

Abstract
Transpositions of mtDNA sequences to the nuclear genome have been documented in a wide variety of individual taxa, but little is known about their taxonomic frequency or patterns of variation. We provide evidence of nuclear sequences homologous to the mtDNA control region in seven species of diving ducks (tribe Aythyini). Phylogenetic analysis places each nuclear sequence as a close relative of the mtDNA haplotypes of the specie(s) in which it occurs, indicating that they derive from six independent transposition events, all occurring within the last ≈1.5 million years. Relative-rate tests and comparison of intraspecific variation in nuclear and mtDNA sequences confirm the expectation of a greatly reduced rate of evolution in the nuclear copies. By representing mtDNA haplotypes from ancestral populations, nuclear insertions may be valuable in some phylogenetic analyses, but they also confound the accurate determination of mtDNA sequences. In particular, our data suggest that the presumably nonfunctional but more slowly evolving nuclear sequences often will not be identifiable by changes incompatible with function and may be preferentially amplified by PCR primers based on mtDNA sequences from related taxa.
The transposition of mtDNA sequences into the nuclear genome has been documented in a wide variety of taxa and for many different mitochondrial genes (1–12). This phenomenon confirms the plausibility of a transfer of mitochondrial genes to the nucleus that is suggested by the endosymbiotic theory of mitochondrial origin (1, 2, 13–15) and may serve as a model for examining mechanisms of transposition, gene duplication, and viral infection (2–4, 16, 17). Insertions of mtDNA sequences into the nucleus, however, also present a potentially serious problem for molecular studies in systematics and population biology, an issue that has received attention in several recent studies (5–8). Direct sequencing of PCR products that include both mitochondrial and nuclear sequences may yield sequences with numerous ambiguities (5, 9), or worse, lead to error in phylogenetic analyses through the unwitting inclusion of paralogous nuclear sequences instead of mitochondrial sequences for some taxa.
Most reports of mtDNA sequences in the nucleus have considered a single taxon and often a single individual, leaving questions about intra- and interspecific variation in nuclear insertions largely unanswered. We addressed these questions as part of population and phylogenetic studies of the tribe Aythyini, a closely related group of diving ducks, in which we found evidence of nuclear sequences homologous to the mtDNA control region. Phylogenetic analysis of nuclear and mtDNA sequences suggests multiple, independent transposition events of relatively recent origin within this small group of species. This pattern suggests a high frequency of these events and differs markedly from two recent phylogenetic analyses of nuclear insertions, in which nuclear sequences in a number of taxa originated from a single, more ancient transposition (6, 7). By comparing levels of intraspecific variation in the two sequences, we also directly compared rates of evolution in nuclear and mitochondrial DNAs.
We suggest that any studies involving mtDNA sequences carefully consider the possibility of nuclear contamination. Particularly problematic for molecular systematics, nuclear sequences are more similar to ancestral states than mtDNA sequences because of their slower rate of evolution and therefore may be preferentially amplified by PCR primers based on sequences of related taxa.
MATERIALS AND METHODS
Samples for genetic analysis included blood, feathers, and web taken from live birds in the wild and captivity, muscle and feathers from hunter-shot birds, and embryos from eggs collected in the field. Genomic DNA was isolated from 0.1 g of muscle or web tissue, 20 μl of whole blood in lysis buffer, or the base of the quill (3 mm) of a single medium-sized (100 mm) feather by digestion with proteinase K and SDS (and dithiothreitol for feathers) followed by phenol/chloroform extraction (18). mtDNA was isolated from 2.5 g of embryonic tissue using sucrose-gradient ultracentrifugation (19).
We determined the sequence of the most variable portion of the control region for 14 pochard species (genera Aythya and Netta) and two outgroup taxa (Cairina scutullata and Marmaronetta angustirostris). An ≈380 nucleotide-pair (np) fragment at the 5′ end of the control region was amplified with primers C1 (L78, 5′-GTTATTTGGTTATGCATATCGTG-3′) and C1R (H493, 5′-AAAATGTGAGGAGGGCGAGG-3′) (L and H numbers refer to the strand and nucleotide position of the 3′ end in the chicken sequence; ref. 20). These primers are located in relatively conserved portions of the control region and work with most ducks (subfamily Anatinae). Gel-purified, double-stranded PCR products were sequenced directly with C1, C1R, and internal primers C1F2 (5′-ACTATCCTACTACGCAAGGAC-3′) and C1R2 (5′-CGATTAGTAAATCCATCTGGTAC-3′).
We designed additional primers to selectively amplify either mitochondrial or nuclear sequences (see Results). These included M1 (5′-GTCCCAGTAATACACATTATTC-3′, located 56 np internal to C1), which was paired with C1R to amplify mtDNA in Aythya valisineria and Aythya ferina; N1 (5′-CTATGGTCCCAGTAATACACACC-3′, 51 np internal to C1) and N1R (5′-TGGGCCTGAAGCTAGTCACG-3′, 37 np internal to C1R), which amplified the nuclear insertion in these species; and RHN1 (5′-CCCTACTATGCAAGGACTAAAC-3′, 85 np internal to C1) and RHN1R (5′-GTCGGACATTATGTGCAAGGA-3′, 57 np internal to C1R), which amplified the nuclear insertion in Aythya americana. For other taxa, we used restriction enzymes chosen on the basis of initial ambiguous sequences to preferentially cut either the mitochondrial or nuclear copy and then determined the sequences of the intact copy and/or fragments. PCR products amplified with primers C1 and C1R were digested with HaeIII (generating three mtDNA and two nuclear fragments in Aythya innotata), MspI (generating two nuclear fragments and leaving the mtDNA product intact in Netta rufina), or AluI (generating two mtDNA and five nuclear fragments in Netta erythrophthalma). Resulting fragments were separated on an agarose gel, purified from gel slices, and sequenced.
Evolutionary relationships among mtDNA control region sequences and homologous nuclear sequences were inferred in parsimony analyses using paup (21). We used heuristic searches with gapmode set to newstate and completed 100 random addition sequence replicates and 100 bootstrap replicates for each analysis. Decay indices for nodes present in strict consensus trees were determined by finding the shortest trees not consistent with constraint trees specifying each node. Sequences were aligned by eye in sequence navigator (Applied Biosystems). (Alignments used in all analyses are available from the authors.)
RESULTS AND DISCUSSION
In A. valisineria, we obtained a single unambiguous sequence for each individual when isolated mtDNA (n = 7) or genomic DNA extracted from tissue (muscle, feather, or web; n = 72) was used in PCR. Genomic DNA extracted from blood (n = 53), however, yielded sequences with ambiguities at the same positions in all individuals (Fig. 1). Given that red blood cells in birds are both nucleated and relatively depauperate in mtDNA (22), we concluded that a relatively high ratio of nuclear DNA resulted in amplification of both mtDNA and homologous nuclear sequences, as has been verified in one avian species (5, 22). On the basis of ambiguous positions in sequences from blood, we designed new PCR primers to discriminate between the putative mitochondrial and nuclear sequences (Fig. 1). Mitochondrial- and nuclear-specific primers yielded two different sequences from each bird, whether genomic DNA from blood or tissue was used. Differences between the two sequences corresponded to ambiguous positions in sequences obtained from blood when using our original, conserved primers.
Figure 1.
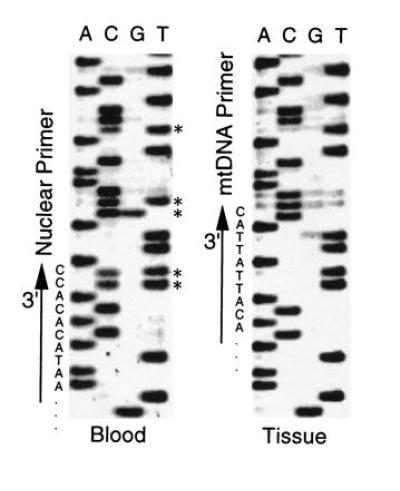
Comparison of L-strand sequences obtained from blood and tissue samples of A. valisineria (positions 40–70 from C1 primer). Asterisks indicate ambiguous positions present in all sequences from blood samples. Locations and 3′-end sequences of L-strand nuclear- and mtDNA-specific primers are shown.
Blood samples of A. ferina, A. americana, Aythya affinis, A. innotata, N. rufina, and Netta erythrophthalma also yielded sequences with ambiguous positions. Ambiguities in A. ferina sequences occurred in the same positions as in A. valisineria, allowing amplification of mtDNA and nuclear sequences using the specific primers designed for A. valisineria. In the remaining species, ambiguities occurred in positions that were unique to each species. We determined putative mtDNA and nuclear sequences for each species by comparing sequences derived from blood with the unambiguous sequences derived from other tissues. We also isolated the nuclear sequence using a species-specific pair of nuclear primers for A. americana or restriction enzymes for A. innotata, N. rufina, and N. erythrophthalma.
To compare levels of variability, we determined mtDNA and nuclear sequences, respectively, for 122 and 6 individuals of A. valisineria, 15 and 5 individuals of A. ferina, and 160 and 8 individuals of A. americana. We found extensive variation in mtDNA sequences: 65, 11, and 80 unique haplotypes in the three species, respectively. In contrast, we found no intraspecific variation in nuclear sequences, except for one ambiguous position in three individuals of A. valisineria, perhaps representing a heterozygous condition. With the exception of this one position, nuclear sequences of A. valisineria were also identical to those of A. ferina. The larger sample of dual sequences from blood samples also showed no evidence of variability in the nuclear sequence. This suggests that nuclear sequences have changed little or not at all since transposition and, in essence, represent mitochondrial haplotypes sampled from ancestral populations.
Phylogenetic analysis of nuclear and mtDNA sequences supports this conclusion. Nuclear sequences are basal to all extant mtDNA haplotypes and are closer than mtDNA sequences to ancestral states (Fig. 2) and to the mtDNA of other species (Table 1). In addition, the identical nuclear sequence shared by A. valisineria and A. ferina branches off before the split of these species’ mtDNA lineages, while the nuclear sequence apparently unique to A. americana branches off after its split with Aythya collaris. Relative-rate tests (24) also indicate a significantly slower rate of evolution in nuclear sequences (P = 0.0001–0.0017 for comparisons of the A. valisineria/A. ferina nuclear sequence with mtDNA haplotypes; P = 0.0016–0.058 for A. americana). Note, however, that nuclear sequences are not necessarily direct ancestors of current mtDNA haplotypes, but may have originated from mtDNA lineages that subsequently went extinct. Much of the divergence between the nuclear copy and the common ancestor of current nuclear and mtDNA sequences probably occurred in the mitochondrial genome prior to transposition.
Figure 2.
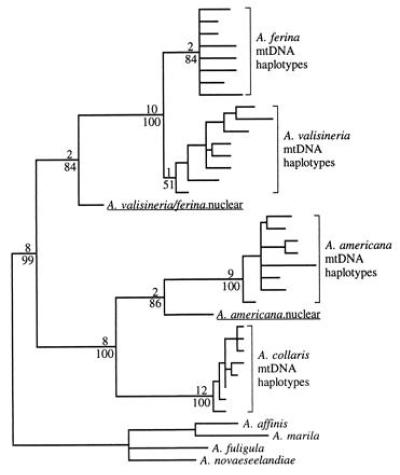
Strict consensus of 1248 shortest trees (288 steps) for mtDNA control region sequences and homologous nuclear sequences found in parsimony analysis assuming equal weights for all character state changes. Branch lengths are proportional to the number of steps along each branch with acctran optimization. Alignment gaps were introduced in one or more taxa at 22 of 392 positions. We excluded positions 1–65 and 366–392 due to missing data in some taxa. The same set of shortest trees was found in all 100 random addition sequence replicates. Decay indices (number of additional steps required in a tree without the node in question) and bootstrap percentages are given above and below branches, respectively. Eight mtDNA haplotypes representing the range of observed variation were selected for A. ferina, A. valisineria, A. americana, and A. collaris. mtDNA haplotypes of A. affinis, Aythya marila, Aythya fuligula, and Aythya novaeseelandiae were designated as the outgroup. These eight species form a clade with no other extant members (see Fig. 3). Because of computational limitations, bootstrap percentages and decay indices were obtained using a smaller data set that included only four mtDNA haplotypes of each of the ingroup taxa.
Table 1.
Corrected percent sequence divergence among mitochondrial control region sequences (.mt) and homologous nuclear sequences (.N) for taxa in Fig. 2
| Taxa | Mean % distance (range)
|
|||||
|---|---|---|---|---|---|---|
| AM.N | AM.mt | VA/FE.N | VA/FE.mt | |||
| Outgroup | 15.8 | < | 18.4 | 13.5 | < | 18.4 |
| (13.8–18.6) | (14.1–23.0) | (10.3–15.7) | (14.1–23.0) | |||
| CO.mt | 7.9 | < | 12.1 | 10.3 | < | 15.4 |
| (7.4–8.4) | (9.1–14.0) | (9.6–11.0) | (12.9–18.3) | |||
| AM.N | 0 | 7.2 | 8.4 | < | 14.9 | |
| (6.1–8.1) | (11.8–17.2) | |||||
| AM.mt | 1.8* | 12.9 | < | 15.0 | ||
| (0.5–3.7)* | (10.9–14.9) | (11.8–19.0) | ||||
| VA/FE.N | 8.4 | < | 12.9 | 0 | 9.7 | |
| (10.9–14.9) | (7.9–11.5) | |||||
| VA/FE.mt | 14.9 | < | 15.0 | 2.5* | ||
| (11.8–17.2) | (11.8–19.0) | (0.3–5.9)* | ||||
We used a = 0.5 in the distance formula (23). Positions with alignment gaps in one or more taxa (22 of 392) were excluded from all comparisons. Outgroup as in Fig. 2. Mitochondrial sequences for A. valisineria (VA) and A. ferina (FE) were combined in a single group for this analysis. AM, A. americana; CO, A. collaris.
Intragroup distances are biased upward by our deliberate selection of divergent mtDNA haplotypes.
Phylogenetic analysis also demonstrates that at least six independent transpositions of mtDNA sequences have occurred in the Aythyini (Fig. 3). Although relationships among sequences are not fully resolved by our data set, each nuclear sequence is closely related to the mtDNA of the specie(s) in which it resides and is separated from other nuclear sequences by two or more well-supported nodes. This pattern contrasts with two recent reports that also analyzed nuclear insertions in a phylogenetic context. In primates (6) and the avian genera Scytalopus and Myornis (7), nuclear sequences within a group of closely related species form a monophyletic group sister to the mtDNA sequences of the same species, suggesting a single, more ancient transposition event followed by divergence among species in the nuclear copy following transposition.
Figure 3.
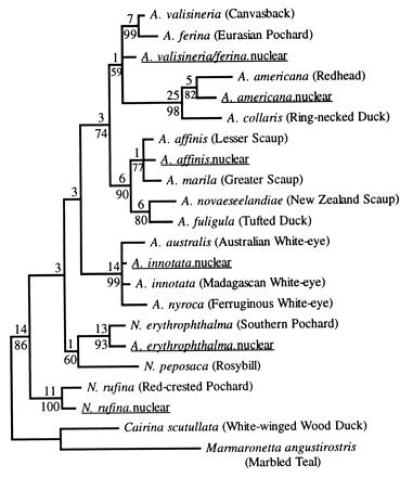
Strict consensus of 13 shortest trees (538 steps) found in a weighted parsimony analysis. The same set of shortest trees was found in all 100 random addition sequence replicates. Decay indices and bootstrap values are as in Fig. 2. Nuclear sequences homologous to the mtDNA control region are underlined. Cairina scutullata and Marmaronetta angustirostris were designated as the outgroup. We specified a step matrix that gave transitions, transversions, and indels weights of 1, 5, and 5, respectively. Note that branch lengths and decay indices reflect the weighted step matrix. Alignment gaps were introduced at 41 of 398 positions, but we excluded positions 127–133, 171–208, and 226–231 due to ambiguities in alignment, and positions 1–15 due to missing data.
Although they include both conserved and highly variable positions, the 380 bases of the control region sequenced in this study are, on average, evolving at about 4.4 times the rate of other mtDNA sequences: mean corrected percent sequence divergence (23) between ingroup and outgroup mtDNA haplotypes in Fig. 2 is 17.7%, compared with 4.0% divergence in cytochrome b sequences of the same taxa (unpublished data) and 3.2 to 4.3% divergence between A. affinis and three of our ingroup taxa based on mtDNA restriction fragment length polymorphism data (25). This suggests a rate of 8.8% divergence per million years for the control region fragment, assuming an overall rate of 2% per million years for waterfowl mtDNA (26). Given estimates of divergence from outgroup taxa (Table 1), and assuming nuclear homologues evolve at a rate of 4.6 × 10−9 substitutions per site per million years following transposition, a rate for nuclear pseudogenes (27), we solved equations 2–4 of Li et al. (27) to estimate the coalescence time of mtDNA and nuclear sequences and the time of transposition. Respectively, these estimates are 1.66 and 1.24 million years ago (Mya) for the nuclear copy shared by A. valisineria and A. ferina, and 1.12 and 0.66 Mya for the nuclear copy in A. americana. Corrected percent sequence divergence between mtDNA and nuclear sequences in N. erythrophthalma (7.8 ± 2.1) was comparable to that in A. americana, but was substantially less in three other species (2.6 ± 0.9 in A. affinis, 2.9 ± 1.0 in A. innotata, and 4.1 ± 1.2 in N. rufina), suggesting even more recent transpositions.
We consider these calculations to provide only very rough estimates of the actual times of transposition. Nonetheless, estimates on the order of 105–106 years before present for the Aythyini are more recent than transposition events documented in most other taxa. Similar estimates for other taxa include 2 Mya in cats (9), >5 Mya in birds (7), 9–43 Mya in humans (17, 28), 13.5 Mya in rats (16), 25 Mya in yeast (10), and 30 Mya in primates (6). The lack of intraspecific variation we observed in nuclear sequences suggests a dramatically slower rate of sequence evolution in the nucleus (29, 30), but is consistent with estimated rates of nuclear pseudogene evolution given these relatively short times since transposition. Given a transposition time of 1.24 Mya for the oldest transposition we studied and 4.6 × 10−9 substitutions per site per million years in the nuclear copy (27), only 1.7 substitutions would be expected in the 292-nucleotide nuclear sequence we surveyed in A. valisineria and A. ferina. One ambiguous position in the nuclear sequence of some individuals suggests that at least one mutation has occurred following transposition.
The high frequency of nuclear integration of mtDNA sequences suggested by our data raises two paradoxical questions. First, why are nuclear copies not observed in all species, and second, why are multiple nuclear copies representing successive transposition events dating back in time (31) not evident within species? We suspect that nuclear homologues of mtDNA sequences are very common, if not ubiquitous, but usually are not amplified from total DNA extracts of mtDNA-rich tissues or isolations of mtDNA. PCR amplification of nuclear homologues may also depend on variation among species or individual samples in the ratio of mtDNA to nuclear DNA or variation in copy number of the nuclear sequence. For example, Lopez et al. (9) characterized a 7.9-kb insertion of mtDNA sequence that is tandemly repeated from 38 to 76 times in the nuclear genome of the cat. White cell counts might be an important source of variation in mtDNA copy number in avian blood samples (22). The slow rate of evolution in the nucleus, however, argues against the proposition that primer sites in unconstrained nuclear copies rapidly accumulate substitutions that make them unavailable to PCR.
Perhaps more problematic is the lack of evidence for multiple transpositions within a species. In this regard, our results are consistent with those for most other taxa in which a single nuclear homologue of a mtDNA gene has been described (however, see ref. 31). As in our study, blood samples yielded only two sequences from each individual in the two previous reports of nuclear copies of mtDNA in birds (5, 7). One possibility is that nuclear copies are periodically overwritten by current mitochondrial sequences through some form of gene conversion (32). Taxa with phylogenetically independent transpositions that had identical flanking regions in the nucleus would strongly support this hypothesis. Based on screenings of gene libraries, however, Fukuda et al. (28) estimated that hundreds of mtDNA-like sequences have accumulated over evolutionary time in the human nuclear genome. These sequences might rapidly become unavailable to PCR if they are inserted in regions with high frequencies of recombination, such that they are soon fragmented, rearranged (4, 10, 17), or lost completely. We also point out that previous reports of mtDNA homologues in the nucleus have given little consideration to the population-level process (i.e., drift) that must follow a transposition event in an individual before a nuclear copy is likely to be detected in a population. Only 1/2Ne of transposition events will become fixed. Populations with intermediate frequencies of a nuclear copy should also be expected (33).
Our results have important implications for mtDNA-based studies of phylogeny and population structure. First, nuclear copies can be expected and will complicate the accurate determination of mtDNA sequences (5–8). Second, given their slower rate of evolution, nuclear copies will not necessarily be identifiable by the presence of insertions, deletions, or stop codons in protein-coding sequences (e.g., see refs. 3, 6, 7, and 31) or changes incompatible with secondary structure in ribosomal or transfer RNAs. Significantly shorter branches in phylogenetic analyses may help to identify older nuclear insertions, but those of very recent origin may be difficult to distinguish from mtDNA heteroplasmy or cross-contamination of samples. Particularly problematic, PCR primers based on sequences of related taxa may preferentially amplify nuclear homologues because mtDNA diverges more rapidly than nuclear DNA from ancestral sequences, particularly for rapidly evolving loci such as the control region. Nonetheless, comparison of sequences derived from isolations of nuclear and mtDNAs or from tissues known to differ in their ratios of nuclear and mtDNA may help to identify nuclear sequences.
Although the unwitting inclusion of nuclear homologues in phylogenetic analyses may introduce error, nuclear copies also present opportunities. Nuclear sequences can provide a sample of extinct mtDNA haplotypes that may be valuable for reconstructing ancestral states or as outgroups in phylogenetic analyses (5, 33). The identical nuclear sequence in A. valisineria and A. ferina provides a very strong signal of the sister relationship of these species and may be the most appropriate outgroup for phylogenetic analysis of their extant mtDNA haplotypes. Presumably nonfunctional, nuclear sequences homologous to the most variable portion of the mtDNA control region also provide perhaps the most direct comparison possible of mutation rates between nuclear and mtDNA, independent of selective constraints.
Acknowledgments
We thank A. Cooper, C. L. Tarr, and C. E. McIntosh for advice and technical assistance. A. Cooper, W. M. Brown, D. P. Mindell, D. O’Foighil, and anonymous reviewers provided helpful comments on the manuscript. Funding was provided by the Smithsonian Institution and by National Science Foundation grants to M.D.S. and R. B. Payne.
Footnotes
Abbreviations: Mya, million years ago; np, nucleotide pair.
References
- 1.Gellissen G, Bradfield J Y, White B N, Wyatt G R. Nature (London) 1983;301:631–634. doi: 10.1038/301631a0. [DOI] [PubMed] [Google Scholar]
- 2.Kemble R J, Mans R J, Gabay-Laughnan S, Laughnan J R. Nature (London) 1983;304:744–747. [Google Scholar]
- 3.Smith M F, Thomas W K, Patton J L. Mol Biol Evol. 1992;9:204–215. doi: 10.1093/oxfordjournals.molbev.a040714. [DOI] [PubMed] [Google Scholar]
- 4.Jacobs H T, Posakony J W, Grula J W, Roberts J W, Xin J-H, Britten R J, Davidson E H. J Mol Biol. 1983;165:609–632. doi: 10.1016/s0022-2836(83)80270-8. [DOI] [PubMed] [Google Scholar]
- 5.Quinn T W. Mol Ecol. 1992;1:105–117. doi: 10.1111/j.1365-294x.1992.tb00162.x. [DOI] [PubMed] [Google Scholar]
- 6.Collura R V, Stewart C-B. Nature (London) 1995;378:485–489. doi: 10.1038/378485a0. [DOI] [PubMed] [Google Scholar]
- 7.Arctander P. Proc R Soc London B. 1995;262:13–19. doi: 10.1098/rspb.1995.0170. [DOI] [PubMed] [Google Scholar]
- 8.van der Kuyl A C, Kuiken C L, Dekker J T, Perizonius W R K, Goudsmit J. J Mol Evol. 1995;40:652–657. doi: 10.1007/BF00160513. [DOI] [PubMed] [Google Scholar]
- 9.Lopez J V, Yuhki N, Masuda R, Modi W, O’Brien S J. J Mol Evol. 1994;39:174–190. doi: 10.1007/BF00163806. [DOI] [PubMed] [Google Scholar]
- 10.Farrelly F, Butow R A. Nature (London) 1983;301:296–301. doi: 10.1038/301296a0. [DOI] [PubMed] [Google Scholar]
- 11.Zullo S, Sieu L C, Slightom J L, Hadler H I, Eisenstadt J M. J Mol Biol. 1991;221:1223–1235. [PubMed] [Google Scholar]
- 12.Blanchard J L, Schmidt G W. J Mol Evol. 1995;41:397–406. doi: 10.1007/BF00160310. [DOI] [PubMed] [Google Scholar]
- 13.Gray M W. Trends Genet. 1989;5:294–299. doi: 10.1016/0168-9525(89)90111-x. [DOI] [PubMed] [Google Scholar]
- 14.Nugent J M, Palmer J D. Cell. 1991;66:473–481. doi: 10.1016/0092-8674(81)90011-8. [DOI] [PubMed] [Google Scholar]
- 15.Cole R A, Slade M B, Williams K L. J Mol Evol. 1995;40:616–621. doi: 10.1007/BF00160509. [DOI] [PubMed] [Google Scholar]
- 16.Hadler H I, Dimitrijevic B, Mahalingam R. Proc Natl Acad Sci USA. 1983;80:6495–6499. doi: 10.1073/pnas.80.21.6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamimura N, Ishii S, Liandong M, Shay J W. J Mol Biol. 1989;210:703–707. doi: 10.1016/0022-2836(89)90103-4. [DOI] [PubMed] [Google Scholar]
- 18.Cooper A. In: Ancient DNA: Recovery and Analysis of Genetic Material from Paleontological, Archaeological, Museum, Medical, and Forensic Specimens. Herrmann B, Herrmann S, editors. New York: Springer; 1994. pp. 149–165. [Google Scholar]
- 19.Tarr C L, Fleischer R C. Auk. 1993;110:825–831. [Google Scholar]
- 20.Desjardins P, Morais R. J Mol Biol. 1990;212:599–634. doi: 10.1016/0022-2836(90)90225-B. [DOI] [PubMed] [Google Scholar]
- 21.Swofford, D. L. (1993) paup: Phylogenetic Analysis Using Parsimony (Illinois Natural History Survey, Champaign, IL), Version 3.1.
- 22.Quinn T W, White B N. In: Avian Genetics: A Population and Ecological Approach. Cooke F, Buckley P A, editors. London: Academic; 1992. pp. 163–198. [Google Scholar]
- 23.Tamura K, Nei M. Mol Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- 24.Mindell D P, Honeycutt R L. Annu Rev Ecol Syst. 1990;21:541–566. [Google Scholar]
- 25.Kessler L G, Avise J C. Syst Zool. 1984;33:370–380. [Google Scholar]
- 26.Shields G F, Wilson A C. J Mol Evol. 1987;24:212–217. doi: 10.1007/BF02111234. [DOI] [PubMed] [Google Scholar]
- 27.Li W-H, Gojobori T, Nei M. Nature (London) 1981;292:237–239. doi: 10.1038/292237a0. [DOI] [PubMed] [Google Scholar]
- 28.Fukuda M, Wakasugi S, Tsuzuki T, Nomiyama H, Shimada K. J Mol Biol. 1985;186:257–266. doi: 10.1016/0022-2836(85)90102-0. [DOI] [PubMed] [Google Scholar]
- 29.Brown W M, George M, Wilson A C. Proc Natl Acad Sci USA. 1979;76:1967–1971. doi: 10.1073/pnas.76.4.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vawter L, Brown W M. Science. 1986;234:194–196. doi: 10.1126/science.3018931. [DOI] [PubMed] [Google Scholar]
- 31.Hu G, Thilly W G. Gene. 1994;147:197–204. doi: 10.1016/0378-1119(94)90065-5. [DOI] [PubMed] [Google Scholar]
- 32.Jacobs H T, Grimes B. J Mol Biol. 1986;187:509–527. doi: 10.1016/0022-2836(86)90330-x. [DOI] [PubMed] [Google Scholar]
- 33.Zischler H, Geisert H, von Haeseler A, Pääbo S. Nature (London) 1995;378:489–492. doi: 10.1038/378489a0. [DOI] [PubMed] [Google Scholar]