

Abstract
Motivation: The elucidation of biological pathways enriched with differentially expressed genes has become an integral part of the analysis and interpretation of microarray data. Several statistical methods are commonly used in this context, but the question of the optimal approach has still not been resolved.
Results: We present a logistic regression-based method (LRpath) for identifying predefined sets of biologically related genes enriched with (or depleted of) differentially expressed transcripts in microarray experiments. We functionally relate the odds of gene set membership with the significance of differential expression, and calculate adjusted P-values as a measure of statistical significance. The new approach is compared with Fisher's exact test and other relevant methods in a simulation study and in the analysis of two breast cancer datasets. Overall results were concordant between the simulation study and the experimental data analysis, and provide useful information to investigators seeking to choose the appropriate method. LRpath displayed robust behavior and improved statistical power compared with tested alternatives. It is applicable in experiments involving two or more sample types, and accepts significance statistics of the investigator's choice as input.
Availability: An R function implementing LRpath can be downloaded from http://eh3.uc.edu/lrpath.
Contact: mario.medvedovic@uc.edu
Supplementary information: Supplementary data are available at Bioinformatics online and at http://eh3.uc.edu/lrpath.
1 INTRODUCTION
The identification of predefined sets of biologically related genes (gene sets) enriched with differentially expressed genes (DEGs) (Tavazoie et al., 1999) has become a routine part of the analysis and interpretation of microarray data (Curtis et al., 2005). Sets of genes associated with the same Gene Ontology (GO) term (Ashburner et al., 2000; Harris et al., 2004) or the same KEGG pathway (Kanehisa et al., 2006) are two commonly used collections of such predefined groups.
The most commonly used approach to identifying enriched sets of genes is based on counting the number of genes in such a set that are also differentially expressed. The statistical significance of such overlap is then established using the Fisher's exact (FE) or χ2-tests. Various web-based or downloadable computer programs utilizing these methods have been developed, such as Onto-Express (Draghici et al., 2003; Khatri et al., 2005), David/EASE (Dennis, Jr et al., 2003; Hosack et al., 2003), the Gostats package of Bioconductor (Gentleman, 2005), GOMiner (Zeeberg et al., 2003, 2005) and FuncAssociate (Berriz et al., 2003). Khatri and Draghici (2005) provided a comparison of several such programs, and (Rivals et al., 2007) presented a thorough review. The inherent limitation of approaches that are based on counts of DEGs is the requirement to choose a specific significance cutoff level to distinguish between genes that are changed versus those that are not. Different threshold choices may lead to dramatically different enriched categories, and thus different biological conclusions (Pan et al., 2005).
Several methods have been proposed to overcome the limitations of such basic procedures (Table 1). BayGO still uses significance counts, but employs a Bayesian framework and accounts for which genes are exclusive to which categories (Vencio et al., 2006). Gene set enrichment analysis (GSEA) uses the complete distribution of differential expressions of all genes, without categorizing them into differentially and non-differentially expressed, to identify enriched gene sets (Subramanian et al., 2005). sigPathway (Tian et al., 2005) determines statistical significance of enrichment by comparing the sum of association measures (standard t-statistics) between genes and phenotype to the distribution of sums under the null hypothesis of no association. A more recent method, ProbCD, has the unique feature of allowing continuous probabilities both for gene significance (differential expression) and category assignment, based on uncertainty (Vencio and Schmulevich, 2007). This method calculates an enrichment statistic based on a k× 2 contingency table with the Goodman−Kruskal gamma, and assesses significance by comparison to a null distribution estimated by permutations. Newton et al. (2007) introduced a random-sets statistical framework which facilitates a unified treatment of methods based on significance counts and methods based on complete distributions of any quantitative gene-level score. The random-sets method detects enriched gene sets by comparing the summary score for the gene list to the distribution of scores for randomly selected sets of the same size. The method is implemented in the allez R-package.
Table 1.
Methods included in comparisons
| Methods | Main statistical test | Input data | User choice for DEG test? |
|---|---|---|---|
| LRpath | Logistic regression-likelihood ratio | Significance levels for all genes | Any |
| FE | FE test | Counts of significant and non-significant genes | Any |
| GSEA | Weighted Kolmo-gorov–Smirnov | Normalized intensities for all genes and all arrays | No |
| ProbCD | Goodman–Kurskal gamma | Significance levels for all genes | Any |
| BayGo | Bayesian 3×2 contingency table | Counts of significant and non-significant genes | Any |
| sigPathway | t-test (2 hypotheses tested) | Normalized intensities for all genes and all arrays, or measures of association | t-test or Wilcoxon rank |
| Random-sets | Score test | Significance levels for all genes | Any |
Here, we introduce and validate a new logistic regression-based method, LRpath, that functionally relates gene set membership status (dependent variable) to the statistical significance of genes' differential expression (independent variable). The basic question asked by LRpath is, ‘Does the odds of a gene belonging to a pre-defined gene set increase as the significance of differential expression increases?’ Logistic regression is a natural extension of the χ2-test, allowing the significance values to remain on a continuous scale and not requiring the use of significance thresholds. We compare the sensitivity and specificity of LRpath to other relevant methods in identifying enriched GO terms using simulated and experimental microarray data. Our simulation study and experimental data analyses were structured so that the true hierarchical GO structure is preserved, thus retaining the natural correlations among categories. The results from experimental data reinforce the simulation findings by additionally preserving the natural correlations among gene expression profiles of experimental microarray data. We circumvent the problem of unknown ‘truth’ in the experimental data comparisons by using two independent datasets examining the same biological phenomenon and compare methods based on the reproducibility of their findings. Our method showed greater reproducibility in identifying enriched GO terms than the other tested methods. Use of the new method is further demonstrated by analyzing a previously published microarray experiment comparing healthy subjects to those with idiopathic pulmonary fibrosis (IPF) (Pardo et al., 2005). Results from two additional datasets are available as Supplementary Material.
2 METHODS
2.1 LRpath model details
Suppose that for a given microarray experiment we have assigned the statistical significance of the comparison of interest to each gene in terms of P-values. Our logistic regression method proceeds as follows. For each category (i.e. gene set) c, the dependent variable y is defined as 1 for genes in c, and 0 for all other genes. We use the significance statistics, defined as −log(P-values), as the explanatory variable x, although a different significance measure could be used. If π is the proportion of genes belonging to the category (y=1) at a specified x value, then π/(1-π) are the corresponding odds that a gene with significance x is a member of this particular category. If the log odds value increases as x increases, then we conclude that the category is associated with the differential expression. Logistic regression is used to model the log-odds of a gene belonging to the specific category as a linear function of the statistical significance x:
where α is the intercept, β is the slope, and both α and β are estimated from the data. The slope parameter, β, corresponds to the change in the log odds of belonging to the specific category for a unit increase in x (or 10-fold decrease in P-value). When β>0, we conclude that the category of interest is ‘enriched’ with DEGs (or conversely that the category is ‘depleted’ if β<0). The evidence in the data that β>0 (or < 0) for a specific category is assessed by calculating the P-value for the null hypothesis that β=0 against the alternative that β≠0 based on the maximum likelihood parameter estimates and the Wald test. The Wald statistic, W, is calculated as:
where 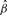 is the maximum likelihood (ML) estimate for
is the maximum likelihood (ML) estimate for 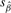 is the standard error of , and the ML is estimated using the iteratively weighted least squares (IWLS) algorithm. For testing β = 0, W can be shown to follow a χ2-distribution with one degree of freedom and the P-value is calculated assuming this null distribution. The P-values from the test of each category c, are then adjusted for multiple testing either by controlling the false discovery rate (FDR) (Benjamini et al., 1995; Storey andTibshirani, 2003), or controlling the family-wise error rate using Bonferroni. Most likely enriched gene sets will be identified based on the P-value, or based on the odds ratio if a ranking independent of category size is desired.
is the standard error of , and the ML is estimated using the iteratively weighted least squares (IWLS) algorithm. For testing β = 0, W can be shown to follow a χ2-distribution with one degree of freedom and the P-value is calculated assuming this null distribution. The P-values from the test of each category c, are then adjusted for multiple testing either by controlling the false discovery rate (FDR) (Benjamini et al., 1995; Storey andTibshirani, 2003), or controlling the family-wise error rate using Bonferroni. Most likely enriched gene sets will be identified based on the P-value, or based on the odds ratio if a ranking independent of category size is desired.
When multiple related comparisons are of interest (e.g. a time course or multiple treatments versus a control) β may be modeled as a vector (β1,…,βn) where each element is the slope at one time point or one treatment. In this case, for each gene set one could test the null hypothesis: βj−βi=0. That is, a combined logistic regression may be performed to identify if a category is significantly more affected by one treatment than another. Alternatively, one could test for each gene set the null hypothesis: β1 =β2 =…=βn =0 (odds ratios = 1 for all time points or dose levels) to determine which categories are affected by any dose level or at any time in the experiment. This type of analysis is illustrated in Supplementary Material.
2.2 Simulation design
Our simulation study imitates 6- and 10-slide, single-channel microarray experiments with three (and five) treated samples and three (five) controls. Gene expression values of DEGs were assigned to human Entrez Gene IDs so that the desired enrichment level of chosen categories was obtained, with the remaining gene expression values assigned to randomly chosen, unique human Entrez Gene IDs. The Entrez Gene IDs were then mapped to all of their assigned GO terms. This allowed the simulations to preserve the actual correlations and gene distributions existing in the structure of the GO database. All simulations were performed using 10 000 ‘genes’, with 500 (5%) genes, or in one set 1000 (10%) genes, designed to be ‘differentially expressed’. The following variables were assessed in the simulations:
Number of DEGs, d: (500, 1000)
Distribution of true fold changes: N(0, 4
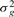 ) (or Uniform([−2.5, −0.5]∪[0.5, 2.5]), where is defined below.
) (or Uniform([−2.5, −0.5]∪[0.5, 2.5]), where is defined below.Number of enriched categories, e: (2, 5).
Level of enrichment, L: (25%, 50%, 75%, 90%) of genes in category are differentially expressed.
Number of array replicates: (3, 5).
The simulations proceed as follows:
For all 10 000 genes, g:
Simulate gene variances,
, assuming equal variance among treatment groups, as random draws from the χ−1(4) distribution.- Without loss of generality, assume that the first d genes are differentially expressed:
- Simulate actual mean log ratios, μg, as random draws from N(0, 4) (or Uniform([−2.5, −0.5]∪[0.5,2.5]).
- For the remaining 10 000-d genes, set the actual mean log ratios, μg=0.
Simulate normalized estimated expression levels as random draws from N(μg
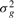 .
.Randomly select e GO terms to be ‘enriched’.
Randomly assign L% of human Entrez Gene IDs from each enriched GO category to DEGs.
Randomly assign unique Entrez IDs to all other gene expression values, including unassigned DEGs, as random draws from all human Entrez IDs represented in GO.
For all GO terms with 10−200 genes (1761 terms), calculate significance statistics (P-values and q-values) for each tested break method.
All compared methods were applied with default parameters with the following exceptions. For GSEA, we permuted genes rather than samples for experiments with less than six replicates (as recommended for small experiments). For BayGO we increased the number of simulations from 100 to 1000 for higher accuracy. For ProbCD, we defined all gene annotation assignments with 100% probability. In the case of allez, we used the z-transformed rankings of the genes based on the statistical significance as input. Since the random-set method allows for the use of any measure of differential expression, we chose z-transformed ranks as the score primarily due to the prominent place that rankings was given in the manuscript describing the procedure, and the fact that the z-transformed-ranks option of allez is stated to improve the z-score quality in the allez documentation. Given the underlying connection between the random-set analysis and the logistic regression (see Section 3), we also directly compared the two procedures using the −log(P-value) as the score for allez and using z-transformed ranks as the input for LRpath.
3 RESULTS
We performed a comparison with the methods in Table 1 using both simulated and experimental data. In the simulation study, we know the truth about enriched GO terms, but the data lacks the natural correlation structure found in experimental data and may have unrealistic distributional properties. On the other hand, in the breast cancer microarray data, the truth is unknown, but the other issue is appropriately addressed. The concordant findings based on simulated and experimental data analyses offer strong evidence that our conclusions are valid and reproducible.
3.1 Simulation study
We applied seven methods Table 1 to each simulated dataset. For FE test, we used five different P-value cutoff levels for DEGs (0.001, 0.01, 0.05, 0.10 and 0.50), and for BayGO we used a 0.01 cutoff. For sigPathway (NTK and NEK hypotheses) (Tian et al., 2005), we use the provided ranking procedure that combines the two hypotheses based on the sum of the two statistics, but separate P-values, because combined P-values are not available. All methods were performed using an R package when available, or R-code downloaded from the original publication's authors otherwise. Because GSEA tests increased and decreased transcript levels separately, we modified the program so that the absolute value of the measure of change is used. The simulated data were first analyzed for detection of DEGs using a standard t-test for input into LRpath, Fisher's exact, ProbCD and BayGO. Results improved when a Bayesian moderated t-statistic (Sartor et al., 2006) was used in place of the t-test for testing differential expression of genes (see the web Supplementary Material).
For each simulation scenario, we simulated 30 datasets and calculated: (i) the average ranks of GO terms ordered by statistical significance and (ii) q-values of enrichment for all GO terms. We compared performances of different methods by comparing the average log-ranking of enriched categories (Fig. 1). To clarify the exact values plotted in Figure 1, we provide the raw data in the Supplementary Material (Table S1). The performance of all methods was strongly affected by the level of enrichment (varied between 25% and 90%).
Fig. 1.

Simulation results: ability to rank enriched GO terms log10-rankings of enriched GO terms were calculated to compare the ability of methods to correctly rank these categories at the top of the list. Thus, lower ranking scores are better. Methods are LRpath, FE with the following three criteria for detecting DEGs (P<0.001, P<0.01, P<0.05, P<0.10 and P<0.50), BayGO, sigPathway (sigPath), allez and ProbCD. Initial four parameter sets (A) used 90%, 75%, 50% and 25% enrichment with DEGs, 500 total DEGs, normally distributed fold changes, two enriched categories and three replicates for treated and control groups. Subsequent groups had the following differences: (B) 1000 DEGs, (C) DEGs with higher fold changes, (D) five enriched GO terms, (E) five replicates. Data shown are averages from 30 simulation runs for each parameter set. LRpath performed significantly better than the next best methods (P=2.2×10−4 compared with FE P<0.05 and P=1.5×10−4 compared with FE P<0.10) using a Wilcoxon rank test.
LRpath performed best overall in ranking the enriched GO terms as most significant. Using the performance ranks of each method across all simulation scenarios (based on values of Table S1), a Wilcoxon rank test was used to test the significance of LRpath's performance over the next best methods, FE P<0.10 and FE P<0.05, and was found to be significant(Wilcoxon rank test P=2.2 × 10−4, as compared with FE P<0.05 and P=1.5×10−4 as compared with FE P<0.10). The performance of FE test varied depending on which P-value cutoff was used to identify DEGs, as previously seen (Pan et al., 2005). For most parameter sets, using a more relaxed cutoff of P<0.05 or 0.10 performed best. Thus, for ranking enriched biological categories using FE test, one would want to apply a less stringent P-value cutoff than would be justified for identifying individual DEGs.
In agreement with Newton et al's finding that FE is more likely to outperform the corresponding averaging method when the level of enrichment is small, we find that at least one FE test outperforms allez for every set at the lowest (25%) enrichment level. Conversely, allez outperforms FE tests more often for the highest level of enrichment. The fact that LRpath still outperforms FE at the low enrichment levels can be attributed to its use of the −log(P-value) rather than z-transformed rankings as input. When the enrichment level is low, the −log(P-value) statistic allows a small number of highly significant genes to drive the enrichment test, whereas the z-transformed rankings does not allow for such strong ‘outliers’. Indeed, additional simulations using the same rankings input for LRpath as for allez resulted in similar poorer performance by LRpath. Conversely, when the default input for LRpath −log(P-value) was used as input for allez, LRpath only slightly outperformed allez based on a Wilcoxon rank test (P=0.014) (Fig. 2).
Fig. 2.

Effect of input statistics on LRpath and allez. Graphed is the average increase in log-rank of enriched GO terms relative to LRpath with –log(P-values) as input, which ranked best. LRpath and allez produced very similar results when given the same input. *P<0.05 from Wilcoxon rank test between allez and LRpath using –log(P). **P<0.05 from Wilcoxon rank test between allez and LRpath using z-transformed gene ranks.
For experiments with more statistical power, as illustrated by our simulations of a 10-slide experiment, using a stricter P-value cutoff for DEG detection may offer better performance. Indeed, using P<0.01 performed better than P<0.05 in ranking GO terms for two of the four parameter sets in the 10-slide experiment, and the performance of the P<0.001 cutoff increased substantially compared with the smaller simulated experiment. As expected, simulating higher actual fold changes for DEGs resulted in overall better performance of all methods.
Simulating twice as many DEGs or increasing the number of enriched categories from two to five had little effect on the differences in methods' performance, although there is some indication of a slight overall decrease in performance among methods. Of the other methods tested, BayGO, allez and FE with the P<0.05, 0.01 or 0.10 cutoff offered the next best performance, depending on parameter values. Because P-values produced by different methods are not directly comparable, we focused on the rankings of enriched gene sets. However, we also offer a comparison of the methods' q-values for enriched GO terms as Supplementary Material (Fig. S1), as well as a measure of bias in the P-values under the hypothesis of no association between GO terms and differential expression (Fig. S2).
3.2 Comparison of results from two breast cancer microarray experiments
We also compared the performance of different methods based on the reproducibility of their findings in two breast cancer datasets. A frequently recurring concern with microarray data is its generalizability. A better method is expected to demonstrate a higher consistency between results obtained from independent experiments studying the same biological phenomenon, despite technical differences. To this end, we examined the consistency of each method's results between two datasets. The first dataset (Sotiriou et al., 2006) consists of human breast carcinoma samples. For this analysis, we used samples from non-treated patients with positive estrogen receptor (ER) status and with histologic grades 1 (29 samples) or 3 (12 samples). The second dataset consisted of the independent samples with positive ER status from another primary breast tumor study, where each sample was also identified as histologic tumor grade 1 (39 samples) or 3 (28 samples) using the Elston−Ellis grading system (Miller et al., 2005).
Preprocessed data was downloaded from the NCBI Gene Expression Omnibus (GEO) public repository (GEO accession GSE2990), and we separately analyzed each dataset for GO categories enriched with genes differentially expressed between the histologic grade 1 and 3 samples. Results from standard t-tests were used for input into LRpath, Bay GO and Fisher's exact. In the first dataset, 10 GO terms were identified with P<0.005 by at least five of the seven methods: cell division, M phase, mitosis, M phase of mitotic cell cycle, spindle organization and biogenesis, regulation of mitosis, condensed chromosome, mitotic checkpoint, cell cycle checkpoint and regulation of progression through cell cycle.
For each method, concordance was measured in two ways: (i) the degree of correlation in significance of GO terms between the two datasets (Fig. 3A) and (ii) the number of overlapping GO terms between the two datasets among top ranked lists (Fig. 3B). The results shown in Figure 3 indicate that LRpath has the greatest consistency between datasets. Consistent with the other analyses, the concordance of FE test between datasets depended on the criteria for DEG detection, with P<0.01 and P<0.10 criteria resulting in greater concordance than that of P<0.50 or P<0.001. Examining the number of overlapping GO terms in the top ranked lists of each method, overall we see LRpath performing best, allez second best and probCD third (although FE with P<0.01 performs as well for the first 20 rankings), FE with the P<0.001 or P<0.50 criteria for DEGs performed worst, and the other methods' performances relatively indistinguishable from each other. All methods except FE with P<0.001 performed markedly better than would be expected by chance, indicating a true signal in answering the question as to what gene sets are enriched between histologic grade 3 versus grade 1 primary breast cancer tumors. The overlapping GO terms among the top 50 ranked for LRpath are listed in Table S3. Notably, this list of 28 GO terms included all 10 of the GO terms identified by at least five methods in the first dataset, and consistent with the findings of Sotiriou et al. (2006), were mainly related to cell-cycle progression. We again looked separately at the performance of the random-set procedure with −log(P-value) as input and results were very similar as in the simulation study. The correlation coefficient increased, when compared to using transformed ranks (from 0.60 to 0.64), but still remained below LRpath's correlation. On the other hand, there was no difference between LRpath and allez when plotting overlapping GO terms by rank (Supplementary Fig. 4).
Fig. 3.

Concordance of methods between two independent Breast Cancer datasets. Reproducibility of the methods (LRpath, FE with cutoffs of 0.50, 0.10, 0.01 and 0.001 for DEGs, BayGO, GSEA, sigPathway, allez and ProbCD, respectively) was tested by measuring the consistency of results across two datasets, both comparing grade 3 to grade 1 tumors. (A)Correlation between datasets for each method. As a measure of significance, the –log(P-values) of GO term enrichment were calculated for each method and dataset separately, and the Pearson correlation coefficients between datasets were calculated. (B) Overlapping enriched GO terms by rank. Ranked lists of GO terms were generated for each method and each dataset separately. The number of overlapping GO terms was calculated between datasets for each method for increasing length of ranked lists.
3.3 Application: results from human IPF dataset
Using a human IPF study (Pardo et al., 2005), we demonstrate the ability of LRpath to implicate important biological pathways and functional groupings missed by the most commonly used analytical approach. The 11 normal and 13 IPF lung tissue samples were analyzed for DEGs using a standard t-test or an empirical Bayes test, Intensity-based moderated t-test (IBMT) (Sartor et al., 2006), and then tested for enriched gene sets using LRpath and FE test. Six KEGG pathways were significant at the FDR<0.05 level using LRpath with IBMT, including altered ‘Cell cycle’, decreased ‘Blood vessel development’ and a decrease in ‘Cytokine−cytokine receptor interaction’ (Table S4). FE test with IBMT resulted in no significant pathway when using a P<0.01 cutoff for DEGs and only 1 (complement and coagulation cascades) when using a P<0.05 cutoff. No enriched KEGG pathways were identified with the t-test in conjunction with Fisher's exact. The significant KEGG pathways identified with LRpath involves several findings consistent with what has been reported in human IPF, and a thorough discussion of these findings is provided as Supplementary Material.
4 DISCUSSION
Identifying predefined gene sets enriched with DEGs has become a routine part of microarray analysis, and provides investigators with greater biological insight than significant gene lists alone. Our aim was to develop a method that (i) does not require the choice of a significance cutoff, (ii) allows the investigator to choose different methods for detecting DEGs, (iii) provides unbiased assessment of statistical significance and (iv) similar to FE test has an intuitive interpretation in terms of odds ratios. The method we developed, LRpath, uses logistic regression to model the relationship between gene set membership and differential expression in terms of odds ratios of enrichment. The basic question addressed by LRpath is whether the odds of a gene belonging to a predefined gene set increases as the significance of differential expression increases. Unlike the χ2-type of methods, our model allows the data resulting from tests of differential expression to remain on a continuous scale. This removes the need to choose a significance cutoff, and has the advantage of taking into account the distribution of significance levels for genes not belonging to, as well as belonging to, the gene set of interest. If expression of genes from a specific biological pathway is affected in the experiment, we would expect that genes with significant P-values are more likely to be members of this pathway than genes with less significant P-values, although we may not know exactly where, or want, to draw a line between ‘significant’ and ‘non-significant’ differential expressions.
Led by the communication from Michael Newton about underlying similarities between the logistic regression and the random-sets framework, we further examined the relationship between the two methods. Our results indicate that the differences between the two procedures, when using the same score are very small. Actually, it can be shown that the random-sets method of allez is nearly identical (see Supplementary Material) to performing logistic regression and using the score test for significance, which is asymptotically equivalent to the statistical test used by LRpath. Thus, small observed differences could be due to small differences in the performance of the score and the Wald tests in this context of logistic regression. Regardless of which of the two procedures are used, one seems to be better off using −log(P-values) instead of using the z-transformed ranks.
We performed an in-depth comparison with other relevant methods using both simulated and experimental data. In the simulation study, we know the exact truth about enriched GO terms, but the simulated data lacks the natural correlation structure found in experimental data and may have unrealistic distributional properties. On the other hand, the exact truth is unknown in the breast cancer microarray data we analyzed, but the other issues are appropriately addressed. This comparison of independent experimental datasets is both inherently free of bias, and addresses the question of which methods provide the most reproducible results. The observed concordance of the results in these different analyses offers strong evidence that our conclusions are valid and reproducible. Results of our simulation study indicate that, as expected, the power to detect enriched GO terms depends greatly on the level of enrichment, and to a lesser extent on several other parameters tested. For FE test, we conclude that both the significance cutoff used to define DEGs and the test used to detect DEGs (data not shown) affect the results of gene enrichment testing. Overall, LRpath performed better than the other methods tested based on all criteria.
Using the concordance between the two independent larger sample breast cancer datasets, we showed that LRpath again resulted in the best performance. The results from these analyses were generally in agreement with results of the simulation study. In both cases, allez and ProbCD performed favorably, and FE with a fairly relaxed cutoff also performed well. ProbCD may offer an additional advantage in situations when the gene set assignments are probabilistic. Newton et al. showed that selection methods (e.g. FE test) and averaging methods (e.g. allez) each have a ‘domain of superiority’ in the space of possible enrichment problems. In general, averaging methods are superior when the differential expression effects are relatively small and are most advantageous when the enrichment level is also high. Based on our results for the breast cancer experiments, it seem likely that these criteria held true, and that the disadvantage of the averaging method can be at least partially offset by using an input measure, such as −log(P-value), that allows a smaller number of highly DEGs to help drive the enrichment process. Further comparisons will be necessary to assess the performance of other gene score measurements as input, such as log-fold change or t-statistics, and to what extent their performances are dataset dependent.
Using the breast cancer (Miller et al., 2005; Sotiriou et al., 2006) and IPF (Pardo et al., 2005) datasets, we also uncovered novel insights into the biological mechanisms of these diseases. In breast cancer, we demonstrate the use of LRpath and other methods to detect consistent GO terms distinguishing histologic grade 3 and grade 1 primary breast tumor samples from two independent datasets. In IPF, we demonstrate the use of LRpath to detect over-represented biological categories not presented in the original analysis and which would not have been identified by FE test (i.e. identifying additional pathways including altered ‘Cell cycle’, decreased ‘Blood vessel development’ and a decrease in ‘Cytokine−cytokine receptor interaction’.)
We have implemented LRpath as an R function (Ihaka et al., 1996, and Supplementary Material) which can be downloaded along with all other Supplementary Material from our supporting website http://eh3.uc.edu/lrpath. The function is designed to automatically test the categories of GO terms or KEGG Pathways, but can be modified for use with user-defined categories. Current implementation accepts as input significance statistics of the investigator's choice and allows for duplicate and missing gene identifiers.
Supplementary Material
ACKNOWLEDGEMENTS
Comments provided by Michael Newton and an anonymous reviewer helped us significantly improve upon the initial version of this article.
Funding: The National Institute of Health (P30ES06096, U01ES015675 and R01HG003749).
Conflict of Interest: none declared.
REFERENCES
- Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, et al. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- Berriz GF, et al. Characterizing gene sets with FuncAssociate. Bioinformatics. 2003;19:2502–2504. doi: 10.1093/bioinformatics/btg363. [DOI] [PubMed] [Google Scholar]
- Curtis RK, et al. Pathways to the analysis of microarray data. Trends Biotechnol. 2005;23:429–435. doi: 10.1016/j.tibtech.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Dennis G, Jr, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(3) [PubMed] [Google Scholar]
- Draghici S, et al. Global functional profiling of gene expression. Genomics. 2003;81:98–104. doi: 10.1016/s0888-7543(02)00021-6. [DOI] [PubMed] [Google Scholar]
- Gentleman RC. Bioconductor package, GOstats vignette. 2005 Available at http://www.bioconductor.org/repository/devel/vignette/GOstats.pdf.
- Harris MA, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–D261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosack DA, et al. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4:R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R, et al. R: a language for data analysis and graphics. J. Comput. Graph. Stat. 1996;5:299–314. [Google Scholar]
- Kanehisa M, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, Draghici S. Ontological analysis of gene expression data: current tools, limitations and open problems. Bioinformatics. 2005;21:3587–3595. doi: 10.1093/bioinformatics/bti565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, et al. Recent additions and improvements to the onto-tools. Nucleic Acids Res. 2005;33:W762–W765. doi: 10.1093/nar/gki472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LD, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects and patient survival. Proc. Natl Acad. Sci. USA. 2005;102:13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton MA, et al. Random-set methods identify distinct aspects of the enrichment signal in gene-set analysis. Ann. of Applied Stat. 2007;1:85–106. [Google Scholar]
- Pan KH, et al. Effects of threshold choice on biological conclusions reached during analysis of gene expression by DNA microarrays. Proc. Natl Acad. Sci. USA. 2005;102:8961–8965. doi: 10.1073/pnas.0502674102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo A, et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005;2 doi: 10.1371/journal.pmed.0020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivals I, et al. Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics. 2007;23:401–407. doi: 10.1093/bioinformatics/btl633. [DOI] [PubMed] [Google Scholar]
- Sartor MA, et al. Intensity-based hierarchical Bayes method improves testing for differentially expressed genes in microarray experiments. BMC. Bioinformatics. 2006;7:538. doi: 10.1186/1471-2105-7-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiriou C, et al. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J. Natl Cancer Inst. 2006;98:262–272. doi: 10.1093/jnci/djj052. [DOI] [PubMed] [Google Scholar]
- Storey JD, et al. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie S, et al. Systematic determination of genetic network architecture. Nat. Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- Tian L, et al. Discovering statistically significant pathways in expression profiling studies. Proc. Natl Acad. Sci. USA. 2005;102:13544–13549. doi: 10.1073/pnas.0506577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vencio RZ, et al. BayGO: Bayesian analysis of ontology term enrichment in microarray data. BMC. Bioinformatics. 2006;7:86. doi: 10.1186/1471-2105-7-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vencio RZ, Schmulevich I. ProbCD: enrichment analysis accounting for categorization uncertainty. BMC. Bioinformatics. 2007;8:383. doi: 10.1186/1471-2105-8-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeberg BR, et al. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeberg BR, et al. High-throughput GoMiner, an ‘industrial-strength’ integrative gene ontology tool for interpretation of multiple-microarray experiments, with application to studies of Common Variable Immune Deficiency (CVID) BMC. Bioinformatics. 2005;6:168. doi: 10.1186/1471-2105-6-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.