

Abstract
We have found conditions for saturation mutagenesis by restriction enzyme mediated integration that result in plasmid tagging of disrupted genes. Using this method we selected for mutations in genes that act at checkpoints downstream of the intercellular signaling system that controls encapsulation in Dictyostelium discoideum. One of these genes, mkcA, is a member of the mitogen-activating protein kinase cascade family while the other, regA, is a novel bipartite gene homologous to response regulators in one part and to cyclic nucleotide phosphodiesterases in the other part. Disruption of either of these genes results in partial suppression of the block to spore formation resulting from the loss of the prestalk genes, tagB and tagC. The products of the tag genes have conserved domains of serine proteases attached to ATP-driven transporters, suggesting that they process and export peptide signals. Together, these genes outline an intercellular communication system that coordinates organismal shape with cellular differentiation during development.
Keywords: Dictyostelium discoideum, saturation mutagenesis, second site suppressors, proteasome, mitogen-activated protein kinase cascade
Development of a well-proportioned multicellular organism relies on concurrent differentiation of individual cells. In Dictyostelium discoideum, terminal differentiation into spores and stalk cells is coordinated in both time and space by an intercellular communication system. Two closely related prestalk genes, tagB and tagC, are necessary to produce a signal that passes from prestalk cells to prespore cells during culmination (ref. 1; G.S. and W.F.L., unpublished data). Null mutations in either gene lead to a cell-autonomous defect in prestalk differentiation such that the signal for sporulation is not produced. Strains carrying these mutations aggregate normally and differentiate into the initial prestalk cells and prespore cells. However, the prestalk cells fail to differentiate further or form the tips that normally appear after mound formation. Therefore, prespore cells never receive the signal to encapsulate. However, sporulation of mutant cells can be induced by developing them in chimeric mixtures where the wild-type cells in the apical prestalk region (PST-A cells) can generate the signal for encapsulation (1).
Both tagB and tagC encode composite proteins in which a serine–protease homology domain is contiguous with a domain homologous to ATP-driven transporters. Combinations of proteases and peptide transporters have been previously found to function in intercellular communication systems in both yeast and mammals. Processing and secretion of the a mating factor in Saccharomyces cerevisiae involves two proteases, AXL1 and STE23 (2), as well as the transporter, STE6 (3–5). Major histocompatibility complex (MHC) class I presentation of intracellular epitopes on mammalian plasma membranes involves degradation of cytoplasmic proteins by proteasomes and subsequent transport of the peptides by the TAP-1 and TAP-2 transporters (6–12). The primary sequence of TagB and TagC suggests that they are involved in proteolysis and secretion of a peptide signal from prestalk cells that diffuses to adjacent prespore cells (ref. 1; G.S. and W.F.L., unpublished data), but the nature of the signal itself and the mechanism that receives and integrates it in prespore cells are still unknown.
Second-site suppressor analysis is a powerful genetic method that can be used to recognize components of complex genetic pathways. Commonly, genetic suppressors overcome the consequences of the primary mutation by modifying an interacting or a downstream component in the defective pathway. In Dictyostelium, the method of choice for mutagenesis is restriction enzyme mediated integration (REMI) (13), which generates null alleles by insertion of linearized plasmid DNA into restriction sites within genes in vivo. Suppressors generated by null mutations are expected to reveal genes that normally function as negative components downstream of the primary defect. To collect all such mutations that can suppress the block to sporulation in tagB− mutants, we developed a method for saturation mutagenesis of nonvital genes. We selected for spores from 105 independently transformed cells and found several strains in which the signal transduction mechanism that gates encapsulation of prespore cells is no longer dependent on the signal from prestalk cells.
METHODS
Saturation REMI Mutagenesis and Suppressor Screens.
A REMI vector, pBSR1, was made by cloning the blasticidin S resistance cassette (14) into the PvuII site of pGEM-3 (Promega). The BglII site was removed by site directed mutagenesis and a unique BamHI linker replaced the pGEM-3 multiple cloning site. tagB− cells (1) were grown to 4 × 106 cells per ml in liquid HL-5 medium (15) supplemented with 0.2 μg/ml folic acid and 0.6 μg/ml cyanocobalamin (Sigma). Cells were cooled on ice for 15 min, harvested by centrifugation, and resuspended at 1 × 107 cells per ml in ice-cold electroporation buffer (13). Aliquots (20 ml) of cells were mixed with 10 μg/ml of BamHI-linearized pBSR1 DNA and 30 units/ml of DpnII restriction endonuclease (New England Biolabs) immediately before electroporation. Aliquots (0.8 ml) were electroporated in 0.4-cm gap cuvettes (BTX, San Diego) with a Bio-Rad Gene Pulser set at 1.0 kV and 3 μF and transferred immediately into HL-5. Cells were plated at 3 × 106 per 10 cm plate (Falcon) and incubated for 24 h. Blasticidin S (10 μg/ml) was added and cells were incubated for 6 more days. Drug-resistant cells were collected, counted, and plated at 0.5 to 5.0 × 103 cells per 10-cm plate on SM agar in association with Klebsiella aerogenes (15). Plates were screened for mutants with morphologies more advanced than the original tagB− mutant. After 8 days, cells were collected, treated with 0.3% cemulsol NP12 (SFOS, Persan, France) in 20 mM potassium phosphate buffer (pH 6.2), centrifuged, and washed with buffer without detergent. Pellets were resuspended and plated on SM agar in association with K. aerogenes (15) to recover detergent-resistant strains.
Nucleic Acid Manipulation.
Cloning DNA sequences flanking the insertion sites and recapitulation of mutations by homologous recombination were performed as described (13). Riboprobes were made from the respective plasmids that were linearized with the restriction enzyme used for cloning. cDNA molecules were cloned as described (1). DNA sequencing was performed with an Applied Biosystems Prism 377 DNA sequencer. Purification of nucleic acids, Southern blot analysis, and Northern blot analysis were performed as described (16). RNA in situ hybridization was performed as described (17, 18) using cDNA riboprobes as indicated in Fig. 1.
Figure 1.

Sequence analysis of mkcA and regA. (A) Sequenced regions of mkcA and regA genomic DNA are shown. Coding regions are indicated in boxes, and V shapes indicate introns. Plasmid (4.5 kb, not to scale) insertion sites (IS) in the respective mutant strains are indicated as triangles. Solid box in mkcA is the kinase homology domain. Thatched boxes in regA encode the response regulator homology domain, and checked boxes encode the cyclic nucleotide phosphodiesterase homology domain. Gray boxes under the genes indicate cDNA probes. (B) Sequence similarity between the putative protein kinase domain of mkcA and those of the mitogen-activating protein (MAP) kinase cascade genes PAK65 (Swiss-Prot Protein Sequence Data Bank no. P35465P35465) and STE20 (GenBank accession no. L04655L04655). (C) Sequence similarity between the putative response regulator domain of regA and those of the bacterial response regulators cheY [National Center for Biotechnology Information (NCBI) no. 145525], ntrC (Swiss-Prot Protein Sequence Data Bank no. P10576P10576), and pleD (NCBI no. 1119215). (D) Sequence similarity between the putative cyclic nucleotide phosphodiesterase domain of regA and those of a rat phosphodiesterase (r.PDE; NCBI no. 436012) and a bovine calmodulin stimulated phosphodiesterase (b.PDE; NCBI no. 533781). Conserved regions are underlined, and amino acid numbers are indicated on the left.
Quantitation of Sporulation.
Cells were developed on nitrocellulose filters as described (16). Sporulation efficiency was determined by detergent treatment of 36- to 48-h developed cells and plating on SM agar in association with K. aerogenes (15). The number of plaques in the bacterial lawn indicated the number of viable spores.
RESULTS
Saturation REMI Mutagenesis and Selection for Suppressors.
Using a plasmid carrying the blasticidin resistance gene, we found that it was possible to generate 105 independent transformants by electroporating BamHI-cut plasmid DNA along with the restriction enzyme DpnII into about 109 tagB null cells. Because each electroporation introduced DNA and enzyme into 8 × 107 cells, we had to carry out 110 electroporations. However, this could be accomplished within a few hours. The cells were then plated and subjected to selection for resistance to blasticidin S. We recovered 105 independent drug resistant survivors, indicating that the efficiency of transformation was about 1:104. This technique can be used to generate populations with near saturation mutagenesis of the nonvital genes in Dictyostelium that could be screened for any desired developmental genes.
A population of 105 independent blasticidin S-resistant transformants of the tagB− strain was plated on SM agar in association with K. aerogenes to allow each transformant to generate a clone which could develop. The clones were visually screened after a week and 12 were picked because they formed multicellular structures more advanced than the mounds formed by tagB null strains. The remainder of the transformants were collected to select for those that could form spores by resuspending them in 0.3% cemulsol which kills cells but not spores. Spores were pelleted from the detergent lysates and plated on SM agar plates in association with K. aerogenes. Three to 7 days later, plaques appeared in the bacterial lawns, representing suppressor strains that sporulated and survived the detergent treatment. Six independently derived strains were isolated and examined for the presence of spores by phase contrast microscopy and tested for spore viability following detergent treatment. Genomic DNA fragments flanking the insertion sites in these strains were cloned and used as probes for Southern blot analysis (data not shown). Four of the six sporulation suppressors were found to have suffered an insertion into one gene, regA, indicating that the original population carried multiple disruptions of this developmental gene. Each of the other two sporulation suppressors, mkcA and STB18, was found only once. Another gene, prtC, was found in two independent suppressor strains that were picked by visual inspection of the surviving transformants. However, neither of these strains carrying partial morphological suppressors formed spores. prtC is a novel Dictyostelium gene homologous to the human C2-proteasome subunit (19) and its sequence was deposited in the GenBank data base. Our recovery of multiple independent insertions in both regA and in prtC indicate that the mutagenesis procedure resulted in near saturation.
Sporulation in Suppressors of tagB.
Mutational insertions in mkcA or in regA were recapitulated by homologous recombination in a fresh tagB null host as well as in tagC null cells and in wild-type AX4 cells. Sporulation efficiencies of the respective strains are shown in Table 1. To determine the sporulation efficiency of the parental tagB null or tagC null strains, we developed 1010 or 109 cells, respectively, and selected for spores following detergent treatment. In neither case did we detect a single viable spore. In the mkcA− tagB− double mutant, about one cell per aggregate (105 cells) had encapsulated, and the same ratio was observed when the mkcA gene was disrupted in a tagC null background (Table 1). Although this sporulation efficiency is low, it represents at least 104-fold increase relative to the parental strain. Disruption of regA in a tagB null or in a tagC null background resulted in higher levels of sporulation, up to 30% of the wild-type levels in the case of the tagB− regA− double mutants (Table 1). The number of spores produced by the single regA− or mkcA− mutants were almost indistinguishable from the wild-type number (Table 1). While disruption of mkcA results in only partial suppression of the sporulation defect in tagB− or tagC− strains, disruption of regA results in very effective suppression. The results presented in Table 1 indicate that in the wild type, both mkcA and regA function as negative regulators of sporulation. This is consistent with the notion that suppressors generated by insertional mutagenesis should reveal genes that normally function as negative components in the mutated pathway.
Table 1.
Sporulation efficiency
| Genotype | Viable spores per aggregate, 105 cells |
|---|---|
| Wild type | 105 |
| mkcA− | 105 |
| regA− | 5 × 104 |
| tagB− | 0 |
| tagC− | 0 |
| tagB− mkcA− | 1 |
| tagC− mkcA− | 1 |
| tagB− regA− | 3 × 104 |
| tagC− regA− | 7 × 103 |
As indicated above, the first step of mutagenesis resulted in near saturation. The second step of selection for spores was dependent on the sporulation efficiency. Therefore, the difference in sporulation efficiency between the mkcA− tagB− double mutants and the regA− tagB− double mutants (Table 1) may account for the difference in the number of independent suppressor strains we found after the detergent selection step.
Sequence Similarity of mkcA and regA to Signal Transduction Genes.
The gene structures of mkcA and regA are shown in Fig. 1A. Genomic and cDNA clones were sequenced and the deduced amino acid sequences were compared with various protein data bases using blast (20). Fig. 1B shows that mkcA is a member of the MAP kinase cascade gene family which includes the yeast STE20 and the mammalian PAK65 genes. The predicted carboxy terminus of MkcA contains a kinase homology region (Fig. 1B). The amino terminus does not contain previously recognized sequences, and as in STE20 (21), it may be important for regulating the kinase activity.
In regA we found two domains with significant similarity to two distinct gene families. The 5′ end of regA encodes a response-regulator domain (Fig. 1 A and C) similar to those of the bacterial genes ntrC, cheY, and pleD (22, 23). The 3′ end is similar to members of the cyclic nucleotide phosphodiesterase gene family (24) (Fig. 1 A and D), but is clearly distinct from pdsA, the only other published phosphodiesterase gene of Dictyostelium (25). Regulation of cyclic nucleotide phosphodiesterase activity by protein serine-threonine kinases has been demonstrated in many systems (26), and regulation of catalytic enzyme activity by a two-component system was found in the bacterial methylesterase CheB (27). It is therefore likely that the structural linkage between the response regulator and the phosphodiesterase domains in regA signifies a similar functional relationship.
A phosphodiesterase regulated by a histidine kinase in a two-component system could function in previously unsuspected signal transduction pathways mediated by the internal levels of cyclic nucleotides. Our data do not directly indicate a connection between the roles of mkcA and regA. They may function independently or be linked in a pathway, but because a correlation has been established between MAP kinase cascades and two-component systems in other eukaryotes (28–32), it is not implausible that mkcA and regA act as components of a network that functions to gate sporulation.
Effects of mkcA and regA on Cell Type Differentiation.
Null mutations in either tagB or tagC lead to a cell autonomous defect in prestalk differentiation and a 5-fold reduction in the expression of the prestalk gene ecmA. There is no significant effect on expression of the spore coat gene cotB, indicating that, although spores are not made, differentiation of prespore cells is essentially normal (1). We compared the levels of ecmA and cotB gene expression as well as the expression of another prestalk gene, ecmB, in the parental strains and in the respective mkcA or regA null strains. Northern blot analysis revealed that in both mkcA nulls and in regA nulls the level of prestalk gene expression was markedly reduced whereas the expression of the prespore marker cotB was not affected (Fig. 2A). A similar pattern was observed in the suppressor double mutants (data not shown). Despite the low level of expression of the prestalk genes, we were able to detect their mRNAs by in situ hybridization. In mkcA null cells there was no significant difference from the wild-type pattern of accumulation of either ecmA or ecmB mRNA (Fig. 2B). In regA null strains the stalks were short and the lower cup structure was compromised as seen by the lack of hybridization with ecmB (arrow in Fig. 2B). The level of ecmB expression in the upper cup was also reduced (Fig. 2B). The reduced level of prestalk gene expression in both mkcA− and regA− strains and the defects in stalk formation in regA− strains (Fig. 2) indicate that mkcA and regA have functions in prestalk differentiation as well as in encapsulation of prespore cells. We therefore determined the cell type specificity and the developmental regulation of the regA and mkcA gene expression.
Figure 2.
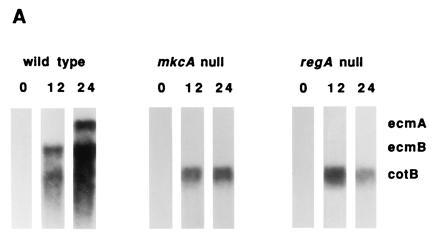
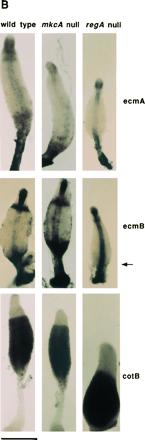
Expression of cell type-specific genes in mkcA null or regA null strains. (A) RNA from wild-type cells and from mutants was prepared at 0, 12, and 24 h of development as indicated. Northern blots were hybridized with cotB, a prespore-specific gene probe and with ecmA and ecmB, two prestalk-specific gene probes. (B) Wild-type or mutant cells were developed to early culmination and subjected to in situ RNA hybridization with riboprobes for ecmA, ecmB, and cotB as indicated. Arrow shows the lower cup. (Bar = 0.2 mm.)
Developmental Regulation of mkcA and regA mRNA Expression.
cDNA probes from mkcA and from regA (Fig. 1A) were used to follow the accumulation of the respective mRNA during development of wild-type cells. The 3.0-kb mkcA mRNA was expressed at constant levels during growth and throughout development (Fig. 3A). The 3.0-kb regA mRNA was weakly expressed in vegetative cells. By 4 h it accumulated to its maximal level which persisted throughout development (Fig. 3A). Using RNA in situ hybridization to wild-type cells we found that mkcA was uniformly expressed throughout the prespore and the prestalk regions at the finger stage as well as during culmination (Fig. 3B). regA mRNA was uniformly distributed at the finger stage, but during culmination it was enriched in the prestalk upper cup region (Fig. 3B). The results presented in Figs. 2 and 3 support the possibility that mkcA and regA can affect sporulation in a cell autonomous manner and may have an additional function in prestalk cells.
Figure 3.
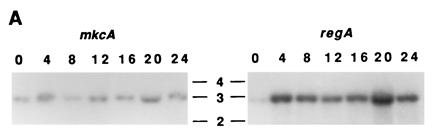
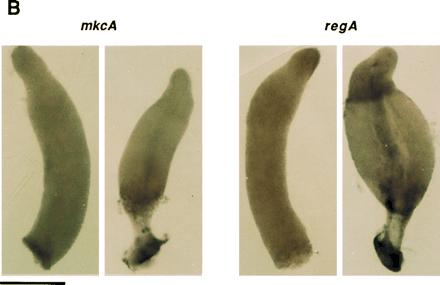
Regulation and cell type specificity of mkcA and regA expression. (A) RNA was prepared from developing wild-type cells at 4-h intervals and analyzed by Northern blot analysis using cDNA riboprobes for mkcA or regA (Fig. 1) as indicated. Time (h) is indicated above the lanes; size (kb) is indicated between blots. (B) Wild-type cells were developed to the finger stage or to early culmination and subjected to in situ RNA hybridization with cDNA riboprobes for mkcA or regA as indicated. (Bar = 0.2 mm.)
DISCUSSION
mkcA and regA were selected as suppressors of the tagB− intercellular signaling defect, implying that they normally function as negative regulators of sporulation to coordinate encapsulation with culmination. The inhibition in prespore cells is lifted when prestalk cells enter culmination and release a signal by a mechanism that depends on tagB and tagC. This gating mechanism, coupling morphological progression with cellular differentiation and sporulation, was previously implied by the expression pattern of the sporulation-specific gene spiA (33).
By primary sequence, tagB and tagC are likely to act as proteases as well as membrane transporters to facilitate the secretion of a peptide signal from prestalk cells (ref. 1; G.S. and W.F.L., unpublished data). mkcA and regA are related to protein kinase signal transduction genes. Systems that combine peptide processing and membrane transport in signal generation and kinase cascades in signal transduction have been observed in other organisms. Table 2 demonstrates similarities between molecular strategies in intercellular signaling systems of Dictyostelium, yeast, and mammals. The precursor protein for the yeast mating pheromone a factor is the gene product of MFA1 and MFA2. The precursor is processed by two proteases, AXL1 and STE23 and secreted by the multiple drug resistance (MDR)-related gene product of STE6 in a cells (2, 5). The peptide interacts with the STE3 membrane receptor on α cells and the signal is integrated by a MAP kinase cascade that includes the mkcA homologue STE20 (34). Recognition of intracellular antigens by cytotoxic T lymphocytes (CTL) in mammals is mediated by a similar mechanism. Cytoplasmic proteins are processed into small peptides by proteasomes (6) and transported into the pre-Golgi by the MDR-related heterodimeric TAP1–TAP2 transporter. The peptides associate with an MHC class I molecule and are eventually presented on the cell surface (7–12). Ligation of the CTL T-cell receptor with MHC class I-associated peptides leads to activation of a MAP kinase cascade via activation of the Lck and Raf-1 protein kinases (35). Thus, the signaling system we have discovered in Dictyostelium is known to function in other eukaryotes and may serve as a general mechanism for peptide-based signaling.
Table 2.
Proteases, transporters, and protein kinases in intercellular signaling
| Organism | Signal generation
|
Signal
integration
|
||||
|---|---|---|---|---|---|---|
| Signal source | Peptide precursor | Proteolytic processing | MDR-related transporter | Receiving cell | Signal transduction | |
| Dictyostelium discoideum | Prestalk cells | Unknown | Serine protease domains of TagB and TagC | MDR domains of TagB and TagC | Prespore cells | mkcA regA |
| Saccharomyces cerevisiae | a cells | pro-a-factor (MFA1, MFA2) | AXL1, STE23 | STE6 | α cells | STE3 receptor STE20–MAP kinase cascade |
| Mammals | MHC class I positive cells | Cytoplasmic proteins | Proteasome | TAP1–TAP2 heterodimer | Cytotoxic T lymphocytes | T-cell receptor Lck Raf-1 MAP kinase cascade |
Assuming that regA encodes a cAMP phosphodiesterase, then our results indicate that cAMP may be directly involved in triggering encapsulation. A model consistent with all the available genetic and structural evidence can be considered in which signal peptides are generated and exported by the TagB/C complex in prestalk cells as they undergo terminal differentiation. The signal diffuses to adjacent prespore cells where it activates a histidine kinase of the two-component system that includes RegA as the second component. Phosphorylation of RegA inactivates its phosphodiesterase activity leading to an increase in the levels of cAMP such that cAMP-dependent protein kinase (PKA) and other cAMP-dependent processes necessary for encapsulation are activated. Sporulation then follows in short order. This model is supported by the observation that direct addition of the membrane permeable cAMP analog, 8-Bromo-cAMP, triggers encapsulation in dissociated cells where signaling from prestalk cells is precluded (36–38). Moreover, we have found that 8-Bromo-cAMP can trigger encapsulation in tagB− cells where signaling from prestalk cells is genetically precluded (G.S. and W.F.L., unpublished data).
Acknowledgments
We thank Dr. R. Kessin for helpful discussions, Danny Fuller for constructing pBSR1, and Negin Iranfar for sequence analyses. G.S. was supported by California Division–American Cancer Society Fellowship S-4-94. R.E. was supported by a MEC/Fulbright Scholarship. This work was supported by National Institutes of Health Grant HD30892.
Footnotes
Abbreviations: REMI, restriction enzyme mediated integration; MAP, mitogen-activating protein; MHC, major histocompatibility complex; MDR, multiple drug resistance.
Data deposition: The sequences reported in this paper have been deposited in the GenBank data base [accession numbers U60086U60086 (tagC), U60168U60168 (prtC), U60169U60169 (mkcA), and U60170U60170 (regA)].
References
- 1.Shaulsky G, Kuspa A, Loomis W F. Genes Dev. 1995;9:1111–1122. doi: 10.1101/gad.9.9.1111. [DOI] [PubMed] [Google Scholar]
- 2.Adames N, Blundell K, Ashby M N, Boone C. Science. 1995;270:464–467. doi: 10.1126/science.270.5235.464. [DOI] [PubMed] [Google Scholar]
- 3.Kuchler K, Sterne R E, Thorner J. EMBO J. 1989;8:3973–3984. doi: 10.1002/j.1460-2075.1989.tb08580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGarth J P, Varshavsky A. Nature (London) 1989;340:400–404. doi: 10.1038/340400a0. [DOI] [PubMed] [Google Scholar]
- 5.Michaelis S. Semin Cell Biol. 1993;4:17–27. doi: 10.1006/scel.1993.1003. [DOI] [PubMed] [Google Scholar]
- 6.Ciechanover A. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 7.Townsend A R M, Gotch F M, Davey J. Cell. 1985;42:457–467. doi: 10.1016/0092-8674(85)90103-5. [DOI] [PubMed] [Google Scholar]
- 8.Trowsdale J, Hanson I, Mockridge I, Beck S, Townsend A, Kelly A. Nature (London) 1990;348:741–744. doi: 10.1038/348741a0. [DOI] [PubMed] [Google Scholar]
- 9.Deverson E V, Gow I R, Coadwell W J, Monaco J J, Butcher G W, Howard J C. Nature (London) 1990;348:738–741. doi: 10.1038/348738a0. [DOI] [PubMed] [Google Scholar]
- 10.Monaco J J, Cho S, Attaya M. Science. 1990;250:1723–1726. doi: 10.1126/science.2270487. [DOI] [PubMed] [Google Scholar]
- 11.Spies T, Bresnahan M, Bahram S, Arnold D, Blanck G, Mellins E, Pious D, DeMars R. Nature (London) 1990;348:744–747. doi: 10.1038/348744a0. [DOI] [PubMed] [Google Scholar]
- 12.Spies T, Cerundolo V, Colonna M, Cresswell P, Townsend A, DeMars R. Nature (London) 1992;355:644–646. doi: 10.1038/355644a0. [DOI] [PubMed] [Google Scholar]
- 13.Kuspa A, Loomis W F. Proc Natl Acad Sci USA. 1992;89:8803–8807. doi: 10.1073/pnas.89.18.8803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adachi H, Hasebe T, Yoshinaga K, Ohta T, Sutoh K. Biochem Biophys Res Commun. 1994;205:1808–1814. doi: 10.1006/bbrc.1994.2880. [DOI] [PubMed] [Google Scholar]
- 15.Sussman M. Methods Cell Biol. 1987;28:9–29. doi: 10.1016/s0091-679x(08)61635-0. [DOI] [PubMed] [Google Scholar]
- 16.Shaulsky G, Loomis W F. Dev Biol. 1993;160:85–98. doi: 10.1006/dbio.1993.1288. [DOI] [PubMed] [Google Scholar]
- 17.Shaulsky G, Loomis W F. Dev Biol. 1996;174:214–220. doi: 10.1006/dbio.1996.0067. [DOI] [PubMed] [Google Scholar]
- 18.Escalante R, Loomis W F. Dev Biol. 1995;171:262–266. doi: 10.1006/dbio.1995.1278. [DOI] [PubMed] [Google Scholar]
- 19.Silva-Pereira I, Bey F, Coux O, Scherrer K. Gene. 1992;120:235–242. doi: 10.1016/0378-1119(92)90098-a. [DOI] [PubMed] [Google Scholar]
- 20.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 21.Ramer S W, Davis R W. Proc Natl Acad Sci USA. 1993;90:452–456. doi: 10.1073/pnas.90.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stock J B, Ninfa A J, Stock A M. Microbiol Rev. 1989;53:450–490. doi: 10.1128/mr.53.4.450-490.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hecht G B, Newton A. J Bacteriol. 1995;177:6223–6229. doi: 10.1128/jb.177.21.6223-6229.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McAllister-Lucas L M, Sonnenburg W K, Kadlecek A, Seger D, Trong H L, Colbran J L, Thomas M K, Walsh K A, Francis S H, Corbin J D, Beavo J A. J Biol Chem. 1993;268:22863–22873. [PubMed] [Google Scholar]
- 25.Lacombe M L, Podgorski G J, Franke J, Kessin R H. J Biol Chem. 1986;261:16811–16817. [PubMed] [Google Scholar]
- 26.Beltman J, Sonnenburg W K, Beavo J A. Mol Cell Biochem. 1993;127/128:239–253. doi: 10.1007/BF01076775. [DOI] [PubMed] [Google Scholar]
- 27.Lupas A, Stock J. J Biol Chem. 1989;264:17337–17342. [PubMed] [Google Scholar]
- 28.Maeda T, Wurgler-Murphy S M, Saito H. Nature (London) 1994;369:242–245. doi: 10.1038/369242a0. [DOI] [PubMed] [Google Scholar]
- 29.Maeda T, Takekawa M, Saito H. Science. 1995;269:554–558. doi: 10.1126/science.7624781. [DOI] [PubMed] [Google Scholar]
- 30.Kieber J, Rothenberg M, Roman G, Feldmann K, Ecker J. Cell. 1993;72:427–441. doi: 10.1016/0092-8674(93)90119-b. [DOI] [PubMed] [Google Scholar]
- 31.Chang C, Kwok S F, Bleecker A B, Meyerowitz E M. Science. 1993;262:539–544. doi: 10.1126/science.8211181. [DOI] [PubMed] [Google Scholar]
- 32.Hua J, Chang C, Sun Q, Meyerowitz E M. Science. 1995;269:1712–1714. doi: 10.1126/science.7569898. [DOI] [PubMed] [Google Scholar]
- 33.Richardson D L, Loomis W F, Kimmel A R. Development (Cambridge, UK) 1994;120:2891–2900. doi: 10.1242/dev.120.10.2891. [DOI] [PubMed] [Google Scholar]
- 34.Herskowitz I. Cell. 1995;80:187–197. doi: 10.1016/0092-8674(95)90402-6. [DOI] [PubMed] [Google Scholar]
- 35.Gupta S, Weiss A, Kumar G, Wang S, Nel A. J Biol Chem. 1994;269:17349–17357. [PubMed] [Google Scholar]
- 36.Kay R R. Development (Cambridge, UK) 1989;105:753–759. [Google Scholar]
- 37.Maeda M. Dev Growth Differ. 1988;30:573–587. doi: 10.1111/j.1440-169X.1988.00573.x. [DOI] [PubMed] [Google Scholar]
- 38.Richardson D L, Hong C B, Loomis W F. Dev Biol. 1991;144:269–280. doi: 10.1016/0012-1606(91)90421-x. [DOI] [PubMed] [Google Scholar]