore, PTHrP-(67–86) was confirmed to stimulate placental calcium transport, and PTHrP-(67–86) is not known to activate the PTH/PTHrP receptor, so it was anticipated that placental calcium transport might be unaffected by deletion of the PTH/PTHrP receptor gene. In fact, relative calcium transport was increased in the HOM, PTH/PTHrP receptor gene-deleted fetuses. This finding suggests the possibility that PTHrP may have up-regulated placental calcium transport, perhaps in response to the fetal hypocalcemia. This further confirms the hypothesis that the stimulation of placental calcium transport by PTHrP is not mediated by the PTH/PTHrP receptor.
Thus, it is likely that a receptor specific for the mid-region of PTHrP is expressed in the placenta. Further evidence for a receptor that recognizes a mid-molecular region of PTHrP includes the stimulation by PTHrP-(67–86) of a rise in intracellular calcium, epidermal growth factor-receptor mRNA and fibronectin (but not cAMP accumulation) in a squamous cell cancer line (38), and stimulation of bicarbonate excretion by PTHrP-(1–141) and PTHrP-(1–108), but not PTHrP-(1–34), in the isolated, perfused rat kidney (39). Studies of the posttranslational processing of the PTHrP mRNA made in transfected cell lines suggest that the mid-molecular forms of PTHrP likely encompass amino acids 38–94 (40, 41).
The relative hypocalcemia of HOM, PTHrP gene-deleted fetuses is consistent with, but probably not solely due to, the reduction in 45Ca transport that was observed in this study. The fetal blood calcium level most likely is determined not only by transport of calcium across the placenta but by flux of calcium in and out of the developing skeleton, excretion of calcium by the fetal kidney, and reabsorption of calcium from the amniotic fluid. The latter processes might result in part from the actions of PTH and/or PTHrP on the PTH/PTHrP receptor. This is further suggested by the findings that fetuses HOM for absence of the PTH/PTHrP receptor gene are hypocalcemic in utero and that the reduction in ionized calcium is greater than that due to loss of PTHrP. It is, therefore, plausible that the relative hypocalcemia of the HOM, PTHrP-deleted fetuses results from the failure of PTHrP to act on the PTH/PTHrP receptor in kidney and bone, in addition to the absence of PTHrP action on the receptor in the placenta that recognizes the mid-region of PTHrP. The hypocalcemia in PTH/PTHrP receptor gene-deleted fetuses may result from the combined loss of PTH and PTHrP action in bone and kidney, despite intact or increased placental calcium transfer. These findings also indicate that placental calcium transport is only one determinant of fetal blood calcium because hypocalcemia is present in HOM, PTH/PTHrP receptor gene-deleted fetuses, despite intact or increased placental calcium transport.
In summary, this study shows that HOM, PTHrP gene-ablated mice have relative fetal hypocalcemia, obliterated maternal–fetal calcium gradient, and reduced placental transport of calcium. The calcium transport abnormalities were reversed acutely by PTHrP-(1–86) and PTHrP-(67–86) injection but not by PTHrP-(1–34) or PTH. HOM, PTH/PTHrP receptor gene-deleted fetuses are also hypocalcemic but have increased placental calcium transfer. These findings demonstrate that PTHrP is an important regulator of fetal calcium physiology, acting at least partly by stimulating a novel receptor to increase placental calcium transport. The findings also show that the PTH/PTHrP receptor in tissues other than placenta (probably bone and kidney) is an important determinant of fetal calcium homeostasis.
Acknowledgments
We thank the following for invaluable suggestions, advice, and technical support: Alison Pirro, Alison Smith, Julia McLaughlin, Jennifer Heymont, and Drs. Dale Goad and Susan MacDonald. We also thank Prof. Anthony D. Care for helpful discussions and suggestions. This work was supported by National Institutes of Health Grant DK47038. C.S.K. was supported by a fellowship award from the Medical Research Council of Canada.
Footnotes
Abbreviations: PTH, parathyroid hormone; PTHrP, PTH-related peptide; HOM, homozygous; 1,25-D, 1,25-dihydroxyvitamin D; HET, heterozygous.
This work was presented in part at the 1995 Annual Meeting of the American Society for Bone and Mineral Research, Baltimore, Sept. 9–13, 1995, and the 1996 International Congress of Endocrinology, San Francisco, June 12–13, 1996.
References
- 1.Fisher G J, Kelley L K, Smith C H. Am J Physiol. 1987;252:C38–C46. doi: 10.1152/ajpcell.1987.252.1.C38. [DOI] [PubMed] [Google Scholar]
- 2.Borke J L, Caride A, Verma A K, Kelley L K, Smith C H, Penniston J T, Kumar R. Am J Physiol. 1989;257:C341–C346. doi: 10.1152/ajpcell.1989.257.2.C341. [DOI] [PubMed] [Google Scholar]
- 3.David L, Anast C S. J Clin Invest. 1974;54:287–296. doi: 10.1172/JCI107764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garel J M, Barlet J P. Pediatr Res. 1976;10:749–754. doi: 10.1203/00006450-197608000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Hollis B W, Pittard W B. J Clin Endocrinol Metab. 1984;59:652–657. doi: 10.1210/jcem-59-4-652. [DOI] [PubMed] [Google Scholar]
- 6.Seki K, Furuya K, Makimura N, Mitsui C, Hirata J, Nagata I. J Perinat Med. 1994;22:189–194. doi: 10.1515/jpme.1994.22.3.189. [DOI] [PubMed] [Google Scholar]
- 7.Chalon S, Garel J M. Reprod Nutr Dev. 1983;23:567–573. [PubMed] [Google Scholar]
- 8.Durand D, Barlet J P, Braithwaite G D. Reprod Nutr Dev. 1983;23:235–244. doi: 10.1051/rnd:19830208. [DOI] [PubMed] [Google Scholar]
- 9.Halloran B P, De Luca H F. Arch Biochem Biophys. 1981;209:7–14. doi: 10.1016/0003-9861(81)90251-4. [DOI] [PubMed] [Google Scholar]
- 10.Miller S C, Halloran B P, DeLuca H F, Jee W S. Calcif Tissue Int. 1983;35:455–460. doi: 10.1007/BF02405076. [DOI] [PubMed] [Google Scholar]
- 11.Garel J M. Horm Metab Res. 1972;4:131–132. doi: 10.1055/s-0028-1097089. [DOI] [PubMed] [Google Scholar]
- 12.Rubin L P, Posillico J T, Anast C S, Brown E M. Pediatr Res. 1991;29:201–207. doi: 10.1203/00006450-199102000-00020. [DOI] [PubMed] [Google Scholar]
- 13.Weatherley A J, Ross R, Pickard D W, Care A D. Placenta. 1983;4:271–277. doi: 10.1016/s0143-4004(83)80006-x. [DOI] [PubMed] [Google Scholar]
- 14.Care A D, Caple I W, Abbas S K, Pickard D W. Placenta. 1986;7:417–424. doi: 10.1016/s0143-4004(86)80029-7. [DOI] [PubMed] [Google Scholar]
- 15.Rodda C P, Kubota M, Heath J A, Ebeling P R, Moseley J M, Care A D, Caple I W, Martin T J. J Endocrinol. 1988;117:261–271. doi: 10.1677/joe.0.1170261. [DOI] [PubMed] [Google Scholar]
- 16.Moseley J M, Kubota M, Diefenbach-Jagger H, Wettenhall R E, Kemp B E, Suva L J, Rodda C P, Ebeling P R, Hudson P J, Zajac J D, Martin T J. Proc Natl Acad Sci USA. 1987;84:5048–5052. doi: 10.1073/pnas.84.14.5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abbas S K, Pickard D W, Rodda C P, Heath J A, Hammonds R G, Wood W I, Caple I W, Martin T J, Care A D. Q J Exp Physiol. 1989;74:549–552. doi: 10.1113/expphysiol.1989.sp003303. [DOI] [PubMed] [Google Scholar]
- 18.Care A D, Abbas S K, Pickard D W, Barri M, Drinkhill M, Findlay J B, White I R, Caple I W. Q J Exp Physiol. 1990;75:605–608. doi: 10.1113/expphysiol.1990.sp003437. [DOI] [PubMed] [Google Scholar]
- 19.Barlet J P, Davicco M J, Coxam V. J Endocrinol. 1990;127:33–37. doi: 10.1677/joe.0.1270033. [DOI] [PubMed] [Google Scholar]
- 20.Barlet J P, Davicco M J, Coxam V. In: Calcium Regulating Hormones and Bone Metabolism. Cohn D V, Gennari C, editors. Amsterdam: Elsevier; 1992. pp. 124–128. [Google Scholar]
- 21.Robinson N R, Sibley C P, Mughal M Z, Boyd R D. Pediatr Res. 1989;26:109–115. doi: 10.1203/00006450-198908000-00008. [DOI] [PubMed] [Google Scholar]
- 22.Shaw A J, Mughal M Z, Maresh M J, Sibley C P. J Endocrinol. 1991;129:399–404. doi: 10.1677/joe.0.1290399. [DOI] [PubMed] [Google Scholar]
- 23.Karaplis A C, Luz A, Glowacki J, Bronson R T, Tybulewicz V L, Kronenberg H M, Mulligan R C. Genes Dev. 1994;8:277–289. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- 24.Lanske B, Karaplis A C, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize L, Ho C, Abou-Samra A B, Jüppner H, Segre G V, Kronenberg H M. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- 25.Karaplis A C, Yasuda T, Hendy G N, Goltzman D, Banville D. Mol Endocrinol. 1990;4:441–446. doi: 10.1210/mend-4-3-441. [DOI] [PubMed] [Google Scholar]
- 26.Krukowski M, Smith J J. Biol Neonate. 1976;29:148–161. doi: 10.1159/000240859. [DOI] [PubMed] [Google Scholar]
- 27.Math F, Davrainville J L. Experientia. 1979;35:1355–1356. doi: 10.1007/BF01964005. [DOI] [PubMed] [Google Scholar]
- 28.Wallach S, Verch R L. J Am Coll Nutr. 1984;3:69–74. doi: 10.1080/07315724.1984.10720038. [DOI] [PubMed] [Google Scholar]
- 29.Symonds H W, Sansom B F, Twardock A R. Res Vet Sci. 1972;13:272–275. [PubMed] [Google Scholar]
- 30.Ramberg C F, Jr, Delivoria-Papadopoulos M, Crandall E D, Kronfeld D S. J Appl Physiol. 1973;35:682–688. doi: 10.1152/jappl.1973.35.5.682. [DOI] [PubMed] [Google Scholar]
- 31.Potts J T, Jr, Bringhurst F R, Gardella T, Nussbaum S, Segre G, Kronenberg H. In: Endocrinology. DeGroot L J, editor. Philadelphia: Saunders; 1995. pp. 920–966. [Google Scholar]
- 32.MacIsaac R J, Caple I W, Danks J A, Diefenbach-Jagger H, Grill V, Moseley J M, Southby J, Martin T J. Endocrinology. 1991;129:757–764. doi: 10.1210/endo-129-2-757. [DOI] [PubMed] [Google Scholar]
- 33.Ferguson J E, Seaner R, Bruns D E, Redick J A, Mills S E, Jüppner H, Segre G V, Bruns M E. Am J Obstet Gynecol. 1994;170:1018–1024. doi: 10.1016/s0002-9378(94)70095-8. [DOI] [PubMed] [Google Scholar]
- 34.Ferguson J E, Gorman J V, Bruns D E, Weir E C, Burtis W J, Martin T J, Bruns M E. Proc Natl Acad Sci USA. 1992;89:8384–8388. doi: 10.1073/pnas.89.17.8384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Senior P V, Heath D A, Beck F. J Mol Endocrinol. 1991;6:281–290. doi: 10.1677/jme.0.0060281. [DOI] [PubMed] [Google Scholar]
- 36.Usdin T B, Gruber C, Bonner T I. J Biol Chem. 1995;270:15455–15458. doi: 10.1074/jbc.270.26.15455. [DOI] [PubMed] [Google Scholar]
- 37.Karperien M, van Dijk T B, Hoeijmakers T, Cremers F, Abou-Samra A B, Boonstra J, de Laat S W, Defize L H. Mech Dev. 1994;47:29–42. doi: 10.1016/0925-4773(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 38.Ellis A G, Adam W R, Martin T J. J Endocrinol. 1990;126:403–408. doi: 10.1677/joe.0.1260403. [DOI] [PubMed] [Google Scholar]
- 39.Orloff, J. J., Kats, Y., Mitnick, M., Gasalla-Herraiz, J. & Isales, C. M. (1993) J. Bone Miner. Res. 8, Suppl. 1, S133.
- 40.Orloff J J, Reddy D, de Papp A E, Yang K H, Soifer N E, Stewart A F. Endocr Rev. 1994;15:40–60. doi: 10.1210/edrv-15-1-40. [DOI] [PubMed] [Google Scholar]
- 41.Wu T L, Vasavada R C, Yang K H, Massfelder T, Ganz M, Abbas S K, Care A D, Stewart A F. J Biol Chem. 1996;271:24371–24381. doi: 10.1074/jbc.271.40.24371. [DOI] [PubMed] [Google Scholar]