

Abstract
Calorie restriction (CR) prevents many age-associated diseases and prolongs the lifespan. CR induces multiple metabolic and physiologic modifications, including anti-inflammatory, antioxidant, and neuroprotective effects that may be beneficial in multiple sclerosis (MS). The present studies sought to determine whether CR or increased calorie intake alters the course of experimental autoimmune encephalomyelitis (EAE), the leading animal model for MS. SJL and C57BL/6 mice were subjected to 40% CR beginning at 5 weeks of age. After 5 weeks of CR, EAE was induced by immunizing with proteolipid protein in SJL mice and with myelin oligodendrocyte glycoprotein in C57BL/6 mice. Clinical, histologic, and immunologic features of EAE were compared with mice fed ad libitum and to SJL mice fed a high-fat, high-calorie diet. CR ameliorated clinical EAE in both mouse strains with less severe inflammation, demyelination, and axon injury. No suppression of immune function was observed. A high-calorie diet did not alter the EAE course. CR was associated with increased plasma levels of corticosterone and adiponectin and reduced concentrations of IL-6 and leptin. The CR-induced hormonal, metabolic, and cytokine changes observed in our studies suggest a combined anti-inflammatory and neuroprotective effect. CR with adequate nutrition and careful medical monitoring should be explored as a potential treatment for MS.
Keywords: autoimmunity, inflammation, adipokines, corticosteroids, multiple sclerosis
INTRODUCTION
Multiple sclerosis (MS) is a complex disease involving CNS inflammation, demyelination, and axonal damage. MS is common, with a prevalence of one to two per 1000 in North America. Several partially effective therapies are now U.S. Food and Drug Administration (FDA)-approved, but 25–50% of MS patients are suboptimal responders to these immunomodulatory therapies [1]. Several promising, new treatments also have significant risks. Thus, more effective but less risky therapies are still greatly needed.
Experimental autoimmune encephalomyelitis (EAE) is the leading animal model for MS. EAE is an autoimmune disease initiated by myelin-reactive T cells, but other components of the immune system, including monocytes/macrophages, B cells, antibodies, cytokines, and chemokines, play roles in its pathogenesis [2,3,4]. Although EAE is not a perfect model for MS, it has been instrumental in better understanding the disease. Moreover, murine EAE was crucial in the development of three FDA-approved medications for MS (Copaxone, Tysabri, Novantrone) [5].
Several studies have shown that calorie restriction (CR) without malnutrition prevents many age-associated, chronic diseases and prolongs the lifespan of mammals. Long-term CR causes many metabolic and physiologic changes that would be expected to be beneficial in MS. These include enhanced endogenous glucocorticoid production [6], reduced inflammation [7], Sirtuin 1 induction [8], increased CNS levels of neurotrophic factors [9, 10], decreased serum leptin, and increased adiponectin concentration [11, 12]. Moreover, CR has been reported to decrease mortality and reduce inflammatory responses in animal models of acute inflammation [13] and autoimmunity [14, 15].
Based on the reported anti-inflammatory and neuroprotective effects of CR, we undertook calorie modification studies in two mouse models of EAE, each of which bears similarities to a subtype of MS. To determine the effects of chronic CR on the clinical course of EAE, extended experiments were performed in SJL mice, which display a relapsing-remitting clinical course, and in C57BL/6 mice, which display a chronic, nonremitting course. We also tested the effects of increased calorie intake on EAE by feeding SJL mice with a high-fat (HF) diet. In the present studies, EAE was less severe in mice subjected to 40% CR, whereas a HF diet did not alter EAE. An important advantage of CR in our studies, especially if it is to be translated safely to humans with MS, was that it did not lead to immune system suppression.
MATERIALS AND METHODS
Mice
Female SJL/J and C57BL/6J mice (Jackson Labs, Bar Harbor ME, USA) were obtained at 4 weeks of age, individually housed and maintained in microisolator cages in accordance with university and National Institutes of Health (NIH) guidelines. Baseline daily food intake was tracked for 1 week before mice were assigned randomly to calorie modification or normal feeding.
Calorie modification
Beginning at 5 weeks of age, after 1 week of acclimation and determination of food intake, control mice continued to be fed regular chow (4.3% fat; PicoLab Rodent Diet 20 #5053, Purina, St. Louis, MO, USA) ad libitum, and CR mice were fed 40% less than the baseline intake. In Experiments 1 and 2, a third group was fed ad libitum with a HF diet (23% fat; Test Diet #58V8, Purina). Following 5 weeks on the assigned diet, mice were immunized to induce EAE as described below. Mouse body weights were recorded throughout the experiment, and clinical scores were recorded daily after immunization.
EAE induction
Mice were immunized s.c. with 50 μg proteolipid protein (PLP)139–151 or myelin oligodendrocyte glycoprotein (MOG)35–55 (PLP aa 139–151: HCL GKW LGH PDK F; MOG aa 35–55: MEV GWY RSP FSR VVH LYR NGK; Sigma Genosys, Woodlands, TX, USA), emulsified in IFA containing 60 μg/SJL mouse or 50 μg/B6 mouse Mycobacterium tuberculosis, strain H37RA. In all but one experiment, mice also received 200 ng/SJL mouse or 300 ng/B6 mouse pertussis toxin (PTX; List Biological Laboratories, Campbell, CA, USA) i.v. at the time of immunization and 72 h later. In adoptive transfer experiments, spleen cells were stimulated in vitro (4×106 cells/ml) with the indicated antigen (10 μg/ml). After 2 days in culture, 2.5 × 107 cells were transferred i.v. in naïve recipient mice. The development of EAE was followed and graded on a scale of 0–5 as described previously [16].
Corticosterone radioimmunoassay
Serial blood samples were obtained at baseline after 4 weeks of calorie modification preimmunization, before onset of EAE, and during clinical disease. Plasma samples were obtained by a heparinized capillary tube via retro-orbital plexus puncture at 10 a.m. at each time-point. Plasma corticosterone levels were quantified using the ImmuChem double-antibody corticosterone kit for rodents (MP Biomedicals, Orangeburg, NY, USA).
Body composition measurements
Body composition was determined using a portable PIXImus small animal densitometer in randomly selected mice from each dietary group. This device uses dual energy X-ray absorptiometry to assay adipose mass, as well as lean tissue mass and bone mineral content.
Proliferation assays
Spleen and lymph node cells were isolated from immunized mice and cultured at 2.5 × 106 cells/ml in complete RPMI-1640 medium, 5% FBS, with or without 10 μg/ml antigen (PLP139–151 or MOG35–55). Incorporation of 3H-thymidine (0.5 μCi/well) during the final 18 h was counted (Betaplate 1205, Wallac, Gaithersburg, MD, USA), and the stimulation index was calculated as the ratio between cpm with antigen and cpm in medium alone.
Cytometric bead array (CBA)
Spleens from SJL and C57BL/6 mice were removed at Day 29 postimmunization (p.i.). Single-cell suspensions were prepared, and cells were cultured with medium alone or with PLP139–151 or MOG35–55 (10 μg/ml). Aliquots of cell culture supernatant were removed at 48, 72, and 96 h of culture and frozen at –80° for subsequent analysis. IL-12p70, TNF-α, IFN-γ, MCP-1, IL-10, and IL-6, present in cell culture supernatants, were analyzed by CBA (BD Biosciences, Franklin Lakes, NJ, USA).
Cytokine and adipokine ELISAs
Spleen and lymph node cells were isolated from immunized mice and cultured at 2.5 × 106 cells/ml with the indicated peptide antigen at 10 μg/ml in complete RPMI 1640 with 5% FBS. Cell culture supernatants were collected at the indicated times and frozen at –80°C until assayed for IFN-γ by QuantikineM ELISA (R&D Systems, Minneapolis, MN, USA). Leptin and adiponectin were measured in plasma using commercially available kits (Linco Research, St. Charles, MO, USA). Plasma levels of IL-6 were analyzed by ELISA (R&D Systems) in visibly nonhemolyzed samples.
CNS histology
Mice were perfused with buffered 4% paraformaldehyde, CNS tissue-dissected, and embedded in paraffin. Thick sections (4 μm) on slides were stained with H&E to assess inflammation and Luxol Fast Blue (LFB) to assess demyelination. Scoring for inflammation and demyelination was done in a blinded manner by a single observer (A. H. Cross) on a scale of 0–5, as previously published [16]. For inflammation, the scoring system was: 0 = no inflammation; 1 = a few inflammatory cells in the leptomeninges; 2 = organization of inflammatory cells around blood vessels; 3 = extensive perivascular cuffing with extension into the underlying parenchyma; 4 = large regions of white matter inflammation extending into the parenchyma covering between one-fourth and one-half cord white matter; 5 = extensive inflammation covering more than one-half cord white matter. For demyelination, the following scale was used: 0 = none; 1 = a few subpial fibers affected; 2 = partial rim of subpial involvement, not into the parenchyma; 3 = extension beyond the subpial region into the parenchyma; 4 = large regions of white matter involvement, less than one-half cord cross-section; 5 = one-half or more than one-half cord white matter cross-section involved.
Immunohistochemistry for axonal damage
The distribution of injured axons was identified with double-staining experiments using antibodies to nonphosphorylated neurofilament (SMI-32 at 1:200; Sternberger Monoclonals, Luthersville, MD, USA) and myelin basic protein (MBP; at 1:200; Zymed Laboratories Inc., South San Francisco, CA, USA). Primary antibodies were applied overnight at 4°C degree in 3% FBS/PBS. Next, sections were incubated with fluorescently labeled secondary antibodies for 1 h at room temperature (Invitrogen, Carlsbad, CA, USA) and mounted with Vectashield medium (Vector H-1500, Vector Labs, Burlingame, CA, USA). Sections were examined with a Nikon Eclipse 80i microscope. Images were captured with a Photometrics charged-coupled device digital camera using MetaMorph image acquisition software (Universal Imaging Corp., Downington, PA, USA).
Statistical analyses
Parametric data were analyzed, with two-tailed t-test or ANOVA (for three groups) and Welch’s correction factor applied for unequal variances. Tukey correction factor was applied when comparing selected pairs within groups when there were more than two. For nonparametric data, the Mann-Whitney test or a Kruskal-Wallis test was used. Kaplan-Meier analysis was used to compare time with death from EAE in Experiment 1. Differences in EAE incidence and death rate between groups were analyzed by χ2. P< 0.05 was considered significant.
RESULTS
Weights and body composition of mice undergoing calorie modification
At the time of immunization to induce EAE, after 5 weeks on the assigned dietary regimen, the 10- to 11-week-old CR mice weighed significantly less than control and HF groups in all experiments (Fig. 1A and Table 1). As typically observed in EAE, mouse weights dropped immediately prior to clinical onset and gradually returned toward normal in control and HF mice after the acute phase of EAE. In contrast, body weights of CR mice were stably low throughout the experiment with a slight decline at disease peak (Fig. 1A). To examine changes in fat mass and body composition as a result of CR, densitometry scans were performed on representative mice immediately prior to immunization. Although total fat was reduced significantly in CR mice (mean±sd: 2.8 g±0.7) compared with animals fed ad libitum (4.2 g±0.8; P=0.01, two-tailed t-test), the percentage of body fat was not significantly different between groups (23%±4.5 for control mice and 20%±4.7 for CR mice; P=0.45).
Fig. 1.

CR ameliorates the clinical course of EAE in SJL and C57BL/6 mice. (A) Body weights of control mice (•), mice fed with HF diet (▪), and CR mice (○) throughout the experiment before and after immunization (values are mean±sd). The weight changes shown are representative of four experiments. The dotted vertical line indicates the day of immunization. (B) Experiment 1. Survival was increased significantly in CR mice (80%) compared with control (23%) and HF (30%) groups (P=0.02 CR vs. control by χ2). (C) Experiment 3. CR regimen reduces the clinical severity of relapsing-remitting EAE in SJL mice compared with the ad libitum diet in control mice. (D) Experiment 4. CR ameliorates the clinical course of chronic-progressive EAE in C57BL/6 mice. (C and D) Values are mean ± sem; clinical scores were calculated as the average of all animals in each group.
TABLE 1.
Summary of EAE Clinical Characteristics
| Strain | Incidence | Mortality | Weight at immunization | Day of onset | Maximum clinical scorea | Cumulative clinical scoreb | Relapse ratec | |
|---|---|---|---|---|---|---|---|---|
| Expt. 1 | SJL | |||||||
| Control | 10/10 | 7/9 (77%) | 19.5 ± 0.9d | 11.2 ± 0.6 | 5 (4–5) | NA | NA | |
| HF | 10/10 | 7/10 (70%) | 22.2 ± 2.7d | 11.6 ± 0.8 | 5 (2.5–5) | NA | NA | |
| CR | 9/10 | 2/10 (20%)e | 15.7 ± 2d | 11.1 ± 1.1 | 4 (0–5)f | NA | NA | |
| Expt. 2 | SJL | |||||||
| Control | 13/13 | 5/13 (38%) | 18.6 ± 0.9g | 10.7 ± 0.9 | 4 (3–5) | 29.4 ± 18 | 1.7 | |
| HF | 12/12 | 5/12 (41%) | 21.3 ± 1.6g | 11.5 ± 1.6 | 4.2 (3.5–5) | 30.8 ± 5.6 | 1.1 | |
| CR | 11/11 | 3/11 (27%) | 12.4 ± 0.6g | 20.5 ± 4.6h | 4 (3–5) | 18.8 ± 10 | 0.5 | |
| Expt. 3 | SJL | |||||||
| Control | 11/11 | 0/11 | 18.4 ± 0.9 | 12.7 ± 1.3 | 4 (3–4) | 45.8 ± 10.1 | NA | |
| (45.8 ± 80.7) | ||||||||
| CR | 7/11 | 0/11 | 13.4 ± 0.8i | 15.5 ± 3.4j | 2.5 (0–4)k | 19.5 ± 17.2l | NA | |
| (30.7 ± 9.8)j | ||||||||
| Expt. 4 | C57BL/6J | |||||||
| Control | 9/9 | 4/9 (44%) | 19 ± 0.7 | 12.7 ± 4.3 | 4 (3.5–5) | 44.3 ± 11.7 | NR | |
| (44.3 ± 11.7) | ||||||||
| CR | 8/9 | 2/9 (22%) | 13.5 ± 0.4i | 18.1 ± 4.3m | 3.5 (0–5) | 22.2 ± 15.4j | NR | |
| (25.9 ± 13)n |
Values are means ± sd;
median (range);
cumulative clinical score calculated over the first 30 days for all mice or only sick mice (given in parentheses)/group;
average number of relapses per mouse;
Expt. 1, P < 0.05 for control versus HF; P < 0.01 for control versus CR, and P < 0.001 for CR versus HF; ANOVA with Tukey’s multiple comparison test;
P = 0.02 CR versus control death rate by χ2;
CR versus control; P < 0.05, Kruskal-Wallis;
Exp. 2, P < 0.001 for weights in each group compared with each other group; ANOVA with Tukey’s multiple comparison test;
CR is greater than HF and control P < 0.001; ANOVA with Tukey’s multiple comparison test;
P < 0.0001 by t-test;
P = 0.02 by t-test;
P = 0.08 by Mann-Whitney U test;
P = 0.003 by t-test;
P = 0.01 by t-test;
P = 0.03 by t-test. NA, Not available because of high mortality early during the experiment or because follow-up was only 30 days p.i.; NR, non relapsing.
CR ameliorates relapsing-remitting EAE in SJL mice
Three experiments were conducted in SJL mice immunized with PLP139–151 to assess the effects of CR on the course of EAE (summarized in Table 1). In the first experiment, there was a high mortality rate in all mice. However, survival was increased significantly in CR mice (80%) compared with control (23%) and HF (30%) groups (P=0.02 CR vs. control; Fig. 1B). Most mice did not live long enough to allow comparison of relapse rates. In Experiment 2, which had a lower EAE mortality rate, clinical onset of EAE was delayed significantly in CR animals relative to control and HF mice (P<0.001; Table 1). Relapse rate for the CR group was 0.5 compared with 1.1 for controls and 1.7 for HF mice, but this difference did not reach statistical significance. As there were no significant differences between HF and control groups in the first two experiments, mice fed a HF diet were not included in subsequent experiments.
To induce a less-aggressive EAE with a lower mortality rate than in the first two experiments, an immunization protocol that eliminated the adjuvant PTX [17] was used in Experiment 3. No mice in any group died from EAE in this experiment. With the milder overall disease, the beneficial impact of CR on all clinical outcomes was seen more readily (Table 1 and Fig. 1C). EAE incidence was 63% for mice on CR compared with 100% in control mice. In mice that developed EAE, the day of clinical onset was delayed in CR mice versus controls, to 15.5 days p.i. compared with 12.7 days, respectively (P=0.02; Table 1). A clinical course was significantly milder in CR animals with a median maximum clinical score of 2.5 versus 4 in control mice and a cumulative clinical score of 19.5 for CR versus 45.8 for controls (P=0.003; Table 1).
CR ameliorates EAE in C57BL/6 mice
Next, the effects of CR were tested in an EAE model using the C57BL/6 mouse strain, which shows a chronic course of disease. Individually housed C57BL/6 mice were immunized after 5 weeks of an ad libitum diet or 40% CR. Chronic EAE was induced by immunization with MOG35–55 peptide. CR was effective in ameliorating EAE clinical course in this model as well (Fig. 1D). The mortality rate was lower with CR (22% compared with 44% in the control group), although this difference did not reach statistical significance (Table 1). As with the SJL mice, C57BL/6 mice on CR displayed a significant delay in disease onset compared with controls (P=0.01) and a significant reduction of cumulative clinical scores (P=0.02; Table 1). EAE incidence was similar, with eight of nine (88%) CR animals developing disease compared with nine of nine in controls.
Chronic CR decreases CNS inflammation, demyelination, and axonal damage during EAE
The degrees of CNS inflammation and demyelination were evaluated at Day 30 p.i. in the spinal cords of SJL mice in the CR group (n=15) and the normal diet group (n=15). Inflammation and demyelination were graded in a blinded manner. Inflammation was reduced significantly in mice that experienced CR compared with control mice (median: 2.5 in controls and 1.5 in CR mice; P=0.03; Fig. 2G). Notably, in CR tissues, inflammatory cells were localized mainly to the meninges and were rarely observed within the CNS parenchyma (Fig. 2E). In contrast, control mice showed a considerable amount of inflammatory infiltration deeply into the CNS parenchyma, in addition to meningeal infiltration (Fig. 2B). Demyelination was reduced dramatically in mice on a CR regimen compared with controls (median: 3 in controls and 0 in CR mice; P<0.001; Fig. 2H). Extensive areas of demyelination were detectable in control mice, and little demyelination was observed in CR mice (Fig. 2, A and C, compared with D and F).
Fig. 2.

Spinal cord inflammation and demyelination are reduced in CR mice compared with control mice fed ad libitum. Spinal cords were obtained on Day 30 p.i. Representative SJL mice from Experiment 3 for control (A–C) and CR (D–F) groups are shown. LFB staining demonstrates extensive demyelination in the control (A and C) compared with CR (D and F). In the H&E-stained sections, inflammatory cells deeply infiltrated the CNS parenchyma in the control group (B), and they were localized mainly to the meninges in the CR group (E). Black arrows indicate infiltrating inflammatory cells; white arrow indicates demyelination. Original magnification: A and D, 4×; B and C, 20×; E and F, 16×. Original scale bars, 200 μm (A and D) or 50 μm (B, C, E, and F). (G) Inflammation and (H) demyelination quantified in a blinded manner were reduced significantly in CR (▵) compared with control mice (▪; *, P=0.03, by Mann-Whitney test; **, P<0.001, by Mann-Whitney test). Horizontal lines indicate median values.
Axonal damage was detected by immunostaining for SMI-32, which is specific for dephosphorylated, neurofilament H in spinal cords from control and CR mice (Fig. 3A–D). The damaged SMI-32-positive axons were counted in random spinal cord sections by an observer blinded to the treatment group. Fewer SMI-32-positive axons were present in the CR group compared with the control group (mean±sd: 244±210 vs. 718±167, respectively; n=5 per group; P=0.005; Fig. 3E).
Fig. 3.

Spinal cord axonal damage was less in CR mice compared with control mice fed ad libitum. In Experiment 3, spinal cords were taken from mice that were terminally perfused on Day 30 p.i. Representative SJL mice for control (A and B) and CR (C and D) groups are shown (clinical scores were 3.5 for the control mouse and 1.5 for CR). SMI-32, specific for dephosphorylated neurofilament H in damaged axons, is stained green. Antibodies to MBP were used to detect myelin (in red). Higher numbers of SMI-32-positive axons were detected in the spinal cord white matter of controls (A and B) compared with CR (C and D). White arrows indicate SMI-32-positive axons. Original magnification: A, 6.4×; B, 16×; C, 4.8×; and D, 12×. Original scale bars, 200 μm (A and C) or 100 μm (B and D). (E) Axonal damage quantification (n=5 in each group) shows a significant reduction in CR compared with control mice (*, P=0.005, by two-tailed t-test).
Chronic CR does not inhibit T cell priming in the EAE-induction phase
One mechanism through which chronic CR could attenuate EAE is by inhibiting the initial phase of T cell priming against myelin antigens. Splenocytes of immunized control, HF, or CR mice were characterized by flow cytometry at Day 63 p.i. (n=4/group). CD4+ T cells, CD8+ T cells, CD19+ B cells, and CD11b+ monocytes were equally represented in the three treatment groups. Moreover, CD69+CD4+ and CD69+CD8+ T cells, representing recently activated T cells, were not significantly different between groups (Table 2).
TABLE 2.
Phenotypic Characterization of Splenocytes
| CD4+ (%) | CD8+ (%) | CD19+ (%) | CD11b+ (%) | CD4+CD69+ (%) | CD8+CD69+ (%) | |
|---|---|---|---|---|---|---|
| Control | 30.7 ± 5.4 | 16.4 ± 3.3 | 29 ± 7.8 | 11.5 ± 2.4 | 17.2 ± 4 | 13.1 ± 3.6 |
| HF | 33.2 ± 3.4 | 17.8 ± 2.2 | 29.4 ± 6.4 | 6.8 ± 0.3 | 16.9 ± 2.5 | 8.5 ± 1.6 |
| CR | 27.1 ± 2.5 | 16.0 ± 3.3 | 26 ± 6.9 | 13 ± 3.6 | 15.2 ± 2.3 | 8.8 ± 1.6 |
Values are mean ± sem; splenocytes were isolated at Day 63 p.i. (n = 4 per group).
To investigate whether mice subjected to chronic CR were able to fully mount a cellular immune response directed against myelin antigens, proliferation and cytokine production in response to in vitro stimulation with PLP139–151 was assessed for cells isolated 10 days p.i. from draining lymph nodes of SJL controls and mice assigned to a HF or CR diet. No significant differences in antigen-specific proliferation and IFN-γ production were demonstrated amongst the three groups after 72 h of stimulation in vitro (Fig. , 4 A and B). Proliferation and cytokine production in response to PLP139–151 in vitro was also tested in splenocytes isolated from SJL mice 30 days after immunization (Fig. , 4 C and D). IL-12p70, TNF-α, IFN-γ, MCP-1, IL-10, and IL-6 production was assayed in cell culture supernatants. No significant differences in proliferation and cytokine production were detected between spleen cells from CR versus control mice in two experiments. Similarly, antigen-specific lymphocyte proliferation and cytokine production were compared for CR-treated versus control C57BL/6 mice with chronic EAE. As with SJL mice, no differences were detected between control and CR groups (data not shown). The levels of IL-10 in supernatants varied greatly between mice and were not statistically different between groups (Fig. 4D).
Fig. 4.

Chronic CR does not inhibit T cell priming in the EAE-induction phase. In vitro proliferation rate (A) and IFN-γ production (B) in response to PLP139–151 by cells isolated 10 days p.i. from draining lymph nodes (axillary and inguinal) did not show any significant differences amongst SJL controls, mice assigned to a HF or CR (n=2/group; all mice were Clinical Grade 0 except one in the control group that was Grade 2). Proliferation and cytokine production in response to PLP139–151 by splenocytes isolated from SJL mice 30 days p.i. was also tested (n=5/group; control mice were at clinical scores ranging from 2 to 4, two CR mice were at score 0, two were at 2, and one at score 1; C and D). Neither splenocyte proliferation in vitro nor cytokine production showed significant differences between the control and CR groups. Values are mean ± sem. These data are representative of four experiments.
Lymphocytes derived from CR mice transfer EAE to naïve recipients
To determine whether cells from immunized mice in each dietary group were similarly encephalitogenic, spleen cells from mice in the CR group, the HF group, and the control group were removed and activated 48 h in vitro with PLP139–151. Next, 2.5 × 107 cells were transferred i.v. to naïve, syngeneic recipients, who were observed for the development of EAE. Antigen-activated splenocytes from mice in each group were able to transfer EAE to naive recipients in a comparable manner. The recipient of CR cells developed EAE on Day 7 post-transfer (PT), progressing to score 4.0 by Day 9 PT. The two HF cell recipients got sick on Day 8 PT (maximum score 4.0) and 26 PT (maximum score 2.0). Only one of two recipients of control mouse cells developed EAE, which was on Day 9 PT (maximum score 3). Thus, it was concluded that the phase of T cell priming occurred normally in CR mice and that they were fully capable of mounting an immune response against myelin antigen.
Corticosterone levels are increased by CR
SJL female mice in the control and CR groups were bled prior to initiation of the specified diet. These baseline plasma samples were compared with samples obtained at three other time-points: after 4 weeks of the assigned diet (preimmunization), prior to EAE onset following immunization with PLP139–151 (Preclinical Day 10 p.i.), and during clinical EAE (Days 16–19 or 30 p.i.). All blood draws were performed at 10 a.m. to control for circadian variations in corticosterone secretion. Plasma corticosterone levels were not significantly different between the groups at baseline (Fig. 5A). Four weeks of CR significantly elevated corticosterone (mean±sem: 40.7 ng/ml±17.6 in controls vs. 226.9±50.4 in CR; P=0.01), and these high levels were maintained in the CR mice at the later time-points. After immunization, corticosterone levels also rose in the control mice so that no significant differences were noted between groups in the preclinical phase. During clinical EAE, mean corticosterone levels in the CR mice were again significantly higher compared with the control group (76.5 ng/ml±31.7 in controls and 197.4±44.7 in CR; P=0.04; Fig. 5A).
Fig. 5.

CR alters plasma levels of corticosterone, leptin, and adiponectin. SJL mice were bled at different time-points during the experiment: prior to initiation of the specified diet (Baseline), after 4 weeks of the assigned diet (Pre-immunization), prior to EAE onset following immunization with PLP139–151 (Preclinical; Day 10 p.i.), and during clinical EAE (Days 16–19 or 30 p.i.). All blood draws were performed at 10 a.m. to control for circadian variations. Corticosterone (A), leptin (B), and adiponectin (C) plasma levels were not significantly different at baseline. (A) After 4 weeks of CR (Pre-immunization) corticosterone was elevated significantly compared with controls (P=0.01; by t-test with Welch’s correction). During clinical EAE, CR mice also had significantly elevated corticosterone levels over the control group (P=0.04; by t-test; n=6 for baseline and preimmunization; n=3 for preclinical; and n=8 for clinical). (B) At preimmunization, leptin levels were significantly lower in CR mice compared with the control mice (P=0.01 by t-test). During clinical EAE, leptin was significantly lower in CR than controls (P=0.02; by t-test with Welch’s correction). Within the control group and the CR group, leptin levels dropped progressively after immunization for EAE (n=7 for baseline, preimmunization, and preclinical; n=15 for clinical; at the preclinical stage, all mice were at score 0; at clinical stage, all controls and 11/15 CR mice displayed clinical EAE signs). (C) Adiponectin was significantly higher in CR than controls during clinical EAE (P=0.05; by t-test with Welch’s correction; n=6 for baseline and preimmunization; n=3 for preclinical; and n=11 for clinical; at the preclinical stage, all mice were at score 0; at clinical stage, all controls and 4/11 CR mice displayed clinical EAE signs). *, P ≤ 0.02; **, P ≤ 0.05.
Blood leptin levels are decreased by CR
Plasma levels of the adipokine leptin were evaluated at the same time-points as for corticosterone. At baseline, no differences in plasma leptin levels were seen between the control and CR groups (Fig. 5B). After 4 weeks on the assigned diet, CR mice had significantly lower levels of leptin compared with controls (mean±sem: 8 ng/ml±1 for controls vs. 4.9±0.5 for CR; P=0.01). Following immunization, leptin levels decreased significantly in control and CR groups (preimmunization vs. preclinical; P=0.04 by t-test for both groups), concomitant with the initial weight loss preceding EAE onset. From the preclinical to the clinical stage, leptin levels dropped even further in the CR group (P<0.05). During acute clinical EAE, leptin levels were significantly lower in CR mice compared with controls (2.8 ng/ml±0.8 in controls vs. 0.6±0.1 in CR; P=0.02; Fig. 5B).
Blood adiponectin levels are increased by CR
Adiponectin levels were measured in the same control and CR samples used for corticosterone and leptin assays. At baseline, there were no differences in plasma adiponectin between groups. Adiponectin levels increased in CR mice following 1 month of CR, but differences did not reach statistical significance (mean±sem: 7.4 ng/ml±0.4 in controls vs. 9.6±1.2 in CR) at preimmunization. Adiponectin levels in CR mice were significantly higher than controls during clinical EAE (6.2 ng/ml±0.5 for controls vs. 7.5±0.2 for CR; P=0.05; Fig. 5C). In normally fed, control mice, adiponectin levels declined after immunization in the preclinical stage (P=0.02) compared with preimmunization and baseline levels. In contrast, adiponectin levels did not decline significantly in CR mice at the preclinical stage compared with baseline or preimmunization.
IL-6 levels in blood are reduced by CR during EAE
IL-6 increases systemically during acute-phase responses and is a proinflammatory cytokine critical for EAE induction. Thus, we quantitated IL-6 in blood samples obtained from SJL mice at time of sacrifice on Day 30 p.i. (n=5 for CR; n=9 for controls) by ELISA. IL-6 levels were significantly lower in the CR-treated mice compared with control animals fed ad libitum (P=0.02; Fig. 6).
Fig. 6.
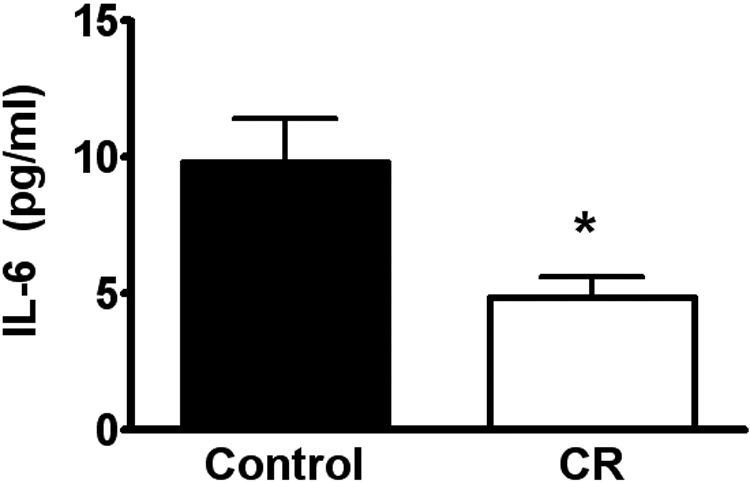
IL-6 plasma levels are reduced by CR during clinical EAE. Blood samples were obtained at time of sacrifice at Day 30 p.i. from mice fed ad libitum or CR SJL mice. Serum IL-6 levels were measured by ELISA (n=5 for CR; n=9 for controls). IL-6 levels were significantly lower in the CR-treated mice compared with controls (*, P=0.02; two-tailed t-test with Welch’s correction).
DISCUSSION
The studies reported here demonstrate that a regimen of chronic CR was effective in ameliorating the clinical course of a relapsing-remitting and a chronic EAE model. The clinical results were confirmed by less-severe CNS pathology in the CR mice. It is well known that CR is associated with a complex spectrum of metabolic, hormonal, and neuroendocrine modifications, many of which are potentially protective during EAE. In these studies, we show that CR was associated with increased plasma levels of corticosterone and adiponectin, along with reduced concentrations of IL-6 and leptin. These and perhaps other changes as a result of CR likely shifted the balance toward an anti-inflammatory milieu able to counteract EAE pathogenic processes.
Esquifino and colleagues [18, 19] have published the only two papers reporting the effects of CR in EAE using the monophasic Lewis rat model. In those studies, calories were restricted by 33% or 66%, and EAE was totally inhibited in the latter group. Rats undergoing the severe regimen of 66% CR had depressed immune function with fewer T cells in the lymphoid organs, impaired proliferation, and cytokine production [19]. In contrast, our data from mice experiencing 40% CR showed amelioration of EAE but without depression of T cell proliferation or cytokine production in response to the myelin antigen. Thus, suppression of the immune response did not explain the CR-mediated inhibition of clinical and histologic autoimmune disease in the present studies. As the ultimate goal of these studies is to determine whether CR might be translated safely to humans with MS, the lack of a demonstrable suppressive effect on the immune system in our studies was encouraging. CR with adequate nutrition has not been associated with depression of the immune function and increased susceptibility to infections. In contrast, in mice and nonhuman primates, long-term CR has been shown to improve T cell function, to delay immune senescence, and to reduce susceptibility to infectious diseases [20, 21].
Our data suggest that CR attenuated EAE by acting on the effector phase of the disease, which includes myelin-specific lymphocyte entry into the CNS and a subsequent cascade of inflammatory events leading to demyelination and axonal damage. One mechanism by which CR may act is via enhanced endogenous corticosteroid production. Corticosterone is the most abundant glucocorticoid in mice and is released cyclically. After 1 month, CR mice in our study had significantly higher corticosterone levels than mice on a normal diet. Glucocorticoids have broad inhibitory effects on the immune system and inflammatory gene expression [22]. Endogenous glucocorticoids greatly affect EAE severity. Activation of the hypothalamic-pituitary-adrenal axis during clinical disease is crucial for the recovery from EAE, as suppression of this response results in lethal disease. Furthermore, treatment with exogenous glucocorticoids blocks EAE [23,24,25]. Administration of synthetic corticosteroids is the standard treatment to treat MS exacerbations in humans [26], but endogenous corticosteroid production, such as that caused by CR [27], is attractive, as it would avoid many side-effects seen with exogenous steroid administration.
A second mechanism by which CR may improve EAE is the combined anti-inflammatory effects of significantly lower IL-6 and higher adiponectin in CR-treated mice. These findings are consistent with prior CR studies in mice [7, 12]. As IL-6 can be produced by adipose tissue [28], the reduction in plasma IL-6 levels in CR animals might be a result of the reduction in body fat associated with CR. IL-6 is a critical cytokine for EAE induction, as EAE cannot be induced in IL-6−/− mice [29]. IL-6, when present together with TGF-β, has been demonstrated to inhibit the generation of T regulatory cells (Tregs) and to induce instead the differentiation of IL-17-producing T (Th17) cells. The latter is now accepted as the main population of pathogenic T cells driving EAE [30].
Our results also showed that plasma levels of adiponectin were elevated significantly in CR animals during clinical EAE. Adiponectin is an important insulin-sensitizing cytokine produced by adipocytes [31]. Blood levels of adiponectin increase with weight loss. Adiponectin also has anti-inflammatory actions, which include reduced production of TNF-α and IL-6 and an induction of IL-10R and IL-1R antagonist [32]. Interestingly, adiponectin-deficient mice show an up-regulation of endothelial adhesion molecules and increased leukocyte-endothelium interactions [12, 33].
An additional potential mechanism for the CR-mediated, beneficial effects on EAE is the suppression of endogenous leptin production. Leptin is an adipocyte-secreted hormone, and its serum concentration is reduced by fasting and increased by overfeeding. In addition to influencing food intake and metabolism, leptin is involved in inflammation and autoimmunity. In our experiments, leptin levels declined progressively after EAE induction in CR and control animals compared with preimmunization, similar to what has been reported by others [34]. Leptin reduction could be related to the weight loss, which precedes EAE onset and could also represent an endogenous regulatory mechanism to control inflammation during EAE. Leptin induces T cell proliferation, promotes Th1 cell differentiation, and induces proinflammatory cytokine production [35]. Leptin-deficient mice are resistant to induction of active and adoptively transferred EAE [34, 36]. It has been reported that lymphocyte infiltrates in the CNS of EAE-affected mice showed in situ production of leptin [34]. We found elevated leptin mRNA in spinal cords of mice with EAE, which was less elevated in CR mice (data not shown). Taken together, these findings suggest an involvement of leptin in CNS inflammation during EAE. In MS patients, it has been reported that leptin is increased in serum and cerebrospinal fluid, with levels that correlate positively with IFN-γ secretion and inversely with the percentage of circulating Tregs [37].
In conclusion, our hypothesis is that CR benefits EAE through multiple metabolic and cytokine/adipokine changes, leading to a reduced inflammatory response. In our studies, CR increased plasma levels of corticosterone and adiponectin, both of which are known to be anti-inflammatory, and decreased plasma levels of leptin and IL-6, which in contrast, are considered proinflammatory. Other possible mechanisms include the CR-associated increase in ghrelin [38], a peptide that has been shown to inhibit the expression of proinflammatory cytokines [39] and some pathologic inflammatory conditions [40].
Environmental factors are believed to play roles in the pathogenesis of MS, which is far more common in the Westernized world, with higher prevalence seen in association with increased intake of saturated fats of animal origin [41]. Although patients and clinicians have speculated that diet may alter the course of MS [42], few randomized, controlled studies of dietary alterations in MS have been published, and none involve CR [43]. An effective and safe dietary intervention would be an attractive treatment for MS patients, given the side-effects associated with most current treatments. CR, with adequate nutrition, can be accomplished safely in humans with proper monitoring and provides multiple beneficial, metabolic effects, including improved insulin sensitivity, lower low-density lipoprotein, cholesterol, and blood pressure, and importantly, a powerful, anti-inflammatory action [44] [45]. The findings reported here, along with data from human CR studies, suggest that CR with adequate nutrition should be tested as a treatment for MS.
Acknowledgments
This work was supported by Clinical Nutrition Research Unit grant DK56351, Washington University School of Medicine, the Frala Osherow Fund for MS Research, and the Barnes-Jewish Hospital Foundation. L. P. is a Fellow of the National MS Society USA (FG-1665-A-1) and was supported in part by Fondazione Italiana Sclerosi Multipla (FISM; 2004/B/4). J. L. S. was a Kirschstein-National Research Service Award Fellow of the NIH during these studies. A. H. C. was supported in part by the Manny and Rosalyn Rosenthal–Dr. John L. Trotter MS Center Chair in Neuroimmunology. The authors thank Dr. John O. Holloszy and Dr. Luigi Fontana for advice, Bob Mikesell, Michael Ramsbottom, Hsiao-Fang Liang, and Dr. Neville Rapp for technical assistance, Trey Coleman and Dr. Clay Semenkovich for assistance with the densitometry studies, and Dr. Kathryn Trinkaus for biostatistics consultation.
References
- Rudick R A, Lee J C, Simon J, Ransohoff R M, Fisher E. Defining interferon β response status in multiple sclerosis patients. Ann Neurol. 2004;56:548–555. doi: 10.1002/ana.20224. [DOI] [PubMed] [Google Scholar]
- Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–1971. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- Benveniste E N. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med. 1997;75:165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- Elhofy A, Kennedy K J, Fife B T, Karpus W J. Regulation of experimental autoimmune encephalomyelitis by chemokines and chemokine receptors. Immunol Res. 2002;25:167–175. doi: 10.1385/IR:25:2:167. [DOI] [PubMed] [Google Scholar]
- Steinman L, Zamvil S S. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. 2006;60:12–21. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]
- Chacon F, Esquifino A I, Perello M, Cardinali D P, Spinedi E, Alvarez M P. 24-Hour changes in ACTH, corticosterone, growth hormone, and leptin levels in young male rats subjected to calorie restriction. Chronobiol Int. 2005;22:253–265. doi: 10.1081/cbi-200053522. [DOI] [PubMed] [Google Scholar]
- Spaulding C C, Walford R L, Effros R B. Calorie restriction inhibits the age-related dysregulation of the cytokines TNF-α and IL-6 in C3B10RF1 mice. Mech Ageing Dev. 1997;93:87–94. doi: 10.1016/s0047-6374(96)01824-6. [DOI] [PubMed] [Google Scholar]
- Guarente L, Picard F. Calorie restriction—the SIR2 connection. Cell. 2005;120:473–482. doi: 10.1016/j.cell.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ware M, Mattson M P. Reversal of behavioral and metabolic abnormalities, and insulin resistance syndrome, by dietary restriction in mice deficient in brain-derived neurotrophic factor. Endocrinology. 2003;144:2446–2453. doi: 10.1210/en.2002-0113. [DOI] [PubMed] [Google Scholar]
- Maswood N, Young J, Tilmont E, Zhang Z, Gash D M, Gerhardt G A, Grondin R, Roth G S, Mattison J, Lane M A, Carson R E, Cohen R M, Mouton P R, Quigley C, Mattson M P, Ingram D K. Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson’s disease. Proc Natl Acad Sci USA. 2004;101:18171–18176. doi: 10.1073/pnas.0405831102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederich R C, Lollmann B, Hamann A, Napolitano-Rosen A, Kahn B B, Lowell B B, Flier J S. Expression of ob mRNA and its encoded protein in rodents. Impact of nutrition and obesity. J Clin Invest. 1995;96:1658–1663. doi: 10.1172/JCI118206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Miura J, Lu L X, Bernier M, DeCabo R, Lane M A, Roth G S, Ingram D K. Circulating adiponectin levels increase in rats on caloric restriction: the potential for insulin sensitization. Exp Gerontol. 2004;39:1049–1059. doi: 10.1016/j.exger.2004.03.024. [DOI] [PubMed] [Google Scholar]
- Matsuzaki J, Kuwamura M, Yamaji R, Inui H, Nakano Y. Inflammatory responses to lipopolysaccharide are suppressed in 40% energy-restricted mice. J Nutr. 2001;131:2139–2144. doi: 10.1093/jn/131.8.2139. [DOI] [PubMed] [Google Scholar]
- Kubo C, Gajar A, Johnson B C, Good R A. The effects of dietary restriction on immune function and development of autoimmune disease in BXSB mice. Proc Natl Acad Sci USA. 1992;89:3145–3149. doi: 10.1073/pnas.89.7.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthukumar A, Sun D, Zaman K, Barnes J L, Haile D, Fernandes G. Age associated alterations in costimulatory and adhesion molecule expression in lupus-prone mice are attenuated by food restriction with n-6 and n-3 fatty acids. J Clin Immunol. 2004;24:471–480. doi: 10.1023/B:JOCI.0000040918.92219.d1. [DOI] [PubMed] [Google Scholar]
- Cross A H, Misko T P, Lin R F, Hickey W F, Trotter J L, Tilton R G. Aminoguanidine, an inhibitor of inducible nitric oxide synthase, ameliorates experimental autoimmune encephalomyelitis in SJL mice. J Clin Invest. 1994;93:2684–2690. doi: 10.1172/JCI117282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begolka W S, Vanderlugt C L, Rahbe S M, Miller S D. Differential expression of inflammatory cytokines parallels progression of central nervous system pathology in two clinically distinct models of multiple sclerosis. J Immunol. 1998;161:4437–4446. [PubMed] [Google Scholar]
- Esquifino A I, Cano P, Jimenez V, Cutrera R A, Cardinali D P. Experimental allergic encephalomyelitis in male Lewis rats subjected to calorie restriction. J Physiol Biochem. 2004;60:245–252. doi: 10.1007/BF03167069. [DOI] [PubMed] [Google Scholar]
- Esquifino A I, Cano P, Jimenez-Ortega V, Fernandez-Mateos M P, Cardinali D P. Immune response after experimental allergic encephalomyelitis in rats subjected to calorie restriction. J Neuroinflammation. 2007;4:6. doi: 10.1186/1742-2094-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaoudi I, Warner J, Fischer M, Park B, Hill B, Mattison J, Lane M A, Roth G S, Ingram D K, Picker L J, Douek D C, Mori M, Nikolich-Zugich J. Delay of T cell senescence by caloric restriction in aged long-lived nonhuman primates. Proc Natl Acad Sci USA. 2006;103:19448–19453. doi: 10.1073/pnas.0606661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolich-Zugich J, Messaoudi I. Mice and flies and monkeys too: caloric restriction rejuvenates the aging immune system of non-human primates. Exp Gerontol. 2005;40:884–893. doi: 10.1016/j.exger.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Adcock I M, Ito K, Barnes P J. Glucocorticoids: effects on gene transcription. Proc Am Thorac Soc. 2004;1:247–254. doi: 10.1513/pats.200402-001MS. [DOI] [PubMed] [Google Scholar]
- Mason D. Genetic variation in the stress response: susceptibility to experimental allergic encephalomyelitis and implications for human inflammatory disease. Immunol Today. 1991;12:57–60. doi: 10.1016/0167-5699(91)90158-P. [DOI] [PubMed] [Google Scholar]
- Bolton C, O'Neill J K, Allen S J, Baker D. Regulation of chronic relapsing experimental allergic encephalomyelitis by endogenous and exogenous glucocorticoids. Int Arch Allergy Immunol. 1997;114:74–80. doi: 10.1159/000237646. [DOI] [PubMed] [Google Scholar]
- Dowdell K C, Gienapp I E, Stuckman S, Wardrop R M, Whitacre C C. Neuroendocrine modulation of chronic relapsing experimental autoimmune encephalomyelitis: a critical role for the hypothalamic-pituitary-adrenal axis. J Neuroimmunol. 1999;100:243–251. doi: 10.1016/s0165-5728(99)00211-8. [DOI] [PubMed] [Google Scholar]
- Barnes D, Hughes R A, Morris R W, Wade-Jones O, Brown P, Britton T, Francis D A, Perkin G D, Rudge P, Swash M, Katifi H, Farmer S, Frankel J. Randomized trial of oral and intravenous methylprednisolone in acute relapses of multiple sclerosis. Lancet. 1997;349:902–906. doi: 10.1016/s0140-6736(96)06453-7. [DOI] [PubMed] [Google Scholar]
- Sabatino F, Masoro E J, McMahan C A, Kuhn R W. Assessment of the role of the glucocorticoid system in aging processes and in the action of food restriction. J Gerontol. 1991;46:B171–B179. doi: 10.1093/geronj/46.5.b171. [DOI] [PubMed] [Google Scholar]
- Hoene M, Weigert C. The role of interleukin-6 in insulin resistance, body fat distribution and energy balance. Obes Rev. 2008;9:20–29. doi: 10.1111/j.1467-789X.2007.00410.x. [DOI] [PubMed] [Google Scholar]
- Okuda Y, Sakoda S, Bernard C C, Fujimura H, Saeki Y, Kishimoto T, Yanagihara T. IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int Immunol. 1998;10:703–708. doi: 10.1093/intimm/10.5.703. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom T B, Oukka M, Weiner H L, Kuchroo V K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman M L, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol. 2005;115:911–919. doi: 10.1016/j.jaci.2005.02.023. [DOI] [PubMed] [Google Scholar]
- Ouedraogo R, Gong Y, Berzins B, Wu X, Mahadev K, Hough K, Chan L, Goldstein B J, Scalia R. Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. J Clin Invest. 2007;117:1718–1726. doi: 10.1172/JCI29623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna V, Di Giacomo A, La Cava A, Lechler R I, Fontana S, Zappacosta S, Matarese G. Leptin surge precedes onset of autoimmune encephalomyelitis and correlates with development of pathogenic T cell responses. J Clin Invest. 2003;111:241–250. doi: 10.1172/JCI16721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matarese G, Moschos S, Mantzoros C S. Leptin in immunology. J Immunol. 2005;174:3137–3142. doi: 10.4049/jimmunol.174.6.3137. [DOI] [PubMed] [Google Scholar]
- Matarese G, Di Giacomo A, Sanna V, Lord G M, Howard J K, Di Tuoro A, Bloom S R, Lechler R I, Zappacosta S, Fontana S. Requirement for leptin in the induction and progression of autoimmune encephalomyelitis. J Immunol. 2001;166:5909–5916. doi: 10.4049/jimmunol.166.10.5909. [DOI] [PubMed] [Google Scholar]
- Matarese G, Carrieri P B, La Cava A, Perna F, Sanna V, De Rosa V, Aufiero D, Fontana S, Zappacosta S. Leptin increase in multiple sclerosis associates with reduced number of CD4(+)CD25+ regulatory T cells. Proc Natl Acad Sci USA. 2005;102:5150–5155. doi: 10.1073/pnas.0408995102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Youm Y H, Nakata C, Dixit V D. Chronic caloric restriction induces forestomach hypertrophy with enhanced ghrelin levels during aging. Peptides. 2007;28:1931–1936. doi: 10.1016/j.peptides.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit V D, Schaffer E M, Pyle R S, Collins G D, Sakthivel S K, Palaniappan R, Lillard J W, Jr, Taub D D. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J Clin Invest. 2004;114:57–66. doi: 10.1172/JCI21134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rey E, Chorny A, Delgado M. Therapeutic action of ghrelin in a mouse model of colitis. Gastroenterology. 2006;130:1707–1720. doi: 10.1053/j.gastro.2006.01.041. [DOI] [PubMed] [Google Scholar]
- Alter M, Yamoor M, Harshe M. Multiple sclerosis and nutrition. Arch Neurol. 1974;31:267–272. doi: 10.1001/archneur.1974.00490400081010. [DOI] [PubMed] [Google Scholar]
- Schwarz S, Leweling H. Multiple sclerosis and nutrition. Mult Scler. 2005;11:24–32. doi: 10.1191/1352458505ms1119oa. [DOI] [PubMed] [Google Scholar]
- 43.Farinotti, M., Simi, S., Di Pietrantonj, C., McDowell, N., Brait, L., Lupo, D., Filippini, G. (2007) Dietary interventions for multiple sclerosis. Cochrane Database Syst. Rev.Jan. 24, CD004192. [DOI] [PubMed] [Google Scholar]
- Fontana L, Klein S. Aging, adiposity, and calorie restriction. JAMA. 2007;297:986–994. doi: 10.1001/jama.297.9.986. [DOI] [PubMed] [Google Scholar]
- Meyer T E, Kovacs S J, Ehsani A A, Klein S, Holloszy J O, Fontana L. Long-term caloric restriction ameliorates the decline in diastolic function in humans. J Am Coll Cardiol. 2006;47:398–402. doi: 10.1016/j.jacc.2005.08.069. [DOI] [PubMed] [Google Scholar]