

Abstract
Amyotrophic lateral sclerosis (ALS) is a spontaneous, relentlessly progressive motor neuron disease, usually resulting in death from respiratory failure within 3 years. Variation in the genes SOD1 and TARDBP accounts for a small percentage of cases, and other genes have shown association in both candidate gene and genome-wide studies, but the genetic causes remain largely unknown. We have performed two independent parallel studies, both implicating the RNA polymerase II component, ELP3, in axonal biology and neuronal degeneration. In the first, an association study of 1884 microsatellite markers, allelic variants of ELP3 were associated with ALS in three human populations comprising 1483 people (P = 1.96 × 10−9). In the second, an independent mutagenesis screen in Drosophila for genes important in neuronal communication and survival identified two different loss of function mutations, both in ELP3 (R475K and R456K). Furthermore, knock down of ELP3 protein levels using antisense morpholinos in zebrafish embryos resulted in dose-dependent motor axonal abnormalities [Pearson correlation: −0.49, P = 1.83 × 10−12 (start codon morpholino) and −0.46, P = 4.05 × 10−9 (splice-site morpholino), and in humans, risk-associated ELP3 genotypes correlated with reduced brain ELP3 expression (P = 0.01). These findings add to the growing body of evidence implicating the RNA processing pathway in neurodegeneration and suggest a critical role for ELP3 in neuron biology and of ELP3 variants in ALS.
INTRODUCTION
Spontaneous, relentlessly progressive motor neuron degeneration occurs in several diseases of humans. The commonest adult onset human motor neuron disease is amyotrophic lateral sclerosis (ALS), which usually results in death from respiratory muscle weakness within 3 years. In 5–10% of cases there is a family history of ALS and about a quarter of these are attributable to mutation in the superoxide dismutase (SOD1) or TAR-DNA binding protein 43 (TARDBP) genes. The genetic contribution to sporadic ALS is largely unknown, but candidate gene association studies have revealed SOD1 mutations in 1–7% of cases and TARDBP mutations in 0.5–5% (1–3).
Single-nucleotide polymorphism (SNP) based genome-wide association studies have been inconclusive. One small study has not shown a significant association (4). A larger study using a DNA pooling approach to prioritize SNPs has identified ALS-associated variants in an uncharacterized gene, FLJ10986 (5). By combining with other data sets, a Dutch study has identified ALS-associated variants in the genes ITPR2 and DPP6 (6,7). The combination of some of the Dutch study samples with further samples from an Irish population detected DPP6 as the most strongly associated variant but this did not reach statistical significance (8). Nevertheless, the appeal of genome-wide association studies is that any genetic association will provide an insight that would not be possible with a candidate gene approach. Although SNP-based studies are simple to perform and have excellent genomic coverage, microsatellite-based studies provide an alternative view of the genome and may be more likely to detect rare variants (9). Similarly, mutagenesis in small organisms followed by screening for neurodegeneration phenotypes may reveal genes critical for motor neuron function that are not found by other methods. We therefore performed two independent studies to identify genes important in neuronal function or survival: the first, a microsatellite-based genetic association study of ALS in humans and the second, a mutagenesis screen in Drosophila. In both cases, variants of the same gene, elongator protein 3 (ELP3) were identified as critical for axonal biology, and this was supported by further functional studies.
RESULTS
Human association study
Demographic features of the study populations are shown in Supplementary Material, Table S1. We used a multistage design to examine a population from the UK with 1884 microsatellite markers, which were then ranked by strength of association (Supplementary Material, Figs S1–S4) and followed-up by replication studies in two other populations from the USA and Belgium and fine mapping using SNPs. Four markers were followed-up, two on chromosome 3 and two on chromosome 8, each pair about 1 Mb apart. We used permutation to correct for the multiple testing inherent in examination of multiple microsatellite alleles as instituted in the program CLUMP. Alleles of D8S1820, a 15-allele marker, were associated with ALS (P = 1.96 × 10−9) (Supplementary Material, Table S2). At the end of the permutation procedure, CLUMP had grouped the alleles of D8S1820 into two groups: alleles 1, 6, 10, 14 and 15 (hereafter called the protection-associated alleles), and the remaining alleles (hereafter called the risk-associated alleles) (Supplementary Material, Table S3). To better understand the risk associated with the two allelic groups, we performed a 2 × 2 χ2 test for independence of the allelic groups with ALS. We again confirmed a highly significant association in an overall analysis stratified for the populations, with an odds ratio of 0.46, 95% CI 0.35–0.60, P = 8.94 × 10−9 (Table 1). Each study population also showed the association with a similar odds ratio (Breslow–Day test for homogeneity P = 0.42). Bioinformatics analysis with the programs ePCR and BLAT confirmed a unique location of D8S1820 in intron 10 of the ELP3 gene.
Table 1.
Alleles of marker D8S1820 as grouped by CLUMP, analyzed by χ2 test
| Allelic ratios (cases, controls; protection-associated: risk-associated) | Odds ratio (95% CI) | P-value | n (cases, controls) | |
|---|---|---|---|---|
| UK | 36:538, 64:516 | 0.54 (0.35–0.83) | 0.004 | 287, 290 |
| USA | 45:563, 60:344 | 0.46 (0.30–0.69) | 1.41 × 10−4 | 304, 202 |
| Belgium | 12:368, 39:381 | 0.32 (0.16–0.62) | 3.96 × 10−4 | 190, 210 |
| Total: stratified test | As for each country | 0.46 (0.35–0.60) | 8.95 × 10−9 | 781, 702 |
| Total: unstratified test | 93:1469, 163:1241 | 0.48 (0.37–0.63) | 4.34 × 10−8 | 781, 702 |
Counts and association results for alleles of marker D8S1820 analyzed as a bi-allelic system of protection-associated alleles against risk-associated alleles. Alleles were classified after permutation testing by CLUMP.
The extent of genomic coverage by our microsatellite selection is difficult to estimate. The markers had a mean spacing of 1.5 Mb and a median spacing of 0.67 Mb covering all autosomes and the X chromosome, 46% targeted to candidate regions and 54% targeted to gene-dense regions, but we expect that there will be large genomic regions not included in this analysis.
The relationship of linkage disequilibrium between SNPs and microsatellite alleles is complex and often weak for some alleles, but may extend long distances (9). Consequently, translating a microsatellite allelic association into an SNP or haplotype association can be difficult. To examine patterns of linkage disequilibrium in the region as a prelude to fine mapping, we analyzed D8S1820 alleles in the Utah CEPH (CEU) HapMap samples (http://www.hapmap.org). As expected, we observed a complex pattern of linkage disequilibrium with neighbouring SNPs (data not shown).
To search for a functional variant, we selected 61 tag-SNPs in and around the ELP3 gene for fine-mapping studies in the study populations (Supplementary Material, Table S4). We observed the same linkage disequilibrium pattern in each population (Fig. 1, Supplementary Material, Figs S5 and S6). One SNP, rs13268953, showed weak association with ALS (stratified P = 0.029, unstratified P = 0.030), but this did not survive Bonferroni correction for multiple testing, nor was it significantly associated with ALS in the individual study populations. No other SNPs showed association.
Figure 1.

A scatterplot of the linkage disequilibrium between common alleles of microsatellite D8S1820 and neighboring SNPs in controls of the study population. The position of the ELP3 gene is shown as a red bar below the X-axis. Marker D8S1820 is marked by the central vertical arrow and rs12682496 by the right vertical arrow. Because of the multi-allelic nature of microsatellite markers it is difficult to show patterns of LD using the conventional triangle plots used for SNPs (but see Supplementary Material, Figs S5 and S6). This graph plots the pairwise LD between each SNP and the common microsatellite alleles, with the strength of LD represented by the P-value for a χ2 test of association. As can be seen, the pattern of LD with neighboring SNPs is complex, the strength of LD varies for different alleles, and LD may extend long distances.
To search for a haplotypic association, we first simplified the microsatellite information by examination of the individual allelic associations. This showed that the signal came most strongly from allele 6, which was under-represented in cases compared with controls (case frequency 0.027, controls 0.057). We then sought a two-marker haplotype with allele 6, testing each SNP in turn using the omnibus haplotype test implemented in PLINK (10). The haplotype with marker rs12682496 gave an omnibus P-value of 2.31 × 10−6 (corrected for 61 SNPs, P = 1.41 × 10−4), with the allele 6-rs12682496 C haplotype being strongly associated with ALS (P = 1.05 × 10−6). This haplotype was also associated with ALS in each of the study populations.
Mutagenesis screen in Drosophila
In parallel, and independently of the genetic association study, we performed a forward ethyl methanesulphonate (EMS)-based mutagenesis screen in Drosophila using eyFLP technology (11) to discover genes involved in synaptic transmission and neuronal survival or development (12). We retained 138 mutants with defective ‘on’ and ‘off’ transients as candidate mutants (12). Mutations identified using this screening strategy usually affect well-characterized processes involved in presynaptic function, including exocytosis, endocytosis and neuronal survival (13,14).
One of the complementation groups encompassed two lethal alleles showing striking electroretinogram (ERG) phenotypes. The photoreceptor layer depolarized less in response to a one second light pulse and the on and off transients were also dramatically reduced when compared to controls, suggesting abnormal neuronal communication (Fig. 2A and B).
Figure 2.

Drosophila ELP3 mutants identified in a screen for defects in neuronal communication. (A and B) ERG recordings and quantification of ‘on’ and ‘off’ (arrowheads in A) amplitudes of control and ELP3 mutant eyes. Black: on- and grey off-transients. Controls n = 48, elp31 n = 59, elp32 n = 57. Error bars indicate standard error of the mean. t-test: Control-elp31, on: P = 1.86 × 10−16, off: P = 1.78 × 10−14; control-elp32, on: P = 1.58 × 10−19, off: P = 7.90 × 10−10. (C) Confocal microscopy showing the photoreceptor axon projections in the medulla labeled with anti-chaoptin. In the mutants photoreceptors arrive and synapse in the medulla, but the synapses are not properly organized in rows. Scale bars 20 µm. (D) Mapping of ELP3 mutations. KG P-elements used for fine-mapping located in the 24C5-8 cytological region. Numbers under markers are tested/recombinant flies. (E) Recombination distances (cM) for the three P-elements closest to the mutant phenotype (lethality). The position of ELP3 in relation to KG05280 is indicated with a red asterisk and the 20 kb sequenced region as a grey bar. (F) ELP3 mutations (arrows). No mutations were found in surrounding genes. (G) Schematic representation of fly ELP3 (552aa) and mutations elp31 and elp32.
To analyze the integrity of the homozygous mutant photoreceptors, we labeled them with anti-chaoptin (mAb 24B10), an antibody that labels the photoreceptor membrane. In both mutants the R7 and R8 photoreceptors projected into the medulla, but the projection pattern was disrupted and the array of photoreceptor terminals was disordered (Fig. 2C). These data suggest that the defects in neuronal communication we identified might arise, at least in part, from altered axonal targeting and synaptic development.
We mapped the mutants to cytological interval 24F2 of the Drosophila genome and found close linkage to P-element KG05280. We confirmed this with a complementation test using a cytologically mapped deficiency uncovering several genes in this region (Fig. 2D) (15). Sequencing of 20 kb genomic DNA surrounding KG05280 (Fig. 2E) revealed mutations R475K (elp31) and R456K (elp32) in Drosophila ELP3 (Fig. 2F). This gene is highly homologous to human ELP3, being 82% identical and 91% similar. Both mutated arginines are conserved across species and are part of the signature sequence of the GCN5-related acetyl transferase (GNAT) domain of the enzyme (Fig. 2G). In a second complementation test, P{GawB}-NP0434, a lethal transposon molecularly mapped to the ELP3 gene, failed to complement elp31 and elp32, further confirming that the lethality of the mutants and the lesions in ELP3 mapped to the same locus. Taken together, these data independently identify ELP3 as a critical regulator of axon targeting and synaptic communication, and suggest the GNAT domain plays a significant role in this process. Furthermore, the data suggest that it is a loss of function of ELP3 that leads to neuronal defects.
ELP3 expression in humans and transgenic mice
All genes so far known to play a causal role in ALS are expressed in motor neurons (16). To explore the functional role of ELP3, we examined lumbar spinal cord tissue of people who died of non-neurological conditions by staining with rabbit anti-ELP3 antibody. We observed ELP3 protein in human spinal motor neurons Supplementary Material, Fig. S7).
Since SOD1 mutations remain the most common genetic cause of ALS, we explored the possible changes in ELP3 expression in SOD1-mediated ALS. Western blotting revealed robust expression of ELP3 in the ventral and dorsal part of the spinal cord both in SOD1WT and SOD1G93A transgenic ALS mice (Supplementary Material, Fig. S8). There was no difference in expression between the two spinal cord regions, and no variation of expression in the ventral cord during disease progression. Immunostaining of the ventral horn showed clear ELP3 expression in motor neurons (Supplementary Material, Fig. S9).
To elucidate whether a loss of ELP3 function was indeed the mechanism in the ALS population, we determined levels of ELP3 expression in carriers of the ELP3 genetic variants. In human cerebellar tissue from control individuals, 15% more ELP3 protein was observed in those carrying protection-associated alleles than those carrying risk-associated alleles only (n = 18, t-test P = 0.01, Fig. 3A and B). In motor cortex from individuals with ALS, 59% more ELP3 protein was observed in those carrying protection-associated alleles than those carrying risk-associated alleles only (n = 17, t-test P = 0.01, Fig. 3C and D).
Figure 3.

Western blot analysis of ELP3 protein expression in human control cerebellar tissue and ALS motor cortex. (A) Western blot analysis of cerebellar tissue samples from controls carrying risk-associated alleles (Risk) or at least one protection-associated allele (Protection). (B) Expression of ELP3 protein as a ratio to β-actin in cerebellar tissue from controls carrying risk-associated alleles (triangles, n = 13) or at least one protection-associated allele (diamonds, n = 5). (C) Western blot analysis of ALS motor cortex tissue samples carrying risk-associated alleles (Risk) or at least one protection-associated allele (Protection). (D) Expression of ELP3 protein as a ratio to β-actin in ALS motor cortex samples carrying risk-associated alleles (triangles, n = 9) or at least one protection-associated allele (diamonds, n = 8).
Knockdown of ELP3 in zebrafish
Based on these findings, we next investigated the significance of a loss of ELP3 function in a zebrafish model. The protein sequence of ELP3 is highly conserved in zebrafish, with 91.3% identity and 97.3% similarity to human ELP3. Western blot analysis of zebrafish embryos 30 h post-fertilization injected with an ELP3-specific RNA-blocking ATG morpholino (ATG-MO), showed a dose-dependent lowering of ELP3 protein levels (Fig. 4A). In line with axonal targeting defects in ELP3 mutant Drosophila photoreceptors, injection of zebrafish embryos with 6 ng of ATG-MO demonstrated abnormal branching in motor axons in 67.5% of the cases, compared with 17.8% of embryos injected with 6 ng of control morpholino (Ctr-MO, Fig. 4B and C). Similar findings were obtained on injection of 9 ng of an ELP3-specific splice-site-targeting morpholino (Sp-MO), which also induced dose-dependent abnormal branching in 63.6% of embryos (Table 2). Moreover, at the highest dose tested the axonal length of ventral motor neuron axons was significantly decreased by 14.7% (ATG-MO) and 18.7% (Sp-MO) (Fig. 4D). This effect too was dose-dependent for both morpholinos [Pearson correlation: −0.49, P = 1.83 × 10−12 (ATG-MO) and −0.46, P = 4.05 × 10−9 (Sp-MO)]. Embryos injected with Ctr-MO showed no defects in these parameters compared with buffer-injected embryos.
Figure 4.

Morpholino-induced knockdown of ELP3 affects motor neuron axonal branching and length. (A) Western blot of ELP3 following treatment with Ctr-MO and ATG-MO. Maximal ELP3 knockdown was 44%. (B) ELP3 knockdown by Sp-MO and ATG-MO resulted in increased branching of motor axons (right) compared with control (left). (C) There was a dose-dependent decrease in axonal length of motor neurons for both Sp-MO and ATG-MO. Results show SEM (*P < 0.01; **P < 1.0 × 10−7). P-values at each dose of ATG-MO compared with 6.0 ng Ctr-MO were 3.0 ng: P = 0.0024; 4.5 ng: P = 1.19 × 10−8; 6.0 ng: P = 7.68 × 10−11. P-values at each dose of Sp-MO compared with 6.0 ng Ctr-MO were 6.0 ng: P = 0.0084; 7.5 ng: P = 0.0039; 9.0 ng, P = 6.94 × 10−9. Scale bar 50 µm.
Table 2.
Knockdown of ELP3 in Zebrafish
| Embryos showing >2/20 branching axons (%) a | Odds ratio (CI) compared with Ctr-MO | χ2P-value | Branching axons per embryo (%) b | |
|---|---|---|---|---|
| Ctr-MO (6.0 ng), n = 45 | 17.8 | 4.1 | ||
| ATG-MO (3.0 ng), n = 35 | 48.6 | 4.4 (1.6–12.0) | 0.003 | 8.7 |
| ATG-MO (4.5 ng), n = 61 | 50.8 | 4.8 (1.9–11.9) | 4.89 × 10−4 | 9.4 |
| ATG-MO (6.0 ng), n = 40 | 67.5 | 9.6 (3.5–26.4) | 3.34 × 10−6 | 10.1 |
| Sp-MO (6.0 ng), n = 38 | 28.9 | 1.9 (0.7–5.3) | 0.23 | 5.4 |
| Sp-MO (7.5 ng), n = 37 | 37.8 | 2.8 (1.0–7.8) | 0.04 | 7.3 |
| Sp-MO (9.0 ng), n = 33 | 63.6 | 8.1 (2.9–23.0) | 3.47 × 10−5 | 12.0 |
Table showing knockdown of ELP3 using two different antisense morpholinos results in dose-dependent abnormal branching of primary motor neurons.
aFor ATG-MO, Spearman correlation: 0.33 (P = 5.28 × 10−6), Kruskal–Wallis test P = 5.33 × 10−5; for Sp-MO, Spearman correlation: 0.33 (P = 2.57 × 10−5), Kruskal–Wallis test P = 3.80 × 10−4.
bFor ATG-MO, Spearman correlation: 0.30 (P = 3.21 × 10−5), Kruskal–Wallis test P = 1.35 × 10−4; for Sp-MO Spearman correlation: 0.38 (P = 1.36 × 10−6), Kruskal–Wallis test P = 3.91 × 10−5.
DISCUSSION
This study provides four lines of evidence implicating the RNA polymerase II component ELP3 as critically important to the axonal biology of neurons and supporting the initial observation of involvement in human motor neuron disease. First, in an association study of 1483 individuals, ELP3 was associated with human motor neuron degeneration in the form of ALS in three different populations. Secondly, an independent mutagenesis screen in Drosophila for defects in neuronal communication and survival identified two different loss of function ELP3 mutations that each conferred abnormal photoreceptor axonal targeting and synaptic development, possibly signifying neurodegeneration. Thirdly, knockdown of ELP3 in zebrafish using antisense morpholino technology resulted in a dose-dependent shortening and abnormal branching of motor neurons with no concomitant morphological abnormality. Finally, risk-associated ELP3 alleles were associated with lower brain ELP3 expression in humans. These findings strongly implicate ELP3 in axonal biology and as a gene conferring risk of neuronal degeneration.
The published SNP-based genome-wide association studies in ALS have not detected associated ELP3 variants (4–6), but the protection-associated ELP3 microsatellite variants have a total frequency of ∼11%, so approaches using tag-SNPs might not detect them. Although we too did not see any single SNP associations using a dense set of SNPs in and around ELP3, we did identify a haplotype between microsatellite allele 6 and the C allele of rs12682496, suggesting either that an untyped causal variant lies on this haplotype or the haplotype itself is functional. Although in general microsatellites are not thought to be functional, differences in gene expression may be conferred by polymorphic microsatellites in regulatory regions (17–19) or in coding sequences (20). Consistent with an important genomic function, the D8S1820 microsatellite repeat is conserved within ELP3 in chimpanzees and rhesus monkeys, which suggests that it predates the common ancestor of apes and rhesus monkeys and is therefore at least 25 million years old.
The ELP3 protein is part of the RNA polymerase II complex and is involved in RNA processing (Supplementary Material, Table S5) (21). It contains an Fe4S4 cluster and is involved in histone acetylation (22), RNA elongation (21), modification of tRNA wobble nucleosides (23) and an unknown catalytic function related to free radical reactions. Alteration in RNA processing is an element in the pathophysiology of several motor neuron disorders and neurodegenerative diseases, including ALS (2,24,25), hereditary motor neuronopathies 5 [MIM600794] and 6 [MIM604320], Charcot–Marie–Tooth disease type 2D [MIM601472], spinal muscular atrophy [MIM253300], familial dysautonomia [MIM223900] (26) and spinocerebellar ataxia 7 [MIM164500] (27) (Supplementary Material, Table S5). In addition, trinucleotide repeat neurodegenerative diseases have been proposed to be the result of a disruption of nuclear organization that prevents proper RNA processing.
A possible explanation for ELP3 involvement in motor neuron degeneration comes from its effect on transcription through histone acetylation. Heat shock proteins (HSPs) are molecular chaperones whose expression is increased in response to cellular stress. Motor neurons have a high threshold for activating HSPs, making them particularly vulnerable to stressors, including mutant SOD1 (28). ELP3 directly regulates HSP70 expression by acetylation of histones H3 and H4 (29), and therefore one possible explanation for the association of high-expressing ELP3 alleles with protection from motor neuron degeneration in humans is the ability to increase the transcription of HSP70. Indeed, intraperitoneal injection of HSP70 prolonged the lifespan of G93A SOD1 transgenic mice (30).
The association of ELP3 variants with motor neuron degeneration and axonal biology in general increases the evidence that the RNA processing pathway is of particular importance to neurons, and provides a potential therapeutic target for treatment of ALS.
MATERIALS AND METHODS
Study patients
Three geographically distinct populations were studied, from the UK, Belgium and the US (Supplementary Material, Table S1). All individuals were of European ancestry. Individuals attending specialist ALS clinics in each participating center were invited to participate. The diagnosis of ALS was made according to the El Escorial criteria after full investigation to exclude other causes. Patients with familial ALS were excluded but samples were not routinely screened for SOD1 mutations. Controls were unrelated individuals travelling with the patient. In the UK samples, 31% were blood donors from the same geographical region. The study was ethically approved by the institutional review board of each participating institution.
Genetic association methods
We used a dual strategy to select microsatellite markers. Using the program MaGIC (31) we generated one set of markers to target genes and regions from candidate pathways based on existing hypotheses of ALS causation and a second set to target gene-dense regions throughout the genome. We used an initial DNA pooling strategy in the UK samples to prioritize the microsatellite markers for further study by conventional genotyping of individual DNA samples. DNA pools were made by standard methods (32), with each pool comprising non-overlapping samples grouped by phenotype. The smallest pool comprised 57 individuals and the largest 123. Microsatellite genotypes were analyzed by electrophoresis of fluorescently labeled PCR products using an Applied Biosystems ABI 3100 or 3130XL Genetic Analyzer (UK, Belgium) or LiCor Genotyping System (USA). Pool quality was validated by allele frequency estimation of microsatellite alleles and SNPs (33). The results were ranked in order of statistical significance using a measure that included factors for pooling and genotyping artifacts. We gave highest priority for follow-up to adjacent markers in the top 1% of results. Prioritized markers were genotyped in the individual UK samples for confirmation. Confirmed associations were then genotyped in the individual Belgian samples, and replicated results further validated in the US samples, also by typing each DNA individually. Replicated associations were then analyzed further in the study populations by fine-mapping of relevant loci with SNPs.
The D8S1820 dinucleotide repeat alleles were numbered sequentially from the smallest (90 bp=allele 1) to the largest (118 bp=allele 15) for PCR products amplified using the amplimers at http://www.gdb.org. SNPs were analyzed by fluorescent end-point PCR using a TaqMan assay or the Illumina 317K Human Infinium array.
Protein studies of human tissue
Brain samples
Cerebellar tissue samples were obtained from non-Alzheimer disease control brains from the Alzheimer’s Disease Research Center at Massachusetts General Hospital. ALS tissue samples were obtained from the Medical Research Council Brain Bank at the MRC Centre for Neurodegeneration Research, King’s College London. Brain tissue homogenates were prepared using a hand-held homogenizer in RIPA buffer [50 mM Tris–HCl (pH 8.0), 150 mm NaCl, 1% NP-40, 12 mm deoxycolic acid] containing protease inhibitors (Roche Applied Science, Wellesley, MA). Bradford protein concentration assays were carried out using standard protocols. Rabbit anti-β-actin was obtained from Sigma, St Louis, MO. Blots were incubated with horse-radish peroxidase-coupled secondary anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA) or secondary anti-rabbit alkaline phosphatase-coupled antibody (Sigma). Semi-quantitative analysis of ELP3 and β-actin protein levels was carried out by scanning of western blots and densitometry analysis using Scion Image or ImageQuant software. Each sample was tested between two and eight times.
Spinal cord samples
Paraffin-embedded spinal cord tissue of controls were stained with rabbit anti-ELP3 antibody and visualized using 3,3′-diaminobenzidine tetrahydrochloride (Sigma).
Western blotting
Western blotting was performed using primary rabbit polyclonal anti-ELP3 antibody raised against gel-purified GST-(yeast) ELP3, rabbit polyclonal anti-ELP3 antibody raised against specific peptide sequences of human ELP3 (CPGGPDSDFEYSTQSY and HKVRPYQVELVRRDYV) and rabbit anti-β-actin.
Transgenic mice
B6SJLTgN (SOD1WT) and B6SJLTgN (SOD1G93A)1Gur transgenic mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Spinal cord was dissected and homogenized in RIPA buffer. Western blotting was performed using primary antibodies of rabbit polyclonal anti-ELP3 antibody raised against gel-purified GST-ELP3 and mouse monoclonal anti-β-actin antibody (Sigma). Blots were incubated with either secondary anti-rabbit or anti-mouse alkaline phosphatase-coupled antibody (Sigma). Immunohistochemical studies of spinal cord were performed on transgenic SOD1G93A and age-matched transgenic SOD1WT mice. Fresh frozen sections were co-stained with mouse anti-SMI32 (Sternberger Monoclonals) and rabbit anti-ELP3 antibody. The sections were incubated with either Alexa Fluor 488 anti-mouse or AlexaFluor 555 anti-rabbit secondary antibody (Molecular Probes).
Drosophila methods
Mutagenesis and phenotyping
Flies were grown on standard molasses medium. We performed a forward EMS-based mutagenesis screen using the eyFLP technology (11) to discover genes involved in synaptic transmission and neuronal survival or development (12). Mutant flies were tested using a countercurrent phototaxis assay to retain blind flies, and using ERG field potential recordings of the eye during a light flash, to determine synaptic transmission efficiency (12). In a normal fly eye, six of the eight photoreceptors of each ommatidium (R1–R6) project into the first optic ganglion, the lamina, while the other two (R7 and R8) project deeper into the second optic ganglion, the medulla, in a stereotyped manner. Examination of this projection pattern allows the quick assessment of changes in neuronal targeting or gross synaptic structure (34). KG P-elements for mapping were obtained from the Bloomington Stock Center, IN, USA. The ELP3 mutants yw eyFLP GMRLacZ;elp31 or 2 P{y+}FRT40Aiso/CyO,Kr::Gal4 UAS::GFP and controls yw eyFLP GMRLacZ;P{y+}FRT40Aiso were crossed to yw eyFLP GMRLacZ;cl2L P{w+}FRT40A/CyOP{y+} to create flies with homozygous ELP3 mutant or wild-type control eyes. For sequencing, yw eyFLP GMRLacZ;elp31 or 2 P{y+}FRT40Aiso/CyO,Kr::Gal4 UAS::GFP animals were crossed to yw eyFLP GMRLacZ;P{y+}FRT40Aiso. ERGs and immunohistochemistry on adult brains were performed as described (14,20). The lethal insertion P{GawB}NP0434 that fails to complement mutant ELP3 alleles was obtained from the Kyoto Drosophila Genetic Resource Center, Japan. For sequencing, yw eyFLP GMRLacZ; elp31 or 2 P{y+} FRT40Aiso/CyO, Kr::Gal4 UAS::GFP animals were crossed to yw eyFLP GMRLacZ; P{y+} FRT40Aiso and heterozygous DNA was amplified using PCR, sequenced and analysed with Seqman (DNAStar). To identify the gene mutated, we determined the recombination distance between the lethal lesions and molecularly mapped markers on the chromosome (P-elements).
Immunohistochemistry
Anti-chaoptin serum (mAb 24B10) was obtained from the Developmental Studies Hybridoma Bank and used at a concentration of 1:200; secondary Alexa 555 conjugated antibodies were used at a concentration of 1:200 (Invitrogen). Images were captured using a Radiance BioRad confocal microscope and processed with ImageJ and Photoshop 7.0.
Zebrafish methods
Adult zebrafish and embryos were maintained and staged as described (35). The following morpholinos to knock down the expression of zebrafish ELP3 were obtained from Gene Tools (LLC, Corvallis): An ATG-morpholino targeting the ATG start codon: 5′-TGGCTTTCCCATCTTAGACACAATC-3′ (ATG-MO), a splice morpholino targeting a splice site: 5′-CTCAAGTCACCTGACGTATAAAACA-3′ (Sp-MO). A reversed ATG sequence 5′-CTAACACAGATTCTACCCTTTCGGT-3′ (Ctr-MO) was a control. Morpholinos were injected using a FemtoJet® (Eppendorf). The data shown in this manuscript were obtained by injecting, at the highest doses used, a total of 6, 6 and 9 ng for ATG-MO, Ctr-MO and Sp-MO, respectively. Primary antibodies for western blot analyses of whole embryos 30 h post-fertilization were rabbit anti-Elp3 and anti-α-tubulin (Sigma). The blots were incubated with either secondary anti-rabbit or anti-mouse alkaline phosphatase-coupled antibody (Sigma). Axonal defects were evaluated as described (36). Embryos were scored as affected when two or more axons of the 20 analyzed per embryo (ten rostral, ventral motor nerves per hemisegment along the yolk sac extension) showed branching.
All animal studies were approved by the institutions in which they took place.
Statistical methods
Ranking of microsatellite associations for individual genotyping
DNA pool genotypes were analyzed by a modification of the meta-regression procedure in STATA 8.0 (Stata Inc.) (37). The pool estimate of frequency was analyzed for each allele and the best P-value per marker used to rank the results. The regression equation was F = βcC + βa A + βsS + K, where F was the pool allele frequency, C was 1 for case, 0 for control, A was pool mean age of onset or sample acquisition for a control and S was the proportion of males. A test of the hypothesis that βc = 0 yielded the test statistic. Results were ranked by the size of the statistic. The sampling variance was pq/2N. The main source of error in pooled analysis of microsatellite genotypes is the degree of stutter (false bands of lower intensity representing PCR products in which one or more repeats has been lost) and differential amplification (the degree to which smaller alleles are amplified preferentially to larger alleles in PCR reactions). These errors were estimated from data for 400 microsatellite markers typed in 16 individuals, and the expected resulting error for the pooled genotypes was modeled in Mx (38). This was σ2 = 0.2152 p(1−p)1.476 for trinucleotide and tetranucleotide repeats, and σ2 = 0.1712 p(1−p)1.379 for dinucleotide repeats.
Individual genotyping
Microsatellites are multi-allelic. Because the multiple ways different alleles can be combined increases the chances of finding an association, an unbiased approach is required that accounts for the inherent multiple testing. We therefore used the permutation-based χ2 test implemented in the program CLUMP, which was written specifically for the association analysis of multi-allelic markers (39). Stratified analyses of all populations were performed by Fisher’s method for combining k P-values,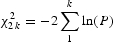 for CLUMP-derived P-values and the Mantel–Haenzsel χ2 test for all other tests. Unstratified analyses were performed by treating the three populations as one. Single SNP and haplotypic association analyses were carried out using PLINK (10) and visualized in Haploview (40). Population structure was analyzed using a χ2 sum statistic for 99 unlinked markers, estimation of Wright’s coefficient FST assuming a single population, and using the program Structure (41).
for CLUMP-derived P-values and the Mantel–Haenzsel χ2 test for all other tests. Unstratified analyses were performed by treating the three populations as one. Single SNP and haplotypic association analyses were carried out using PLINK (10) and visualized in Haploview (40). Population structure was analyzed using a χ2 sum statistic for 99 unlinked markers, estimation of Wright’s coefficient FST assuming a single population, and using the program Structure (41).
Protein expression
Semi-quantitative western blot data were log-transformed to normal. Normality was tested by inspection of standardized residual plots, histograms and the Kolmogorov–Smirnov test (P = 0.15). Equal variances were tested by Levene’s test for homogeneity of variances (P = 0.13). Analysis was by homoscedastic t-test. Two-tailed P-values were reported.
Zebrafish
Zebrafish morpholino experiments were analyzed by χ2 test, Pearson or Spearman correlation and Kruskal–Wallis tests in the program SPSS 13.0 (SPSS Inc., IL, USA).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Medical Research Council (UK) to A.A.-C.; ALS Association to A.A.-C. and R.H.B.; Motor Neurone Disease Association to A.A.-C.; University of Leuven to W.R.; the Belgian government (Interuniversity Attraction Poles Programme 6/43—Belgian State—Belgian Science Policy) to W.R.; VIB to W.R. and P.V.; the Robert Packard Center for ALS Research to W.R.; the Howard Hughes Medical Institute to H.R.H.; The National Institutes of Neurological Disease and Stroke to R.H.B.; the National Institute on Aging to R.H.B.; the Al-Athel ALS Research Foundation to R.H.B.; the ALS Therapy Alliance to R.H.B., the Angel Fund to R.H.B.; Project ALS to R.H.B.; and the Pierre L. de Bourgknecht ALS Research Foundation to R.H.B. For much of this work, A.A.-C. was a Medical Research Council (MRC) Clinician Scientist Fellow and C.L.S. was funded by GKT PhD Studentship. P.S. and H.R.H. were supported by the Howard Hughes Medical Institute. W.R. was supported through the E von Behring Chair for Neuromuscular and Neurodegenerative Disorders. P.V. was supported through a Marie Curie Excellence Grant. Funding to pay the Open Access Charge was provided by the Medical Research Council.
Supplementary Material
ACKNOWLEDGEMENTS
We are indebted to H.J. Bellen, P. Tilkin, RN and A. D’Hondt, RN. We thank Jesper Q. Svejstrup for donation of anti-ELP3 antibody, the Developmental Studies Hybridoma Bank, Iowa for antibodies, the Bloomington Stock Center and Kyoto Genetics Resource Center for flies and Cathryn Lewis and Jean-Marc Gallo for comments.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Simpson C.L., Al-Chalabi A. Amyotrophic lateral sclerosis as a complex genetic disease. Biochim. Biophys. Acta. 2006;1762:973–985. doi: 10.1016/j.bbadis.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Sreedharan J., Blair I.P., Tripathi V.B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J.C., Williams K.L., Buratti E., et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kabashi E., Valdmanis P.N., Dion P., Spiegelman D., McConkey B.J., Velde C.V., Bouchard J.P., Lacomblez L., Pochigaeva K., Salachas F., et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 4.Schymick J.C., Scholz S.W., Fung H.C., Britton A., Arepalli S., Gibbs J.R., Lombardo F., Matarin M., Kasperaviciute D., Hernandez D.G., et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2007;6:322–328. doi: 10.1016/S1474-4422(07)70037-6. [DOI] [PubMed] [Google Scholar]
- 5.Dunckley T., Huentelman M.J., Craig D.W., Pearson J.V., Szelinger S., Joshipura K., Halperin R.F., Stamper C., Jensen K.R., Letizia D., et al. Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N. Engl. J. Med. 2007;357:775–788. doi: 10.1056/NEJMoa070174. [DOI] [PubMed] [Google Scholar]
- 6.van Es M.A., Van Vught P.W., Blauw H.M., Franke L., Saris C.G., Andersen P.M., Van Den Bosch L., de Jong S.W., van ‘t Slot R., Birve A., et al. ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis: a genome-wide association study. Lancet Neurol. 2007;6:869–877. doi: 10.1016/S1474-4422(07)70222-3. [DOI] [PubMed] [Google Scholar]
- 7.van Es M.A., van Vught P.W., Blauw H.M., Franke L., Saris C.G., Van den Bosch L., de Jong S.W., de Jong V., Baas F., van’t Slot R., et al. Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 2008;40:29–31. doi: 10.1038/ng.2007.52. [DOI] [PubMed] [Google Scholar]
- 8.Cronin S., Berger S., Ding J., Schymick J.C., Washecka N., Hernandez D.G., Greenway M.J., Bradley D.G., Traynor B.J., Hardiman O. A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum. Mol. Genet. 2007;17:768–774. doi: 10.1093/hmg/ddm361. [DOI] [PubMed] [Google Scholar]
- 9.Payseur B.A., Place M., Weber J.L. Linkage disequilibrium between STRPs and SNPs across the human genome. Am. J. Hum. Genet. 2008;82:1039–1050. doi: 10.1016/j.ajhg.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newsome T.P., Asling B., Dickson B.J. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 2000;127:851–860. doi: 10.1242/dev.127.4.851. [DOI] [PubMed] [Google Scholar]
- 12.Koh T.W., Verstreken P., Bellen H.J. Dap160/intersectin acts as a stabilizing scaffold required for synaptic development and vesicle endocytosis. Neuron. 2004;43:193–205. doi: 10.1016/j.neuron.2004.06.029. [DOI] [PubMed] [Google Scholar]
- 13.Guo X., Macleod G.T., Wellington A., Hu F., Panchumarthi S., Schoenfield M., Marin L., Charlton M.P., Atwood H.L., Zinsmaier K.E. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 14.Verstreken P., Ly C.V., Venken K.J., Koh T.W., Zhou Y., Bellen H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 15.Zhai R.G., Hiesinger P.R., Koh T.W., Verstreken P., Schulze K.L., Cao Y., Jafar-Nejad H., Norga K.K., Pan H., Bayat V., et al. Mapping Drosophila mutations with molecularly defined P element insertions. Proc. Natl Acad. Sci. USA. 2003;100:10860–10865. doi: 10.1073/pnas.1832753100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasinelli P., Brown R.H. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 17.Fuchs J., Tichopad A., Golub Y., Munz M., Schweitzer K.J., Wolf B., Berg D., Mueller J.C., Gasser T. Genetic variability in the SNCA gene influences{alpha}-synuclein levels in the blood and brain. FASEB J. 2007;22:1327–1334. doi: 10.1096/fj.07-9348com. [DOI] [PubMed] [Google Scholar]
- 18.Hammock E.A., Young L.J. Microsatellite instability generates diversity in brain and sociobehavioral traits. Science. 2005;308:1630–1634. doi: 10.1126/science.1111427. [DOI] [PubMed] [Google Scholar]
- 19.Yim J.J., Ding L., Schaffer A.A., Park G.Y., Shim Y.S., Holland S.M. A microsatellite polymorphism in intron 2 of human Toll-like receptor 2 gene: functional implications and racial differences. FEMS Immunol. Med. Microbiol. 2004;40:163–169. doi: 10.1016/S0928-8244(03)00342-0. [DOI] [PubMed] [Google Scholar]
- 20.Kizawa H., Kou I., Iida A., Sudo A., Miyamoto Y., Fukuda A., Mabuchi A., Kotani A., Kawakami A., Yamamoto S., et al. An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis. Nat. Genet. 2005;37:138–144. doi: 10.1038/ng1496. [DOI] [PubMed] [Google Scholar]
- 21.Winkler G.S., Petrakis T.G., Ethelberg S., Tokunaga M., Erdjument-Bromage H., Tempst P., Svejstrup J.Q. RNA polymerase II elongator holoenzyme is composed of two discrete subcomplexes. J. Biol. Chem. 2001;276:32743–32749. doi: 10.1074/jbc.M105303200. [DOI] [PubMed] [Google Scholar]
- 22.Winkler G.S., Kristjuhan A., Erdjument-Bromage H., Tempst P., Svejstrup J.Q. Elongator is a histone H3 and H4 acetyltransferase important for normal histone acetylation levels in vivo. Proc. Natl Acad. Sci. USA. 2002;99:3517–3522. doi: 10.1073/pnas.022042899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang B., Johansson M.J., Bystrom A.S. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA. 2005;11:424–436. doi: 10.1261/rna.7247705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y.Z., Bennett C.L., Huynh H.M., Blair I.P., Puls I., Irobi J., Dierick I., Abel A., Kennerson M.L., Rabin B.A., et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am. J. Hum. Genet. 2004;74:1128–1135. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greenway M.J., Andersen P.M., Russ C., Ennis S., Cashman S., Donaghy C., Patterson V., Swingler R., Kieran D., Prehn J., et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat. Genet. 2006;38:411–413. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- 26.Anderson S.L., Coli R., Daly I.W., Kichula E.A., Rork M.J., Volpi S.A., Ekstein J., Rubin B.Y. Familial dysautonomia is caused by mutations of the IKAP gene. Am. J. Hum. Genet. 2001;68:753–758. doi: 10.1086/318808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helmlinger D., Hardy S., Sasorith S., Klein F., Robert F., Weber C., Miguet L., Potier N., Van-Dorsselaer A., Wurtz J.M., et al. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum. Mol. Genet. 2004;13:1257–1265. doi: 10.1093/hmg/ddh139. [DOI] [PubMed] [Google Scholar]
- 28.Batulan Z., Shinder G.A., Minotti S., He B.P., Doroudchi M.M., Nalbantoglu J., Strong M.J., Durham H.D. High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J. Neurosci. 2003;23:5789–5798. doi: 10.1523/JNEUROSCI.23-13-05789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han Q., Lu J., Duan J., Su D., Hou X., Li F., Wang X., Huang B. Gcn5- and Elp3-induced histone H3 acetylation regulates hsp70 gene transcription in yeast. Biochem. J. 2008;409:779–788. doi: 10.1042/BJ20070578. [DOI] [PubMed] [Google Scholar]
- 30.Gifondorwa D.J., Robinson M.B., Hayes C.D., Taylor A.R., Prevette D.M., Oppenheim R.W., Caress J., Milligan C.E. Exogenous delivery of heat shock protein 70 increases lifespan in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2007;27:13173–13180. doi: 10.1523/JNEUROSCI.4057-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simpson C.L., Hansen V.K., Sham P.C., Collins A., Powell J.F., Al-Chalabi A. MaGIC: a program to generate targeted marker sets for genome-wide association studies. Biotechniques. 2004;37:996–999. doi: 10.2144/04376BIN03. [DOI] [PubMed] [Google Scholar]
- 32.Sham P., Bader J.S., Craig I., O’Donovan M., Owen M. DNA Pooling: a tool for large-scale association studies. Nat. Rev. Genet. 2002;3:862–871. doi: 10.1038/nrg930. [DOI] [PubMed] [Google Scholar]
- 33.Simpson C.L., Knight J., Butcher L.M., Hansen V.K., Meaburn E., Schalkwyk L.C., Craig I.W., Powell J.F., Sham P.C., Al-Chalabi A. A central resource for accurate allele frequency estimation from pooled DNA genotyped on DNA microarrays. Nucleic Acids Res. 2005;33:e25. doi: 10.1093/nar/gni028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clandinin T.R., Zipursky S.L. Making connections in the fly visual system. Neuron. 2002;35:827–841. doi: 10.1016/s0896-6273(02)00876-0. [DOI] [PubMed] [Google Scholar]
- 35.Westerfield M. The Zebrafish Book. Eugene, Oregon: The University of Oregon Press; 2003. [Google Scholar]
- 36.Lemmens R., Van Hoecke A., Hersmus N., Geelen V., D’Hollander I., Thijs V., Van Den Bosch L., Carmeliet P., Robberecht W. Overexpression of mutant superoxide dismutase 1 causes a motor axonopathy in the zebrafish. Hum. Mol. Genet. 2007;16:2359–2365. doi: 10.1093/hmg/ddm193. [DOI] [PubMed] [Google Scholar]
- 37.Knight J., Sham P. Design and analysis of association studies using pooled DNA from large twin samples. Behav. Genet. 2006;36:665–677. doi: 10.1007/s10519-005-9016-9. [DOI] [PubMed] [Google Scholar]
- 38.Neale M.C., Boker S.M., Xie G., Maes H.H. Mx:. Statistical Modeling. Richmond, VA: Box 980126 MCV; 2003. 23298. [Google Scholar]
- 39.Sham P.C., Curtis D. Monte Carlo tests for associations between disease and alleles at highly polymorphic loci. Ann. Hum. Genet. 1995;59:97–105. doi: 10.1111/j.1469-1809.1995.tb01608.x. [DOI] [PubMed] [Google Scholar]
- 40.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 41.Pritchard J.K., Stephens M., Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.