

Abstract
To clarify genome-wide DNA methylation profiles during multistage renal carcinogenesis, bacterial artificial chromosome array-based methylated CpG island amplification (BAMCA) was performed. Non-cancerous renal cortex tissue obtained from patients with clear cell renal cell carcinomas (RCCs) (N) was at the precancerous stage where DNA hypomethylation and DNA hypermethylation on multiple bacterial artificial chromosome (BAC) clones were observed. By unsupervised hierarchical clustering analysis based on BAMCA data for their N, 51 patients with clear cell RCCs were clustered into two subclasses, Clusters AN (n = 46) and BN (n = 5). Clinicopathologically aggressive clear cell RCCs were accumulated in Cluster BN, and the overall survival rate of patients in Cluster BN was significantly lower than that of patients in Cluster AN. By unsupervised hierarchical clustering analysis based on BAMCA data for their RCCs, 51 patients were clustered into two subclasses, Clusters AT (n = 43) and BT (n = 8). Clinicopathologically aggressive clear cell RCCs were accumulated in Cluster BT, and the overall survival rate of patients in Cluster BT was significantly lower than that of patients in Cluster AT. Multivariate analysis revealed that belonging to Cluster BT was an independent predictor of recurrence. Cluster BN was completely included in Cluster BT, and the majority of the BAC clones that significantly discriminated Cluster BN from Cluster AN also discriminated Cluster BT from Cluster AT. In individual patients, DNA methylation status in N was basically inherited by the corresponding clear cell RCC. DNA methylation alterations in the precancerous stage may generate more malignant clear cell RCCs and determine patient outcome.
Introduction
It is known that DNA hypomethylation results in chromosomal instability as a result of changes in chromatin structure and that DNA hypermethylation of CpG islands silences tumor-related genes in cooperation with histone modification in human cancers (1–5). Accumulating evidence suggests that alterations of DNA methylation are involved even in the early and the precancerous stages (6,7). On the other hand, in patients with cancers, aberrant DNA methylation is significantly associated with poorer tumor differentiation, tumor aggressiveness and poor prognosis (6,7). Therefore, alterations of DNA methylation may play a significant role in multistage carcinogenesis and can become an indicator for carcinogenetic risk estimation and a biological predictor of poor prognosis in patients with cancers. Recently developed array-based technology for accessing genome-wide DNA methylation status (8–10) is now mainly used to identify tumor-related genes silenced by DNA methylation in human cancers. Subclassification of cancers based on DNA methylation status, which may reflect the distinct epigenetic pathways of carcinogenesis, and DNA methylation profiles, which could become the optimum indicator for carcinogenetic risk estimation and prediction of patient outcome, should be further explored in each organ using array-based approaches.
With respect to renal carcinogenesis, we have reported that accumulation of DNA methylation on C-type CpG islands occurs in a cancer-specific but not age-dependent manner (11), even in non-cancerous renal tissue samples obtained from patients with clear cell renal cell carcinomas (RCCs) (6,7,12). Although precancerous conditions in the kidney have been rarely described, from the viewpoint of altered DNA methylation, non-cancerous renal tissues obtained from patients with clear cell RCCs are considered to already be at the precancerous stage in spite of showing no remarkable histological changes and lacking association with chronic inflammation and persistent infection with viruses or other pathogenic microorganisms. Surprisingly, accumulation of DNA methylation on C-type CpG islands in such non-cancerous renal tissues has been shown to be significantly correlated with higher histological grades of the corresponding clear cell RCCs developing in individual patients (6,7,12). However, since in the previous study we examined DNA methylation status on only a restricted number of CpG islands (12), we were unable to conclude that genome-wide DNA methylation alterations in precancerous conditions generate more malignant RCCs. In the previous study, accumulation of DNA methylation on C-type CpG islands in clear cell RCCs themselves was significantly correlated with tumor aggressiveness and poorer patient outcome (12). However, we were unable to conclude that the examined C-type CpG islands are the optimum prognostic indicator for patients with clear cell RCCs.
In this study, in order to clarify genome-wide DNA methylation profiles during multistage renal carcinogenesis, we performed bacterial artificial chromosome array-based methylated CpG island amplification (BAMCA) (13–15) using a microarray of 4361 bacterial artificial chromosome (BAC) clones (16) in normal renal cortex tissue samples, non-cancerous renal cortex tissue samples obtained from patients with clear cell RCC and the corresponding clear cell RCCs.
Materials and methods
Patients and tissue samples
Paired specimens of cancerous tissue (T1–T51) and corresponding non-cancerous renal cortex tissue showing no remarkable histological changes (N1–N51) were obtained from materials surgically resected from 51 patients (RCC1–RCC 51) with primary clear cell RCC. These patients did not receive preoperative treatment and underwent nephrectomy in 1999–2006 at the National Cancer Center Hospital, Tokyo, Japan. There were 34 men and 17 women with a mean (±SD) age of 59 ± 10 years (range 31–81 years). Histological diagnosis was made in accordance with the World Health Organization classification (17). All the tumors were graded on the basis of previously described criteria (18) and classified according to the pathological tumor–node–metastasis (TNM) classification (19). The criteria for macroscopic configuration of RCC (12) followed those established for hepatocellular carcinoma: type 3 (contiguous multinodular type) hepatocellular carcinomas show poorer histological differentiation and a higher incidence of intrahepatic metastasis than type 1 (single nodular type) and type 2 (single nodular type with extranodular growth) hepatocellular carcinomas (20). The presence or absence of vascular involvement was examined microscopically on slides stained with hematoxylin–eosin and elastica van Gieson. The presence or absence of tumor thrombi in the main trunk of the renal vein was examined macroscopically. RCC is usually encapsulated by a fibrous capsule and well demarcated and hardly ever contains fibrous stroma between cancer cells (panel T in Figure 1A). Therefore, we were able to obtain cancer cells of high purity from surgical specimens, avoiding contamination with both non-cancerous epithelial cells and stromal cells.
Fig. 1.

DNA methylation alterations during multistage renal carcinogenesis. (A) Microscopic view of normal renal cortex tissue obtained from a patient without any primary renal tumor (C), non-cancerous renal cortex tissue obtained from a patient with clear cell RCC (N) and clear cell RCC (T). N shows no remarkable histological changes compared with C, i.e. no cytological or structural atypia is evident in N. Since T hardly ever contains fibrous stroma between cancer cells, we were able to obtain cancer cells of high purity, avoiding contamination with stromal cells. Hematoxylin–eosin staining. Original magnification ×20. (B) Scanned array images yielded by BAMCA in C, N and T. Test and reference DNA labeled with Cy3 and Cy5 was cohybridized, respectively. (C) Scattergrams of the signal ratios (test signal:reference signal) yielded by BAMCA in C, N and T. In all eight C samples (C1–C8), the signal ratios of 97% of BAC clones were between 0.67 and 1.5 (red bars). Therefore, in N and T, DNA methylation status corresponding to a signal ratio of <0.67 and >1.5 was defined as DNA hypomethylation and DNA hypermethylation on each BAC clone compared with C, respectively. Even though N did not show any remarkable histological changes compared with C [panels C and N in (A)], many BAC clones showed DNA hypomethylation or hypermethylation. In T, more BAC clones showed DNA hypomethylation or hypermethylation, and the degree of DNA hypomethylation and hypermethylation, i.e. deviation of the signal ratio from 0.67 or 1.5, was increased in comparison with N.
For comparison, eight normal renal cortex tissue samples (C1–C8) were obtained from materials surgically resected from eight patients without any primary renal tumor. These patients included five men and three women with a mean (±SD) age of 61 ± 12 years (range 47–81 years). Six of these patients underwent nephroureterectomy for urothelial carcinomas of the ureter, and the other two patients underwent nephrectomy with resection of retroperitoneal sarcoma around the kidney.
High-molecular weight DNA from these fresh frozen tissue samples was extracted using phenol–chloroform, followed by dialysis. Because DNA methylation status is known to be organ specific (21), the reference DNA for analysis of the developmental stages of clear cell RCC should be obtained from the renal cortex and not from other organs or peripheral blood. Therefore, a mixture of normal renal cortex tissue DNA obtained from six male patients (C9–C14) without any primary renal tumor was used as a reference for analyses of male test DNA samples, and a mixture of normal renal cortex tissue DNA obtained from three female patients (C15–C17) without any primary renal tumor was used as a reference for analyses of female test DNA samples.
This study was approved by the Ethics Committee of the National Cancer Center, Tokyo, Japan.
BAMCA
DNA methylation status was analyzed by BAMCA using a custom-made array (MCG Whole Genome Array-4500) harboring 4361 BAC clones throughout chromosomes 1–22 and X and Y (16), as described previously (13–15). Briefly, 5 μg aliquots of test or reference DNA were first digested with 100 U of methylation-sensitive restriction enzyme SmaI and subsequently with 20 U of methylation-insensitive XmaI. Adapters were ligated to XmaI-digested sticky ends, and polymerase chain reaction was performed with an adapter primer set. Test and reference polymerase chain reaction products were labeled by random priming with Cy3- and Cy5-dCTP (GE Healthcare, Buckinghamshire, UK), respectively, using a BioPrime array CGH genomic labeling system (Invitrogen, Carlsbad, CA) and precipitated together with ethanol in the presence of Cot-I DNA. The mixture was applied to array slides and incubated at 43°C for 72 h. Arrays were scanned with a GenePix Personal 4100A (Axon Instruments, Foster City, CA) and analyzed using GenePix Pro 5.0 imaging software (Axon Instruments) and Acue 2 software (Mitsui Knowledge Industry, Tokyo, Japan). The signal ratios were normalized in each sample to make the mean signal ratios of all BAC clones 1.0.
Statistics
Differences in the average number of BAC clones that showed DNA methylation alterations (DNA hypomethylation and hypermethylation) between non-cancerous renal cortex tissue samples obtained from patients with clear cell RCCs, and the clear cell RCCs themselves, were analyzed using the Mann–Whitney U-test. Differences at P < 0.05 were considered significant. Two-dimensional unsupervised hierarchical clustering analysis of the patients with clear cell RCCs and the BAC clones based on the signal ratios (test signal:reference signal) obtained by BAMCA in non-cancerous renal cortex tissue samples and those in clear cell RCCs were performed using the Expressionist software program (Gene Data, Basel, Switzerland). Correlations between the subclassification of patients with clear cell RCCs yielded by the unsupervised hierarchical clustering based on DNA methylation status of non-cancerous renal cortex tissue samples (Clusters AN and BN) and clinicopathological parameters of the corresponding clear cell RCCs were analyzed using chi-square test. Correlations between the subclassification of patients yielded by the unsupervised hierarchical clustering based on DNA methylation status in clear cell RCCs (Clusters AT and BT) and clinicopathological parameters of the RCCs themselves were analyzed using chi-square test. Survival curves of patients belonging to Clusters AN versus BN and Clusters AT versus BT were calculated by the Kaplan–Meier method, and the differences were compared by the Log-rank test. The Cox proportional hazards multivariate model was used to examine the prognostic impact of the subclassification of patients based on the DNA methylation status of their clear cell RCCs (Clusters AT and BT), histological grade, macroscopic configuration, vascular involvement and renal vein tumor thrombi. Differences at P < 0.05 were considered significant. BAC clones whose signal ratios were significantly different between Clusters AN and BN and Clusters AT and BT were each identified by Wilcoxon test (P < 0.01).
Results
DNA methylation alterations in samples of both cancerous and non-cancerous renal cortex tissue obtained from patients with clear cell RCCs
Figure 1B and C shows examples of scanned array images and scattergrams of the signal ratios (test signal:reference signal), respectively, for normal renal cortex tissue from a patient without any primary renal tumor and both non-cancerous renal cortex tissue and cancerous tissue from a patient with clear cell RCC. In all normal renal cortex tissue samples (C1–C8), the signal ratios of 97% of the BAC clones were between 0.67 and 1.5 (red bars in Figure 1C). Therefore, in non-cancerous renal cortex tissue obtained from patients with clear cell RCCs and the clear cell RCCs themselves, DNA methylation status corresponding to a signal ratio of <0.67 and >1.5 was defined as DNA hypomethylation and DNA hypermethylation of each BAC clone compared with normal renal cortex tissue, respectively. In samples of non-cancerous renal cortex tissue obtained from patients with clear cell RCCs (N1–N51), many BAC clones showed DNA hypomethylation or DNA hypermethylation (panel N of Figure 1C). In clear cell RCCs themselves (T1–T51), more BAC clones showed DNA hypomethylation or DNA hypermethylation, and the degree of DNA hypomethylation and DNA hypermethylation, i.e. deviation of the signal ratio from 0.67 or 1.5, was increased in comparison with non-cancerous renal cortex tissue samples obtained from patients with clear cell RCCs (panel T of Figure 1C). The average number of BAC clones showing DNA hypomethylation increased significantly from non-cancerous renal cortex tissue samples obtained from patients with clear cell RCCs (93 ± 75) to clear cell RCCs (142 ± 74, P = 0.0002). The average number of BAC clones showing DNA hypermethylation also increased significantly in a similar manner (83 ± 73–123 ± 786, P = 0.004).
Unsupervised hierarchical clustering of patients with clear cell RCCs based on DNA methylation status of non-cancerous renal cortex tissue samples
By two-dimensional unsupervised hierarchical clustering analysis based on BAMCA data (signal ratios) for non-cancerous renal cortex tissue samples, the 51 patients with clear cell RCCs were clustered into two subclasses, Clusters AN and BN, which contained 46 and 5 patients, respectively (Figure 2A).
Fig. 2.

Two-dimensional unsupervised hierarchical clustering analysis based on BAMCA data (signal ratios) in non-cancerous renal cortex tissue samples showing no remarkable histological changes (A) and clear cell RCCs (C) and Kaplan–Meier survival curves of patients with clear cell RCCs (B and D). (A) Fifty-one patients with clear cell RCC were hierarchically clustered into two subclasses, Clusters AN (n = 46) and BN (n = 5), based on DNA methylation status of their non-cancerous renal cortex tissue samples. DNA hypomethylation, normomethylation (DNA methylation status corresponding to a signal ratio of between 0.67 and 1.5) and hypermethylation on each BAC clone are shown in green, black and red, respectively. The signal ratio is shown in the color range maps. The cluster trees for patients and BAC clones are shown at the top and left of the panel, respectively. (B) The overall survival rate of patients in Cluster BN (square) defined on the basis of DNA methylation status in their non-cancerous renal cortex tissue samples was significantly lower than that of patients in Cluster AN (circle) (P = 0.0000000613, Log-rank test). (C) Fifty-one patients were hierarchically clustered into two subclasses, Clusters AT (n = 43) and BT (n = 8), based on the DNA methylation status of their clear cell RCCs. (D) The overall survival rate of patients in Cluster BT (square) defined on the basis of DNA methylation status in their clear cell RCCs was significantly lower than that of patients in Cluster AT (circle) (P = 0.0000413, Log-rank test).
Table IA shows the clinicopathological parameters of clear cell RCCs of patients belonging to Clusters AN and BN. The corresponding clear cell RCCs of patients in Cluster BN showed more frequent macroscopically evident multinodular (type 3) growth, vascular involvement and renal vein tumor thrombi and showed higher pathological TNM stages than those in Cluster AN. Figure 2B shows the Kaplan–Meier survival curves of patients belonging to Clusters AN and BN. The period covered ranged from 88 to 2801 days (mean, 1679 days). Three (60%) of the patients in Cluster BN died of recurrent RCC, whereas only one (2%) of the patients in Cluster AN died. The overall survival rate of patients in Cluster BN was significantly lower than that of patients in Cluster AN (Figure 2B).
Table I.
Correlation between the subclassification of patients based on DNA methylation status and the clinicopathological parameters of clear cell RCCs
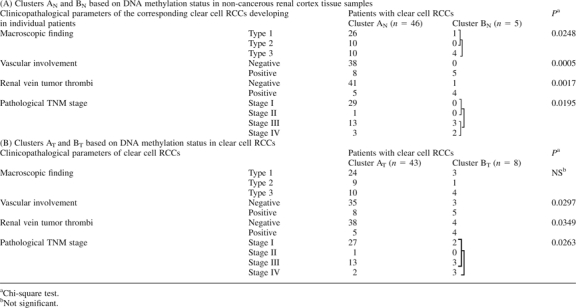 |
Although Cluster AN was divided into three subclusters, AN1 (n = 3), AN2 (n = 19) and AN3 (n = 24) (Figure 2A), there were no significant correlations between these subclusters and any of the clinicopathological parameters examined (data not shown). Even when unsupervised hierarchical clustering was performed separately, based not on signal ratios but on the presence or absence of DNA hypomethylation and the presence or absence of DNA hypermethylation, the majority of patients in Cluster BN were clustered into the same subclass (supplementary Figure S1A and B is available at Carcinogenesis Online).
Wilcoxon test (P < 0.01) revealed that the signal ratios of 1143 BAC clones in non-cancerous renal cortex tissue differed significantly between Clusters AN and BN: e.g. patients belonging to Cluster BN were completely discriminated from patients in Cluster AN by the DNA methylation status of samples of non-cancerous renal cortex tissue for representative BAC clones (Cluster AN versus Cluster BN in Figure 3A) out of the 1143 BAC clones.
Fig. 3.

(A) Scattergrams of the signal ratios in non-cancerous renal cortex tissue samples (Cluster AN versus Cluster BN) and in clear cell RCCs (Cluster AT versus Cluster BT) on representative BAC clones, RP11-71H20, RP11-453D5, RP11-444F15, RP11-3A9 and RP11-87P3. Using the cutoff values (CVs) described in each panel, patients belonging to Cluster BN were completely discriminated from patients in Cluster AN based on the DNA methylation status of non-cancerous renal cortex tissue samples. Using the cutoff value described in each panel, patients belonging to Cluster BT were completely discriminated from patients in Cluster AT based on the DNA methylation status of the clear cell RCCs. When the signal ratios of Cluster BN were lower than those of Cluster AN, the signal ratios of Cluster BT were also lower than those of Cluster AT (RP11-71H20, RP11-453D5, RP11-444F15 and RP11-87P3). When the signal ratios of Cluster BN were higher than those of Cluster AN, the signal ratios of Cluster BT were also higher than those in Cluster AT (RP11-3A9). (B) The signal ratios of non-cancerous renal cortex tissue (N) and clear cell RCC (T) for all 51 patients on a representative BAC clone (RP11-44F3). DNA methylation status in N was basically inherited in the corresponding T developing in the individual patient. Gray bar, patients belonging to Cluster AT; black bar, patients belonging to Cluster BT. The case numbers of patients belonging to Cluster BT (RCC1–RCC8) are also shown on the left side. Patients RCC6–RCC8 did not belong to Cluster BN, but later gained the same DNA methylation profiles as those of patients RCC1–RCC5 during the development of T from N, and joined Cluster BT.
Unsupervised hierarchical clustering based on DNA methylation status of clear cell RCCs
Two-dimensional unsupervised hierarchical clustering analysis based on BAMCA data (signal ratios) for clear cell RCCs was able to group 51 patients into two subclasses, Clusters AT and BT, which contained 43 and eight patients, respectively (Figure 2C).
Table IB shows the clinicopathological parameters of clear cell RCCs of patients belonging to Clusters AT and BT. Clear cell RCCs in Cluster BT showed more frequent vascular involvement and renal vein tumor thrombi and showed higher pathological TNM stages than those in Cluster AT. Figure 2D shows the Kaplan–Meier survival curves of patients belonging to Clusters AT and BT. Three (37.5%) of the patients in Cluster BT died due to recurrent RCCs, whereas only one (2.3%) of the patients in Cluster AT died. The overall survival rate of patients in Cluster BT was significantly lower than that of patients in Cluster AT (Figure 2D). Multivariate analysis revealed that our clustering was a predictor of recurrence and was independent of histological grade, macroscopic configuration, vascular involvement and renal vein tumor thrombi (Table II).
Table II.
Multivariate analysis of the clinicopathological parameters and the subclassification (Clusters AT and BT) based on DNA methylation status of cancerous tissue samples as predictors of recurrence
| Parameters | Hazard ratio (95% CI) | χ2 | P value |
| Histological grade | |||
| Grade 1, 2 or 3 | 1 (Reference) | ||
| Grade 4 | 118.582 (5.186–2711.249) | 8.947 | 0.0028 |
| Macroscopic configuration | |||
| Type 1 | 1 (Reference) | ||
| Type 2 | 5.309 (0.689–40.887) | 2.570 | 0.1089 |
| Type 3 | 0.820 (0.061–11.005) | 0.022 | 0.8808 |
| Vascular involvement | |||
| Negative | 1 (Reference) | ||
| Positive | 1.434 (0.098–20.932) | 0.070 | 0.7920 |
| Renal vein tumor thrombi | |||
| Negative | 1 (Reference) | ||
| Positive | 8.780 (0.429–179.734) | 1.990 | 0.1584 |
| Subclassification based on DNA methylation status | |||
| Cluster AT | 1 (Reference) | ||
| Cluster BT | 8.317 (1.100–62.901) | 4.211 | 0.0402 |
CI, confidence interval.
Although Cluster AT was divided into four subclusters, AT1 (n = 8), AT2 (n = 12), AT3 (n = 13) and AT4 (n = 10) (Figure 2B), there were no significant correlations between these subclusters and any of the clinicopathological parameters examined (data not shown). Even when unsupervised hierarchical clustering was performed separately, based not on signal ratios but on the presence or absence of DNA hypomethylation and the presence or absence of DNA hypermethylation, the majority of patients in Cluster BT were clustered into the same subclass (supplementary Figure S1C and D is available at Carcinogenesis Online).
Wilcoxon test (P < 0.01) revealed that the signal ratios of 1111 BAC clones in clear cell RCCs were differed significantly between Clusters AT and BT. In particular, patients belonging to Cluster BT were completely discriminated from patients belonging to Cluster AT based on the DNA methylation status of 14 BAC clones (Cluster AT versus Cluster BT in Figure 3A). In other words, DNA methylation status of the 14 BAC clones was able to determine whether or not patients in this cohort belonged to Cluster BT, a significant prognostic indicator, with a sensitivity and specificity of 100% using the cutoff values shown in Figure 3A and supplementary Table SI (available at Carcinogenesis Online). DNA methylation status of the 70 BAC clones, including the above 14 BAC clones, was able to determine whether or not the patients in this cohort belonged to Cluster BT, with a sensitivity of 100% and a specificity of 90 or >90%, using the cutoff values shown in supplementary Table SI (available at Carcinogenesis Online).
Comparison between DNA methylation profiles of non-cancerous renal tissue and those of corresponding clear cell RCC
Patients RCC1–RCC5 and patients RCC1–RCC8 were identified as belonging to Clusters BN and BT, respectively, by unsupervised hierarchical clustering based on BAMCA data for non-cancerous renal cortex tissue samples and clear cell RCCs. Namely, Cluster BN (n = 5) was completely included in Cluster BT (n = 8). The 724 BAC clones, the majority of the 1143 BAC clones significantly discriminating Cluster BN from Cluster AN, also discriminated Cluster BT from Cluster AT (Wilcoxon test, P < 0.01). In 311 of the 724 BAC clones, where the average signal ratio of Cluster BN was higher than that of Cluster AN, the average signal ratio of Cluster BT was also higher than that of Cluster AT without exception (Figure 3A). In 413 of the 724 BAC clones, where the average signal ratio of Cluster BN was lower than that of Cluster AN, the average signal ratio of Cluster BT was also lower than that of Cluster AT without exception (Figure 3A). Figure 3B shows the signal ratios of non-cancerous renal cortex tissue samples and clear cell RCCs for all 51 patients for a representative BAC clone (RP11-44F3). In individual patients, DNA methylation status in the non-cancerous renal cortex tissue was basically inherited by the corresponding clear cell RCC (Figure 3B).
Discussion
Many researchers in this field use arrays in which the promoter regions are enriched as probes to identify the genes methylated in cancer cells (8–10). However, the promoter regions of specific genes are not the only target of DNA methylation alterations in human cancers. DNA methylation status in genomic regions not directly participating in gene silencing, such as the edges of CpG islands, may be altered at the precancerous stage before the alterations of the promoter regions themselves occur (22). Genomic regions in which DNA hypomethylation affects chromosomal instability may not be contained in promoter arrays. Moreover, aberrant DNA methylation of large regions of chromosomes, which are regulated in a coordinated manner in human cancers due to a process of long-range epigenetic silencing, has recently attracted attention (23). Therefore, we used a custom-made BAC array (16) that may be suitable, not for focusing on specific promoter regions or individual CpG sites but for overviewing the DNA methylation status of individual large regions among all chromosomes and for subclassifying cancers by hierarchical clustering.
With respect to renal carcinogenesis, several studies of DNA methylation profiles of genes involved in specific signal pathways in clear cell RCCs, such as the p53-signaling (24) and Wnt-signaling (25) pathways, have been performed. However, to our knowledge, there have been no published data on DNA methylation profiles for all chromosomes in clear cell RCCs revealed by array-based technology. In our previous study, we showed that samples of non-cancerous renal cortex tissue from patients with clear cell RCC were already at the precancerous stage with accumulation of DNA methylation on C-type CpG islands, in spite of an absence of marked histological changes (6,7,12). In the present study, genome-wide DNA methylation alterations (both hypomethylation and hypermethylation) in samples of non-cancerous renal cortex tissue from patients with clear cell RCC were confirmed by BAMCA (panel N of Figure 1B and C). We then performed unsupervised hierarchical clustering analysis based on the genome-wide DNA methylation status of the non-cancerous renal cortex tissue samples, and as a result, 51 patients were subclassified into Clusters AN and BN. Corresponding clear cell RCCs showing multinodular growth, vascular involvement, renal vein tumor thrombi and higher pathological TNM stages were found to be accumulated in Cluster BN. Although subclassification of precancerous tissue by unsupervised hierarchical clustering analysis on the basis of genome-wide DNA methylation profiles has never been performed for specific organs, our Clusters AN and BN can be considered clinicopathologically valid.
The significant correlation between genome-wide DNA methylation profiles of samples of non-cancerous renal cortex tissue and aggressiveness of cancers developing in individual patients indicated that it may be possible to estimate the future risk of developing more malignant cancers based on genome-wide DNA methylation status at the precancerous stage. Although kidney biopsy sampling for screening of healthy individuals is not clinically feasible because of its invasive nature, carcinogenetic risk estimation using urine, sputum and other body fluid samples may be a promising approach if optimal indicators can be identified by genome-wide DNA methylation profiling at the precancerous stage in the urinary tract, lung and other organs. Patients belonging to Cluster BN showed poorer outcome than those in Cluster AN, indicating that even patient outcome is determined by DNA methylation status at the precancerous stage.
Although altered DNA methylation on several CpG islands has been reported separately in RCCs (26–28), subclassification of clear cell RCCs, which may reflect the distinct epigenetic pathways of carcinogenesis, has never been established on the basis of genome-wide DNA methylation profiling. Since clear cell RCCs showing a higher incidence of vascular involvement, renal vein tumor thrombi and higher pathological TNM stages were accumulated in Cluster BT, our Clusters AT and BT can be considered clinicopathologically valid. In our previous studies, we examined DNA methylation status on CpG islands for the p16, hMLH 1, VHL and THBS 1 genes, and the methylated in tumor-1, -2, -12, -25 and -31 clones were examined in the same 51 clear cell RCCs (12,29). Correlations between DNA methylation status on each CpG island and our clustering are summarized in supplementary Table SII (available at Carcinogenesis Online). The average number of methylated CpG islands was significantly higher in Cluster BT (2.75 ± 1.67) than in Cluster AT (1.54 ± 0.98, P = 0.01867318). Patients were considered to be positive for the CpG island methylator phenotype when DNA methylation was seen on three or more examined CpG islands, based on previously described criteria (11). The frequency of CpG island methylator phenotype in Cluster BT (62.5%) was significantly higher than that in Cluster AT (16%, P = 0.0174969). Genome-wide DNA methylation alterations consisting of both hypomethylation and hypermethylation of DNA revealed by BAMCA in Cluster BT are associated with regional DNA hypermethylation on CpG islands and participate in malignant progression of clear cell RCCs. Moreover, patients belonging to Cluster BT showed poorer outcome than those in Cluster AT, indicating that prognostication of clear cell RCCs using DNA methylation status as an indicator is a promising approach.
Some RCCs relapse and metastasize to distant organs, even if resection has been considered complete (17,30). Recently, immunotherapy (31) and novel targeting agents (32) have been developed for treatment of RCC. However, unless relapsed or metastasized tumors are diagnosed early by close follow-up, the effectiveness of any therapy is very restricted. Therefore, to assist close follow-up of patients who have undergone nephrectomy and are still at risk of recurrence and metastasis, prognostic indicators have been explored. Multivariate analysis revealed that belonging to Cluster BT was an independent predictor of recurrence. It is known that sarcomatoid RCCs with grade 4 atypia frequently show recurrence (18). However, patients with RCCs showing grade 1–3 atypia also suffer recurrence, and we cannot estimate the risk of recurrence of such RCCs based on known parameters. Belonging to Cluster BT is advantageous even to patients with RCCs showing grade 1–3 atypia because it is a predictor of recurrence that is independent of histological grading. For clinical application, a combination of several BAC clones from the 70 that showed 100% sensitivity and 90 or >90% specificity (including 14 BAC clones showing 100% sensitivity and 100% specificity) can be of optimal prognostic value for patients with clear cell RCCs. Since a sufficient quantity of good-quality DNA can be obtained from each nephrectomy specimen, polymerase chain reaction-based analyses focusing on individual CpG sites are not always required. Array-based analysis that overviews aberrant DNA methylation of each BAC region is immediately applicable to routine laboratory examinations for prognostication after nephrectomy. We are currently attempting to prepare a mini-array harboring some of the 70 BAC clones. The reliability of such prognostication will need to be validated in a prospective study.
We have clarified that genome-wide DNA methylation profiles of non-cancerous renal cortex tissue are inherited by the corresponding clear cell RCC based on the following findings: (i) all patients belonging to Cluster BN were included in Cluster BT; (ii) a majority of the BAC clones characterizing Cluster BN (724 BAC clones) also characterized Cluster BT; (iii) DNA methylation status on such 724 BAC clones of non-cancerous renal cortex tissue in Cluster AN was in accordance with that of clear cell RCCs in Cluster AT and that of non-cancerous renal cortex tissue in Cluster BN was in accordance with that of clear cell RCCs in Cluster BT (Figure 3A) and (iv) DNA methylation status in non-cancerous renal cortex tissue basically corresponded to that in the matching clear cell RCC in each patient (Figure 3B).
Patients RCC6–RCC8 who belonged to Cluster BT but not to Cluster BN may later gain the DNA methylation profiles observed in patients RCC1–RCC5 during the establishment of clear cell RCCs (Figure 3B) and suffer from the same degree of tumor aggressiveness as patients RCC1–RCC5. Although alterations of DNA methylation are considered to be involved even in the precancerous stage in various organs (6,7,33–35), it has not yet been clarified for any organ whether DNA methylation status on only a restricted number of CpG islands is simply altered at such stages or whether genome-wide alterations of DNA methylation status have certain clinicopathological significance. The present unsupervised hierarchical clustering revealed for the first time that DNA methylation alterations in precancerous conditions, which may not occur randomly but are prone to further accumulation of genetic and epigenetic alterations, can generate more malignant cancers and even determine the ultimate patient outcome.
Supplementary material
Supplementary Figure S1 and Tables SI and SII can be found at http://carcin.oxfordjournals.org/
Funding
Grant-in-Aid for the Third Term Comprehensive 10-Year Strategy for Cancer Control from the Ministry of Health, Labor and Welfare of Japan; Grant-in-Aid for Cancer Research from the Ministry of Health, Labor and Welfare of Japan; New Energy and Industrial Technology Development Organization; Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation. Funding to pay the Open Access publication charges for this article was provided by a Grant-in-Aid for the Third Term Comprehensive 10-Year Strategy for Cancer Control from the Ministry of Health, Labor and Welfare of Japan.
Supplementary Material
Acknowledgments
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- BAC
bacterial artificial chromosome
- BAMCA
bacterial artificial chromosome array-based methylated CpG island amplification
- RCC
renal cell carcinoma
- TNM
tumor–node–metastasis
References
- 1.Jones PA, et al. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 2.Eden A, et al. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455. doi: 10.1126/science.1083557. [DOI] [PubMed] [Google Scholar]
- 3.Baylin SB, et al. Epigenetic gene silencing in cancer—a mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer. 2006;6:107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 4.Gronbaek K, et al. Epigenetic changes in cancer. APMIS. 2007;115:1039–1159. doi: 10.1111/j.1600-0463.2007.apm_636.xml.x. [DOI] [PubMed] [Google Scholar]
- 5.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 6.Kanai Y, et al. Alterations of DNA methylation associated with abnormalities of DNA methyltransferases in human cancers during transition from a precancerous to a malignant state. Carcinogenesis. 2007;28:2434–2442. doi: 10.1093/carcin/bgm206. [DOI] [PubMed] [Google Scholar]
- 7.Kanai Y. Alterations of DNA methylation and clinicopathological diversity of human cancers. Pathol. Int. 2008;58:544–558. doi: 10.1111/j.1440-1827.2008.02270.x. [DOI] [PubMed] [Google Scholar]
- 8.Estecio MR, et al. High-throughput methylation profiling by MCA coupled to CpG island microarray. Genome Res. 2007;17:1529–1536. doi: 10.1101/gr.6417007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacinto FV, et al. Discovery of epigenetically silenced genes by methylated DNA immunoprecipitation in colon cancer cells. Cancer Res. 2007;67:11481–11486. doi: 10.1158/0008-5472.CAN-07-2687. [DOI] [PubMed] [Google Scholar]
- 10.Nielander I, et al. Combining array-based approaches for the identification of candidate tumor suppressor loci in mature lymphoid neoplasms. APMIS. 2007;115:1107–1134. doi: 10.1111/j.1600-0463.2007.apm_883.xml.x. [DOI] [PubMed] [Google Scholar]
- 11.Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc. Natl Acad. Sci. USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arai E, et al. Regional DNA hypermethylation and DNA methyltransferase (DNMT) 1 protein overexpression in both renal tumors and corresponding nontumorous renal tissues. Int. J. Cancer. 2006;119:288–296. doi: 10.1002/ijc.21807. [DOI] [PubMed] [Google Scholar]
- 13.Misawa A, et al. Methylation-associated silencing of the nuclear receptor 1I2 gene in advanced-type neuroblastomas, identified by bacterial artificial chromosome array-based methylated CpG island amplification. Cancer Res. 2005;65:10233–10242. doi: 10.1158/0008-5472.CAN-05-1073. [DOI] [PubMed] [Google Scholar]
- 14.Sugino Y, et al. Epigenetic silencing of prostaglandin E receptor 2 (PTGER2) is associated with progression of neuroblastomas. Oncogene. 2007;26:7401–7413. doi: 10.1038/sj.onc.1210550. [DOI] [PubMed] [Google Scholar]
- 15.Tanaka K, et al. Frequent methylation-associated silencing of a candidate tumor-suppressor, CRABP1, in esophageal squamous-cell carcinoma. Oncogene. 2007;26:6456–6468. doi: 10.1038/sj.onc.1210459. [DOI] [PubMed] [Google Scholar]
- 16.Inazawa J, et al. Comparative genomic hybridization (CGH)-arrays pave the way for identification of novel cancer-related genes. Cancer Sci. 2004;95:559–563. doi: 10.1111/j.1349-7006.2004.tb02486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eble JN, et al. Renal Cell Carcinoma. World Health Organization Classification of Tumours. Pathology and Genetics. Tumours of the Urinary System and Male Genital Organs. Lyon: IARC Press; 2004. pp. 10–43. [Google Scholar]
- 18.Fuhrman SA, et al. Prognostic significance of morphologic parameters in renal cell carcinoma. Am. J. Surg. Pathol. 1982;6:655–663. doi: 10.1097/00000478-198210000-00007. [DOI] [PubMed] [Google Scholar]
- 19.Sobin LH, et al. International Union Against Cancer (UICC). TNM Classification of Malignant Tumors, 6th edn. New York, NY: Wiley-Liss; 2002. pp. 193–195. [Google Scholar]
- 20.Kanai T, et al. Pathology of small hepatocellular carcinoma. A proposal for a new gross classification. Cancer. 1987;60:810–819. doi: 10.1002/1097-0142(19870815)60:4<810::aid-cncr2820600417>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 21.Illingworth R, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6:e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maekita T, et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin. Cancer Res. 2006;12:989–995. doi: 10.1158/1078-0432.CCR-05-2096. [DOI] [PubMed] [Google Scholar]
- 23.Frigola J, et al. Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat. Genet. 2006;38:540–549. doi: 10.1038/ng1781. [DOI] [PubMed] [Google Scholar]
- 24.Christoph F, et al. Promoter hypermethylation profile of kidney cancer with new proapoptotic p53 target genes and clinical implications. Clin. Cancer Res. 2006;12:5040–5046. doi: 10.1158/1078-0432.CCR-06-0144. [DOI] [PubMed] [Google Scholar]
- 25.Urakami S, et al. Wnt antagonist family genes as biomarkers for diagnosis, staging, and prognosis of renal cell carcinoma using tumor and serum DNA. Clin. Cancer Res. 2006;12:6989–6997. doi: 10.1158/1078-0432.CCR-06-1194. [DOI] [PubMed] [Google Scholar]
- 26.Ibanez de Caceres I, et al. Identification of novel target genes by an epigenetic reactivation screen of renal cancer. Cancer Res. 2006;66:5021–5028. doi: 10.1158/0008-5472.CAN-05-3365. [DOI] [PubMed] [Google Scholar]
- 27.Costa VL, et al. Quantitative promoter methylation analysis of multiple cancer-related genes in renal cell tumors. BMC Cancer. 2007;7:133. doi: 10.1186/1471-2407-7-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris MR, et al. Functional epigenomics approach to identify methylated candidate tumour suppressor genes in renal cell carcinoma. Br. J. Cancer. 2008;98:496–501. doi: 10.1038/sj.bjc.6604180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arai E, et al. Genetic clustering of clear cell renal cell carcinoma based on array-CGH: its association with DNA methylation alteration and patient outcome. Clin. Cancer Res. 2008;14:5531–5539. doi: 10.1158/1078-0432.CCR-08-0443. [DOI] [PubMed] [Google Scholar]
- 30.Jones J, et al. Genomics of renal cell cancer: the biology behind and the therapy ahead. Clin. Cancer Res. 2007;13:685s–692s. doi: 10.1158/1078-0432.CCR-06-1867. [DOI] [PubMed] [Google Scholar]
- 31.Guida M, et al. Immunotherapy for metastatic renal cell carcinoma: is it a therapeutic option yet? Ann. Oncol. 2007;18(suppl. 6):vi149–52. doi: 10.1093/annonc/mdm245. [DOI] [PubMed] [Google Scholar]
- 32.Patel PH, et al. Targeting von Hippel-Lindau pathway in renal cell carcinoma. Clin. Cancer Res. 2006;12:7215–7520. doi: 10.1158/1078-0432.CCR-06-2254. [DOI] [PubMed] [Google Scholar]
- 33.Kanai Y, et al. Aberrant DNA methylation on chromosome 16 is an early event in hepatocarcinogenesis. Jpn. J. Cancer Res. 1996;87:1210–1217. doi: 10.1111/j.1349-7006.1996.tb03135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eguchi K, et al. DNA hypermethylation at the D17S5 locus in non-small cell lung cancers: its association with smoking history. Cancer Res. 1997;57:4913–4915. [PubMed] [Google Scholar]
- 35.Peng DF, et al. DNA methylation of multiple tumor-related genes in association with overexpression of DNA methyltransferase 1 (DNMT1) during multistage carcinogenesis of the pancreas. Carcinogenesis. 2006;27:1160–1168. doi: 10.1093/carcin/bgi361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.