

Abstract
Myocytes of the failing heart undergo impressive metabolic remodelling. The time line for changes in the pathways for ATP synthesis in compensated hypertrophy is: flux through the creatine kinase (CK) reaction falls as both creatine concentration ([Cr]) and CK activity fall; increases in [ADP] and [AMP] lead to increases in glucose uptake and utilization; fatty acid oxidation either remains the same or decreases. In uncompensated hypertrophy and in other forms of heart failure, CK flux and fatty acid oxidation are both lower; any increases in glucose uptake and utilization are not sufficient to compensate for overall decreases in the capacity for ATP supply and [ATP] falls. Metabolic remodelling is under transcriptional and post-transcriptional control. The lower metabolic reserve of the failing heart contributes to impaired contractile reserve.
Keywords: Energetics, Heart failure, Metabolic remodelling, Familial hypertrophic cardiomyopathy, Adenosine triphosphate
1. Introduction
ATP is required for cell viability and myocardial pump function. Cleavage of the terminal phosphate (a phosphoryl bond) by ATPases [ATP → ADP + inorganic phosphate (Pi)] releases chemical energy that is converted into the work of contraction, ion pumping, synthesis and degradation of large and small molecules, molecular trafficking, indeed all functions of the cell. Because the amount of ATP in the heart is small (∼10 mM, enough for only a few beats) compared with demand (as much as 10 000 times greater), the myocardial cell must continually re-synthesize ATP to maintain normal cardiac pump function and cellular viability. Consequently, the rates of ATP utilization and re-synthesis are very large. The concentration of ATP ([ATP]) is maintained constant, despite large and variable changes in ATP demand.
ATP re-synthesis by fatty acid oxidation in mitochondria is normally sufficient to meet the dynamic demands for chemical energy and is the primary pathway for ATP synthesis. Under conditions of high ATP demand relative to ATP availability, the myocyte recruits additional pathways for ATP synthesis, namely glycolysis and the phosphotransferase reactions catalysed by creatine kinase (CK) and adenylate kinase (AK). The different pathways for ATP supply have different rates of ATP synthesis: phosphoryl transfer via CK is ∼10 times faster than ATP synthesis in mitochondria (∼0.7 mM/s) which is ∼20 times faster than glycolysis.1 The relative contributions of these pathways to overall ATP synthesis change rapidly in response to changes in fuel supply, hormonal and neural signals, availability of substrates and inhibitors of specific enzyme reactions, and by chemical modification of proteins. During acute increases in work in the normal myocardium, glycogen is used,2 more glucose is influxed,3 and phosphocreatine (PCr, the primary energy reserve compound in the heart)4 is used to support the demand for more ATP. To maintain constant [ATP], the sum of the rates of ATP synthesis by the mitochondria, glycolysis and glycogenolysis, and the phosphotransferase reactions matches the sum of rates of ATP utilization by the sarcomere, ion pumps, etc. This flexible dynamic metabolic network is the normal state of the myocyte.
The efficiency of ATP production expressed as the ratio of ATP synthesis rate to O2 consumed (P:O) differs slightly depending on the mix of substrates oxidized: the P:O is ∼15% higher for oxidation of glucose only (a condition that occurs only in severe ischaemia) vs. fatty acids only. On a molar basis, however, much more ATP is produced from fatty acid oxidation than from glucose utilization.5
Phosphoryl transfer between sites of ATP production and utilization occurs by means of metabolic relays via CK, AK, and glycolysis.6–8 The physical association of these metabolic relays with energy-utilizing proteins creates microenvironments or domains whereby phosphoryl groups can be supplied to ATPases without exchange with bulk cytosolic pools, improving the efficiency of ATP supply.6–8
2. The energetic phenotype of the failing heart: an overview
Shown by investigators studying the failing human myocardium and a wide variety of animal models of chronic demand overload using many different tools ranging from genomics to proteomics to metabolomics to classic biochemistry to physiology, it is now known that energy metabolism in myocytes of the failing heart remodels, resulting in a progressive loss of [ATP]. Metabolic remodelling is controlled by energy sensors such as AMP that lead to changes in phosphorylation state (in addition to other chemical modifications) of many proteins for short-term preservation of ATP9 and by activation of transcription factors and co-activators such as peroxisome proliferator-activated receptor γ (PPARγ) co-activator 1 (PGC-1α) that coordinately control long-term remodelling of entire ATP synthesis and utilizing pathways.10–12 The time line for changes in the pathways for ATP synthesis in compensated hypertrophy is: flux through the CK reaction falls as both [Cr] and CK activity fall;13,14 increases in [ADP] and [AMP] due to decreased PCr/Cr lead to increases in glucose uptake and utilization,15 while fatty acid oxidation either remains the same or decreases.16–19 In uncompensated hypertrophy and in other forms of heart failure, CK flux and fatty acid oxidation are both lower;18–21 any increases in glucose uptake and utilization are not sufficient to compensate for overall decreases in the capacity for ATP supply; and [ATP] falls.22,23 Decreased energy reserve by all these pathways contributes to impaired contractile reserve in the failing myocardium. Some of the many recent reviews relevant to the energetics of the failing heart are referenced.2,7–10,12,24–35 Here, we will review the current status of energetics in the failing heart, emphasizing the molecular remodelling that occurs in the myocyte.
3. Heart failure due to increased cost of contraction: familial hypertrophic cardiomyopathy
It is important to emphasize that although the current focus of most studies of ATP metabolism is on the remodelling of the various pathways for ATP synthesis, the ‘metabolic driver’ is the need for ATP by the major ATP-consumers, sarcomeric myosin, and the ion pumps. This is well illustrated by studies defining cardiac energetics of hearts bearing familial hypertrophic cardiomyopathy (FHC)-associated mutations in sarcomeric proteins. Direct measurement of ATP and PCr using 31P NMR spectroscopy in hearts of FHC patients,36–38 hearts of mice bio-engineered to harbour FHC-associated gene mutations in sarcomeric proteins,39–41 and mutant homologous protein isolated from an FHC patient42 all show that the cost of tension development is higher in hearts with FHC-associated mutations in sarcomeric proteins. Hearts bearing FHC-associated mutations in either thin or think filament proteins exhibit the metabolic hallmarks of energy supply/demand imbalance: lower PCr/ATP and higher Pi/ATP.36–38
Is there a common cause for the increased cost of tension development in FHC-associated mutant sarcomeres? Elegant analyses of changes in peptide conformation and dynamics caused by FHC-associated missense mutations where glutamine (Q), leucine (L), or tryptophan (W) replaces arginine (R) at residue 92 (referred to R92) in the tropomyosin-binding domain of cardiac troponin T (cTnT) have shown large mutation-specific differences.43 The structure of this domain determines cTnT–tropomyosin interaction, which in turn determines actin–myosin interaction during cross-bridge cycling.44–46 Thus, these single amino acid mutations at R92 of cTnT disrupt the normal thick–thin filament interactions. Similarly, 3-D reconstructions of smooth muscle myosin bound to actin for wild type and a mutant myosin where glutamine replaces arginine at residue 403 (referred to as R403) show that, unlike the normal fixed actin–myosin loop interaction, the mutant myosin–actin interaction shows disarray.47 Thus, one common consequence of the R92 cTnT and R403 myosin heavy chain mutations is altered actin–myosin interaction leading to increased cost of tension development.36,39–41 While one might expect that perturbation in the actin-binding loop of myosin heavy chain would be the more energy costly mutation, this is not the case. In the same well-controlled experimental protocol using intact mouse hearts, the mutations at R92 cTnT are more energy costly than the R403 mutation in myosin heavy chain.39–41 These results underscore the relatively new discovery of the dynamic role of the thin filament in determining cross-bridge kinetics.44–46 Moreover, as each R92 cTnT mutation (R92Q, L, W) displays a different cost of tension development,39,40 these results also suggest that the consequences of any given mutation cannot be predicted, but must be empirically determined.
Although the energetic phenotype of these FHC models—lower [PCr] and higher [ADP] and [Pi]—is similar to compensated hypertrophied hearts, the hearts are not larger. These energetic and contractile defects are not due to hypertrophy, but rather to altered sarcomere structure and function.36–38 The most common physiological phenotype of hearts bearing FHC-associated missense mutations in sarcomeric proteins is diastolic dysfunction; whether the mutation leads to hypertrophy or to systolic dysfunction is highly variable.36–38 The observation that R403Q mouse hearts demonstrated pure diastolic dysfunction,41 confirmed in patients,48 is a good example of this. A consequence of increased cost of contraction is elevated [ADP] and thus lower chemical driving force, known to slow the cross-bridge cycle and contribute to diastolic dysfunction.49,50
4. Energetic phenotype of the failing heart: lower [ATP] and [PCr]
In the failing human myocardium and in hearts of animal models of severe failure, [ATP] is ∼30% lower than in normal myocardium (Figure 1).27,31,51 The fall in [ATP] occurs in both left and right ventricular myocardium, in widely different species, and due to a variety of aetiologies. The rate of fall in [ATP] is progressive and is due to the loss of the adenine nucleotide pool (Figure 1B).52 It is unknown what controls the magnitude of the fall in [ATP].
Figure 1.

Loss of ATP in the failing heart. (A) 31P NMR spectra from failing human heart showing the loss of PCr and ATP (reprinted from Neubauer et al.96). (B) Data from the pacing dog heart model of heart failure showing the progressive fall in ATP, the progressive loss of the total adenine nucleotide pool (TAN) and the close relationship between ATP and TAN (reprinted from Shen et al.52). (C) Data for ATP (insert) and rate-pressure product (RPP) for wild-type (WT, solid bars) and PGC-1α null mouse hearts (TG, grey bars) at baseline and during inotropic challenge with dobutamine with two different substrate mixes (reprinted with permission from Elsevier from Arany et al.84).
Total [Cr] and consequently [PCr] are lower in both hypertrophied and failing myocardium (Figures 1 and 2).27,31,53,54 31P and 1H NMR spectroscopy and other biochemical tools have been used to show decreased PCr/ATP and decreased absolute levels of PCr and Cr in hypertrophied and failing human myocardium due to a wide variety of aetiologies,13,55 in complete accord with the large number of large and small animal studies.27,31 The decrease in [Cr] occurs earlier and is faster than the fall in [ATP] (compare Figures 1B and 2B).52 Whereas the fall in [ATP] is not more than ∼30%, the decrease in [Cr] (from normal values of 30–45 mM, depending on species) can be as much as 50–70% in severely failing myocardium. Since [ATP] also falls in the failing myocardium, the fall in PCr/ATP underestimates the decrease in [PCr].
Figure 2.

Loss of Cr in the failing heart. (A) [Creatine, Cr] (top) and CK activity (bottom) obtained from biopsy specimens of human myocardium are lower for both accident victims maintained on inotropic support and ventilation and for heart failure patients (reprinted from Nascimben et al.13). (B) Data from the pacing dog heart model of heart failure showing the progressive and rapid fall in Cr (reprinted from Shen et al.52). (C) Product of CK activity and [Cr], an index of energy reserve via the CK reaction, plotted against the increases in left ventricular developed pressure (LVDP, top), change in heart rate (HR, middle), and increases in rate-pressure product (RPP) as an index in contractile reserve (bottom) for TO2 and non-failing Syrian hamster hearts (reprinted with permission from Elsevier from Tian et al.97).
Unlike ATP, which can be made by de novo synthesis pathways in the myocyte, Cr is not made in excitable tissues, but rather accumulates through the action of a Cr transporter. Decreased amount of Cr transporter on the sarcolemma explains the decrease in Cr accumulation in the failing myocardium.56–58 Trafficking of Cr transporter to the plasma membrane is regulated by stress, insulin, growth factors, and mTOR; little is known about Cr transporter regulation in the heart.59
5. Energetic phenotype of the failing heart: metabolic reserve for ATP synthesis by all the major ATP-synthesis pathways is limited
5.1. Decreased metabolic reserve via creatine kinase and adenylate kinase
Decreased [Cr] coupled with decreased CK activity (Vmax) (primarily due to decreases in MM-CK in the cytosol and sMtCK in the mitochondria) combine to limit energy reserve via CK in the hypertrophied and failing heart. In animal models of severe heart failure, ∼30% and ∼60% decreases in Vmax and [Cr], respectively, combine to reduce the unidirectional velocity of the forward CK reaction (CK velfor) by ∼70% (Figure 2C). Recent measurement of CK velfor using saturation transfer NMR in failing human myocardium demonstrates lower CK velfor, by ∼50%,14 as predicted from biochemical analysis of the CK system in human myocardium13 and observed for experimental models.27 Given that the CK velfor is an order of magnitude faster than ATP synthesis by any other ATP-synthesis reaction, the decrease in CK velfor of ∼50% is a major loss of energy reserve. The decreases in [Cr] and CK Vmax are reversible.60,61
Physiological consequences of decreased capacity for phosphoryl transfer via CK and AK are increased cost of mechanical work and decreased contractile reserve, rendering the hearts more susceptible to ischaemic injury. This has been shown in otherwise normal rat hearts in which either CK Vmax or [Cr] was decreased27,31 and in a variety of genetically manipulated mouse hearts.62–66 One demonstration of the greater susceptibility of the CK null mouse heart to failure in response to acute ATP demand is the faster rate of loss of systolic performance during zero-flow ischaemia.67 Another example is shown by studies of myocardial infarction in the rat.68 Unlike control rats which survived acute myocardial infarction, the 24 h mortality of rats with severely compromised CK system was 100%. Hearts from mice unable to synthesize Cr had normal contractile performance at baseline but reduced contractile reserve when challenged with an inotropic agent and increased susceptibility to ischaemic injury.64 A consequence of the disruption of energy transfer relay via CK within the myocyte is well illustrated by increased electrical vulnerability of the heart caused by failure to supply ATP via CK to the KATP channel in MM-CK null mouse hearts.69 In AK null mouse hearts, even though flux through the CK reaction and glycolysis increased to compensate for the loss of AK, more ATP per contraction was used in AK-deficient muscle.70 These results suggest that hearts deficient in phosphotransfer enzymes expend a disproportionate amount of energy to support contraction, a non-sustainable condition.
A gain-of-function strategy was used to test whether increasing Cr transporter protein normalized the cytosolic Cr pool in the mouse heart.71 The myocardial Cr pool increased on average two-fold but, unexpectedly, the fraction of Cr that was phosphorylated was lower by ∼50%, despite normal CK activity. As a consequence of the lower PCr/Cr, cytosolic [ADP] increased and the chemical driving force for ATPase reactions, |ΔG∼ATP|, was lower. These hearts developed left ventricular hypertrophy, dilatation, and contractile dysfunction. These unexpected results support a causal relationship between decreased energy reserve and contractile dysfunction.
The observation that increased [Cr] leads to contractile dysfunction60,71 raises the question of whether the loss of Cr in the failing heart is compensatory or deleterious.52 The notion that loss of Cr could be compensatory seems counter intuitive. Loss of Cr reduces CK velfor, and thus reduces the primary energy buffer in the heart at a time when overall energy supply is compromised. However, loss of Cr also minimizes the increase in free [ADP] and hence maintains a near normal |ΔG∼ATP| required to drive ATPase reactions. Maintaining low cytosolic [ADP] also results in low [AMP], reducing loss of purines from the heart.72 Under these conditions, loss of Cr may be required for cell viability.
5.2. Decreased metabolic reserve via glycolysis
The increase in glucose uptake observed in hypertrophied hearts is explained by increased expression and function of the insulin-independent glucose transporter GLUT1; expression and function of the dominant insulin-regulated glucose transporter GLUT4 is decreased.3,32,73 One mechanism leading to increased glucose uptake and utilization in the hypertrophied myocardium is triggered by the demand for more ATP (Figure 3). Decreases in [PCr] without a concomitant fall in total [Cr], a characteristic of hypertrophied myocardium, lead to increases in [ADP], [AMP], and [Pi]. The increase in [AMP] activates the ‘low-on-fuel’ sensor AMP-activated protein kinase (AMPK).73,74 Consequences of activating AMPK include increasing the activity of proteins in ATP-synthesis pathways (increasing ATP supply) and decreasing the activity of proteins in ATP-consuming pathways (conserving ATP). Key among these are GLUT1 and phosphofructokinase-2 (PFK-2), leading to production of fructose-2,6-Pi2, a potent allosteric activator of the rate-limiting protein for glycolysis, PFK.15 Thus, increased ATP demand manifest as decreased [PCr] in the hypertrophied heart signals an increase in glycolytic flux by two coordinate mechanisms: increasing glucose transport (increasing substrate supply) and activating PFK (increasing utilization).
Figure 3.
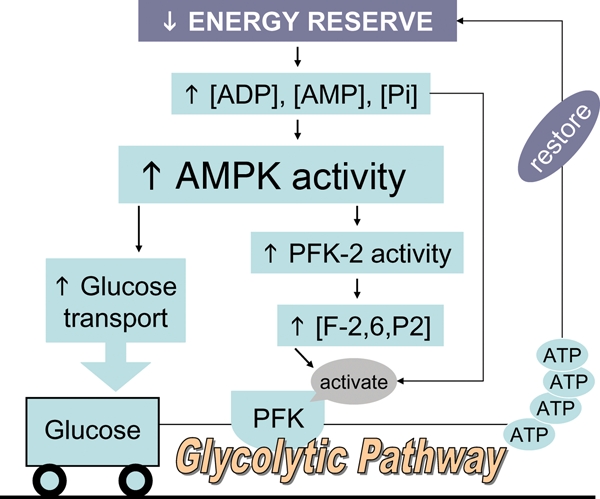
Coordinate control of glycolysis by AMP-activated protein kinase (AMPK). In chronic pressure-overload cardiac hypertrophy in the rat, increased ATP demand signalled as decreased [PCr] leads to an increase in glycolytic flux by two coordinate mechanisms: increasing glucose transport (increasing substrate supply) and activating phosphofructokinase in the glycolytic pathway (increasing utilization), both mediated by AMPK. Redrawn with permission.15
Unlike for control hearts, glucose uptake rates and glycolysis measured in the hypertrophied myocardium of animal models do not increase further during work challenge.75 Importantly, this limitation in metabolic reserve has also been observed in a group of Class I/II patients with dilated cardiomyopathy.23 These results suggest that the presumably adaptive increase in glycolysis is not sufficient to meet ATP demand.
Genetic strategies have been used to test whether increasing glucose utilization further renders hypertrophied hearts more tolerant to chronic haemodynamic overload.76 Transgenic mice with cardiac-specific over-expression of GLUT1 were made to increase basal glucose transport in the heart. Comparing transgenic and wild-type hearts subjected to chronic pressure overload, it was found that increasing myocardial glucose uptake slowed the progression to heart failure and improved survival.76 This study suggests that increasing the capacity for ATP synthesis, in this case via glycolysis, can alter the natural history of heart failure. Increasing glucose uptake further also rescued mouse hearts deficient in the transcriptional activator PPARα, which have a three-fold decrease in fatty acid oxidation and three-fold increase in carbohydrate utilization characteristic of the failing heart.22 PPARα null mouse hearts had reduced contractile reserve, higher than normal myocardial O2 consumption (MVO2) yet produced less ATP, and lost [ATP] with inotropic challenge. Hearts of mice made by crossing the PPARα null mouse with the GLUT1 over expresser mouse sustained increased work without losing [ATP], and MVO2 and ATP synthesis rates returned to normal. These studies suggest that glucose utilization, if sufficiently high, can support and sustain high workload in the failing heart.
5.3. Decreased metabolic reserve via mitochondrial ATP synthesis
O2 is not limiting in the failing myocardium,77,78 and at least in some models, MVO2 is increased.52 Any increase in MVO2, however, is not sufficient to prevent the loss of ATP. Genomic and proteomic studies,79,80 as well as measures of enzyme activities, have shown that proteins involved in fatty acid transport32 and utilization18–21 are down-regulated in failing hearts. Experiments using isolated mitochondria, skinned fibres, isolated hearts,30,81 and in vivo hearts77 all support the conclusion that oxidative capacity and function are reduced in the failing myocardium. Increases in uncoupling proteins81 as well as increases in reactive O2 species and nitric oxide all contribute.30,34,82 Decreased capacity of mitochondrial substrate oxidation in the failing heart leads to decreased cardiac efficiency.52
Fatty acid supply for oxidation is lower in the failing heart. A recent study compared utilization of exogenous vs. endogenous fatty acid in an animal model of pressure overload hypertrophy.18 Fatty acid oxidation rates using exogenous fatty acid were near normal in mild to moderate heart failure, but fell as the disease stage progressed. In contrast, endogenous fats (triacylglycerols) were not oxidized even in early failure. Importantly, as also noted for a group of Class I/II patients with dilated cardiomyopathy,23 oxidation rates did not increase further for either exogenous or endogenous fats with β-adrenergic challenge. Thus, as observed for CK and glycolysis, energy reserve via fatty acid oxidation is also compromised in the failing myocardium.
A potentially important compensatory mechanism for reduced exogenous fatty acid oxidation has recently been identified in an animal model of heart failure due to pressure overload.19 When fatty acid oxidation fell and was uncoupled to tricarboxylic acid cycle flux, tricarboxylic acid cycle flux was unexpectedly sustained by use of glycolytically derived pyruvate through anaplerosis, a pathway that uses pyruvate to supply oxaloacetate downstream from acetyl-CoA in the tricarboxylic acid cycle. Although initially compensatory, because conversion of pyruvate to oxaloacetate via anaplerosis consumes an ATP, this is less efficient than the use of pyruvate through the tricarboxylic acid cycle. The increase in energy cost is not likely to be sustainable. This could be one step in the transition from compensatory hypertrophy to failure.
Genetic tools have been used to define the consequences of altering transcriptional regulators of oxidative metabolism. The transcriptional co-activator PGC-1α, referred to as a ‘master regulator’, controls mitochondrial biogenesis and the synthesis of entire metabolic pathways for ATP synthesis.12 PGC-1α is down-regulated in hypertrophied and failing heart.12,83 PGC-1α null mice have been used to define the consequences of reduced PGC-1α on ATP synthesis and contractile reserve in the heart.84,85 The absence of PGC-1α not only led to reduced gene expression for proteins required for FAO, but their enzyme activities were reduced. [ATP] was decreased by ∼20%, a large decrease similar to that observed in end-stage failing hearts. Importantly, this was the case despite the presence of PGC-1β, which has many overlapping targets with PGC-1α. PGC-1α null hearts had reduced contractile reserve (Figure 1C) and progressed to failure more rapidly than wild-type hearts when subjected to pressure overload.86 Although massive over-expression of PGC-1α led to mitochondrial proliferation to such an extent that the sarcomeres became displaced, leading to cardiomyopathy and heart failure,87 short-term PGC-1α over-expression reversed contractile dysfunction,88 suggesting causative links among PGC-1α expression, mitochondrial biogenesis, ATP synthesis, and contractile performance. Genetic manipulation in the mouse has identified other players in the control of ATP production, such as Tfam89 and Lim protein.90
6. Metabolic remodelling is under transcriptional and post-transcriptional control
The past decade has witnessed an explosion of information identifying the molecular links among physiological and metabolic stimuli and the regulation of gene expression in the myocyte. Not only have the metabolic targets of specific nuclear receptors and DNA-binding transcriptional (co)-activators been identified, but we are also beginning to learn how their signals are amplified and sustained to remodel metabolism. Transcription is activated when transcriptional activators including PPARs, oestrogen receptors (ERRs), retinoid receptors, nuclear respiratory factors, and MEF-2 form protein–protein complexes with PPAR-γ co-activators, PGC-1α and β, tethering PGC-1s to DNA. When complexed with transcriptional activators, PGC-1s activate genes encoding proteins comprising entire metabolic pathways that control ATP synthesis in mitochondria, phosphoryl transfer, glucose uptake and utilization, and, as is recently becoming appreciated, ATP-utilizing proteins. For example,91 the ERRα/PGC-1α complex targets a set of promoters common to genes encoding a wide spectrum of energy producing (FA and glucose uptake, β-oxidation, tricarboxylic acid cycle, and electron transport chain), transferring (sMtCK and adenine nucleotide transporter), and utilizing proteins. In hypertrophied ERRα null mouse hearts, genes for ATP synthesis and transfer were all decreased while the gene encoding the stress protein CK-BB was increased. These experiments support the notion that the normal ERRα/PGC-1α complex is required to blunt the loss of capacity for ATP synthesis in pressure overload hypertrophy.
PGC-1s are themselves regulated. Of the many possible regulators of PGC-1s, it remains unclear which ones operate in the myocardium.10–12 Cyclin-dependent kinases, such as Cdk9 and Cdk7 that play a role in transcriptional elongation, target PGC-1s, thereby conferring additional specificity for the transcriptional control of ATP synthesizing and utilizing reactions.11,92 These are activated in the hypertrophied and failing myocardium, repressing PGC-1α.
Unlike the impressive progress made understanding the transcriptional events that control normal and hypertrophic growth and the development of cardiac dysfunction, much less is known about post-transcriptional control. We do know that the notion that there is a 1-to-1 correspondence in the number of mRNA transcripts and the number of functional proteins is not correct (for example, see ref.60). Post-transcriptional control remains under-studied.
7. Rescuing the failing myocardium
Although space does not allow a full review of this important topic, a few comments will be made. One clinically relevant lesson to be learned from the study of cardiac energetics is that the failing heart has limited energy reserve and, while it can increase work, it does so at a higher cost of contraction. This increases susceptibility to arrhythmia and ischaemic injury. The clinical observations that patients treated with inotropic drugs that increase ATP utilization have poor long-term outcomes can be explained by the lack of energy reserve.24,93 Research into ways of increasing systolic performance or reducing diastolic dysfunction by manipulating sarcomere function without increasing cost of developed tension merits support.
Direct manipulation of adenine nucleotide and Cr pools has been difficult to achieve. Notable in this regard is the report studying experimental right ventricular hypertrophy94 showing that folate treatment protected against loss of adenine nucleotides and diastolic dysfunction. As folate is both readily available and inexpensive, it may be useful in slowing the progression to failure. Another rescue strategy seeks to take advantage of the small increase in the ratio of ATP production to O2 consumed for glucose. Drugs that shift metabolism away from fatty acid oxidation and towards glucose metabolism may improve the efficiency of ATP production by a small amount. It is possible, however, that both glucose and fatty acids are required for the failing heart.95 Much more research needs to be done on this topic.
In any rational strategy, care should be taken to match any metabolic intervention with the stage of disease. The different pathways for ATP synthesis are compromised at different times and to varying extents in the evolution from compensated to uncompensated hypertrophy. Loss of energy reserve supported by CK and AK occurs first and triggers an increase in glycolysis, followed by decreased FAO. Ideally, interventions designed to alter metabolic pathways must be matched to stage of metabolic dysfunction, analogous to NYHA classes.
Funding
This work was supported in part by research funds from the Department of Medicine, Brigham and Women's Hospital and the National Institutes of Health.
Acknowledgement
The author wishes to thank Linda Johnson for her assistance.
Conflict of interest: none declared.
References
- 1.Ingwall JS. ATP and the Heart. Norwell, MA: Kluwer Academic Publishers; 2002. ATP and the heart: an overview; pp. 3–6. [Google Scholar]
- 2.Taegtmeyer H, Wilson CR, Razeghi P, Sharma S. Metabolic energetics and genetics in the heart. Ann N Y Acad Sci. 2005;1047:208–218. doi: 10.1196/annals.1341.019. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J, Duncker DJ, Ya X, Zhang Y, Pavek T, Wei H, et al. Effect of left ventricular hypertrophy secondary to chronic pressure overload on transmural myocardial 2-deoxyglucose uptake. A 31P NMR spectroscopic study. Circulation. 1995;92:1274–1283. doi: 10.1161/01.cir.92.5.1274. [DOI] [PubMed] [Google Scholar]
- 4.Bittl JA, Ingwall JS. Reaction rates of creatine kinase and ATP synthesis in the isolated rat heart. A 31P NMR magnetization transfer study. J Biol Chem. 1985;260:3512–3517. [PubMed] [Google Scholar]
- 5.Ingwall JS. Boston: Kluwer Academic Publishers; 2002. ATP and the Heart. [Google Scholar]
- 6.De Sousa E, Veksler V, Minajeva A, Kaasik A, Mateo P, Mayoux E, et al. Subcellular creatine kinase alterations. Implications in heart failure. Circ Res. 1999;85:68–76. doi: 10.1161/01.res.85.1.68. [DOI] [PubMed] [Google Scholar]
- 7.Dzeja PP, Chung S, Terzic A. Integration of adenylate kinase, glycolytic and glycogenolytic circuits in cellular energetics. In: Saks V, editor. Molecular System Bioenergetics: Energy for Life. Weinheim: Wiley-VCH; 2007. [Google Scholar]
- 8.Ingwall JS. Transgenesis and cardiac energetics: new insights into cardiac metabolism. J Mol Cell Cardiol. 2004;37:613–623. doi: 10.1016/j.yjmcc.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 9.Dyck JRB, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally? J Physiol. 2006;574:95–112. doi: 10.1113/jphysiol.2006.109389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sano M, Izumi Y, Helenius K, Asakura M, Rossi DJ, Xie M, et al. Menage-a-trois 1 is critical for the transcriptional function of PPARγ coactivator 1. Cell Metab. 2007;5:129–142. doi: 10.1016/j.cmet.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1α. Cardiovasc Res. 2008;79:208–217. doi: 10.1093/cvr/cvn098. [DOI] [PubMed] [Google Scholar]
- 13.Nascimben L, Ingwall JS, Pauletto P, Friedrich J, Gwathmey JK, Saks V, et al. Creatine kinase system in failing and nonfailing human myocardium. Circulation. 1996;94:1894–1901. doi: 10.1161/01.cir.94.8.1894. [DOI] [PubMed] [Google Scholar]
- 14.Weiss RG, Gerstenblith G, Bottomley PA. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci USA. 2005;102:808–813. doi: 10.1073/pnas.0408962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nascimben L, Ingwall J, Lorell B, Pinz I, Schulta V, Tornheim K, et al. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension. 2004;44:662–667. doi: 10.1161/01.HYP.0000144292.69599.0c. [DOI] [PubMed] [Google Scholar]
- 16.Degens H, de Brouwer KF, Gilde AJ, Lindhout M, Willemsen PH, Janssen BJ, et al. Cardiac fatty acid metabolism is preserved in the compensated hypertrophic rat heart. Basic Res Cardiol. 2006;101:17–26. doi: 10.1007/s00395-005-0549-0. [DOI] [PubMed] [Google Scholar]
- 17.Lei B, Lionetti V, Young ME, Chandler MP, d'Agostino C, Kang E, et al. Paradoxical downregulation of the glucose oxidation pathway despite enhanced flux in severe heart failure. J Mol Cell Cardiol. 2004;36:567–576. doi: 10.1016/j.yjmcc.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 18.O'Donnell JM, Fields AD, Sorokina N, Lewandowski ED. The absence of endogenous lipid oxidation in early stage heart failure exposes limits in lipid storage and turnover. J Mol Cell Cardiol. 2008;44:315–322. doi: 10.1016/j.yjmcc.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sorokina N, O'Donnell JM, McKinney RD, Pound KM, Woldegiorgis G, LaNoue KF, et al. Recruitment of compensatory pathways to sustain oxidative flux with reduced carnitine palmitoyltransferase I activity characterizes inefficiency in energy metabolism in hypertrophied hearts. Circulation. 2007;115:2033–2041. doi: 10.1161/CIRCULATIONAHA.106.668665. [DOI] [PubMed] [Google Scholar]
- 20.Osorio JC, Stanley WC, Linke A, Castellari M, Diep QN, Panchal AR, et al. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation. 2002;106:606–612. doi: 10.1161/01.cir.0000023531.22727.c1. [DOI] [PubMed] [Google Scholar]
- 21.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 22.Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP, Tian R. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation. 2005;112:2339–2346. doi: 10.1161/CIRCULATIONAHA.105.534594. [DOI] [PubMed] [Google Scholar]
- 23.Neglia D, De Caterina A, Marraccini P, Natali A, Ciardetti M, Vecoli C, et al. Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 2007;293:H3270–H3278. doi: 10.1152/ajpheart.00887.2007. [DOI] [PubMed] [Google Scholar]
- 24.deGoma EM, Vagelos RH, Fowler MB, Ashley EA. Emerging therapies for the management of decompensated heart failure: from bench to bedside. J Am Coll Cardiol. 2006;48:2397–2409. doi: 10.1016/j.jacc.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 25.Fragasso G. Inhibition of free fatty acids metabolism as a therapeutic target in patients with heart failure. Int J Clin Pract. 2007;61:603–610. doi: 10.1111/j.1742-1241.2006.01280.x. [DOI] [PubMed] [Google Scholar]
- 26.Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res. 2007;77:334–343. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- 27.Ingwall JS, Weiss RG. Is the failing heart energy starved? Circ Res. 2004;95:135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 28.Kodde IF, van der Stok J, Smolenski RT, de Jong JW. Metabolic and genetic regulation of cardiac energy substrate preference. Comp Biochem Physiol A. 2006;146:26–39. doi: 10.1016/j.cbpa.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 29.Marin-Garcia J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev. 2008;13:137–150. doi: 10.1007/s10741-007-9079-1. [DOI] [PubMed] [Google Scholar]
- 30.Murray AJ, Edwards LM, Clarke K. Mitochondria and heart failure. Curr Opin Clin Nutr Metab Care. 2007;10:704–711. doi: 10.1097/MCO.0b013e3282f0ecbe. [DOI] [PubMed] [Google Scholar]
- 31.Neubauer S. The failing heart—an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 32.Schwenk RW, Luiken JJ, Bonen A, Glatz JF. Regulation of sarcolemmal glucose and fatty acid transporters in cardiac disease. Cardiovasc Res. 2008;79:249–258. doi: 10.1093/cvr/cvn116. [DOI] [PubMed] [Google Scholar]
- 33.Taha M, Lopaschuk GD. Alterations in energy metabolism in cardiomyopathies. Ann Med. 2007;39:594–607. doi: 10.1080/07853890701618305. [DOI] [PubMed] [Google Scholar]
- 34.Tsutsui H. Mitochondrial oxidative stress and heart failure. Intern Med. 2006;45:809–813. doi: 10.2169/internalmedicine.45.1765. [DOI] [PubMed] [Google Scholar]
- 35.Ventura-Clapier RF, Garnier A, Veksler V. Energy metabolism in heart failure. J Physiol. 2004;555:1–15. doi: 10.1113/jphysiol.2003.055095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crilley JG, Boehm EA, Blair E, Rajagopalan B, Blamire AM, Styles P, et al. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol. 2003;41:1776–1782. doi: 10.1016/s0735-1097(02)03009-7. [DOI] [PubMed] [Google Scholar]
- 37.Jung W, Hoess T, Bunse M, Widmaier S, Sieverding L, Breuer J, et al. Differences in cardiac energetics between patients with familial and nonfamilial hypertrophic cardiomyopathy. Circulation. 2000;101:E121. doi: 10.1161/01.cir.101.12.e121. [DOI] [PubMed] [Google Scholar]
- 38.Jung WI, Sieverding L, Breuer J, Hoess T, Widmaier S, Schmidt O, et al. 31P NMR spectroscopy detects metabolic abnormalities in asymptomatic patients with hypertrophic cardiomyopathy. Circulation. 1998;97:2536–2542. doi: 10.1161/01.cir.97.25.2536. [DOI] [PubMed] [Google Scholar]
- 39.He ZH, Bottinelli R, Pellegrino MA, Ferenczi MA, Reggiani C. ATP consumption and efficiency of human single muscle fibers with different myosin isoform composition. Biophys J. 2000;79:945–961. doi: 10.1016/S0006-3495(00)76349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Javadpour MM, Tardiff JC, Pinz I, Ingwall JS. Decreased energetics in murine hearts bearing the R92Q mutation in cardiac troponin T. J Clin Invest. 2003;112:768–775. doi: 10.1172/JCI15967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG, et al. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Invest. 1998;101:1775–1783. doi: 10.1172/JCI1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keller DI, Coirault C, Rau T, Cheav T, Weyand M, Amann K, et al. Human homozygous R403W mutant cardiac myosin presents disproportionate enhancement of mechanical and enzymatic properties. J Mol Cell Cardiol. 2004;36:355–362. doi: 10.1016/j.yjmcc.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 43.Ertz-Berger BR, He H, Dowell C, Factor SM, Haim TE, Nunez S, et al. Changes in the chemical and dynamic properties of cardiac troponin T cause discrete cardiomyopathies in transgenic mice. Proc Natl Acad Sci USA. 2005;102:18219–18224. doi: 10.1073/pnas.0509181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maytum R, Geeves MA, Lehrer SS. A modulatory role for the troponin T tail domain in thin filament regulation. J Biol Chem. 2002;277:29774–29780. doi: 10.1074/jbc.M201761200. [DOI] [PubMed] [Google Scholar]
- 45.Palm T, Graboski S, Hitchcock-DeGregori SE, Greenfield NJ. Disease-causing mutations in cardiac troponin T: identification of a critical tropomyosin-binding region. Biophys J. 2001;81:2827–2837. doi: 10.1016/S0006-3495(01)75924-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tobacman LS, Nihli M, Butters C, Heller M, Hatch V, Craig R, et al. The troponin tail domain promotes a conformational state of the thin filament that suppresses myosin activity. J Biol Chem. 2002;277:27636–27642. doi: 10.1074/jbc.M201768200. [DOI] [PubMed] [Google Scholar]
- 47.Volkmann N, Lui H, Hazelwood L, Trybus KM, Lowey S, Hanein D. The R403Q myosin mutation implicated in familial hypertrophic cardiomyopathy causes disorder at the actomyosin interface. PLoS ONE. 2007;2:e1123. doi: 10.1371/journal.pone.0001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ho CY, Sweitzer NK, McDonough B, Maron BJ, Casey SA, Seidman JG, et al. Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation. 2002;105:2992–2997. doi: 10.1161/01.cir.0000019070.70491.6d. [DOI] [PubMed] [Google Scholar]
- 49.Tian R, Christe ME, Spindler M, Hopkins JC, Halow JM, Camacho SA, et al. Role of MgADP in the development of diastolic dysfunction in the intact beating rat heart. J Clin Invest. 1997;99:745–751. doi: 10.1172/JCI119220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tian R, Nascimben L, Ingwall JS, Lorell BH. Failure to maintain a low ADP concentration impairs diastolic function in hypertrophied rat hearts. Circulation. 1997;96:1313–1319. doi: 10.1161/01.cir.96.4.1313. [DOI] [PubMed] [Google Scholar]
- 51.Ingwall JS. Energetic basis for heart failure—causes and consequences of decreased ATP and phosphocreatine. In: Mann D, editor. Heart Failure: A Companion to Braunwald's Heart Disease. Philadelphia: Saunders; 2004. pp. 91–108. [Google Scholar]
- 52.Shen W, Asai K, Uechi M, Mathier MA, Shannon RP, Vatner SF, et al. Progressive loss of myocardial ATP due to a loss of total purines during the development of heart failure in dogs: a compensatory role for the parallel loss of creatine. Circulation. 1999;100:2113–2118. doi: 10.1161/01.cir.100.20.2113. [DOI] [PubMed] [Google Scholar]
- 53.Herrmann G, Decherd M. The chemical nature of heart failure. Ann Intern Med. 1939;12:1233–1244. [Google Scholar]
- 54.Ingwall JS. The hypertrophied myocardium accumulates the MB-creatine kinase isozyme. Eur Heart J. 1984;5:129–139. doi: 10.1093/eurheartj/5.suppl_f.129. [DOI] [PubMed] [Google Scholar]
- 55.Ingwall JS, Kramer MF, Fifer MA, Lorell BH, Shemin R, Grossman W, et al. The creatine kinase system in normal and diseased human myocardium. New Engl J Med. 1985;313:1050–1054. doi: 10.1056/NEJM198510243131704. [DOI] [PubMed] [Google Scholar]
- 56.Boehm E, Chan S, Monfared M, Wallimann T, Clarke K, Neubauer S. Creatine transporter activity and content in the rat heart supplemented by and depleted of creatine. Am J Physiol Endocrinol Metab. 2003;284:E399–E406. doi: 10.1152/ajpendo.00259.2002. [DOI] [PubMed] [Google Scholar]
- 57.Neubauer S, Remkes H, Spindler M, Horn M, Weismann F, Prestle J, et al. Down regulation of the Na(+)-creatine co-transporter in failing human myocardium and in experimental heart failure. Circulation. 1999;100:1847–1850. doi: 10.1161/01.cir.100.18.1847. [DOI] [PubMed] [Google Scholar]
- 58.Ten Hove M, Chan S, Lygate C, Monfared M, Boehm E, Hulbert K, et al. Mechanisms of creatine depletion in chronically failing rat heart. J Mol Cell Cardiol. 2005;38:309–313. doi: 10.1016/j.yjmcc.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 59.Strutz-Seebohm N, Shojaiefard M, Christie D, Tavare J, Seebohm G, Lang F. PIKfyve in the SGK1 mediated regulation of the creatine transporter SLC6A8. Cell Physiol Biochem. 2007;20:729–734. doi: 10.1159/000110433. [DOI] [PubMed] [Google Scholar]
- 60.Shen W, Spindler M, Higgins M, Jin N, Gill R, Bloem L, et al. The fall in creatine levels and creatine kinase isozyme changes in the failing heart are reversible: complex post-transcriptional regulation of the components of the CK system. J Mol Cell Cardiol. 2005;39:537–544. doi: 10.1016/j.yjmcc.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 61.Park SJ, Zhang J, Ye Y, Ormaza S, Liang P, Bank AJ, et al. Myocardial creatine kinase expression after left ventricular assist device support. J Am Coll Cardiol. 2002;39:1773–1779. doi: 10.1016/s0735-1097(02)01860-0. [DOI] [PubMed] [Google Scholar]
- 62.Saupe KW, Spindler M, Hopkins JC, Shen W, Ingwall JS. Kinetic, thermodynamic, and developmental consequences of deleting creatine kinase isoenzymes from the heart. Reaction kinetics of the creatine kinase isoenzymes in the intact heart. J Biol Chem. 2000;275:19742–19746. doi: 10.1074/jbc.M001932200. [DOI] [PubMed] [Google Scholar]
- 63.Saupe KW, Spindler M, Tian R, Ingwall JS. Impaired cardiac energetics in mice lacking muscle-specific isoenzymes of creatine kinase. Circ Res. 1998;82:898–907. doi: 10.1161/01.res.82.8.898. [DOI] [PubMed] [Google Scholar]
- 64.ten Hove M, Lygate CA, Fischer A, Schneider JE, Sang AE, Hulbert K, et al. Reduced inotropic reserve and increased susceptibility to cardiac ischemia/reperfusion injury in phosphocreatine-deficient guanidinoacetate-N-methyltransferase-knockout mice. Circulation. 2005;111:2477–2485. doi: 10.1161/01.CIR.0000165147.99592.01. [DOI] [PubMed] [Google Scholar]
- 65.Tian R, Ingwall JS. Energetic basis for reduced contractile reserve in isolated rat hearts. Am J Physiol. 1996;270:H1207–H1216. doi: 10.1152/ajpheart.1996.270.4.H1207. [DOI] [PubMed] [Google Scholar]
- 66.Zweier JL, Jacobus WE, Korecky B, Brandejs-Barry Y. Bioenergetic consequences of cardiac phosphocreatine depletion induced by creatine analogue feeding. J Biol Chem. 1991;266:20296–20304. [PubMed] [Google Scholar]
- 67.Hopkins J, Miao W, Ingwall J. Paradoxical effects of creatine kinase deletion on systolic and diastolic function in ischemia. Circulation. 1998;98:I–758. [Google Scholar]
- 68.Horn M, Remkes H, Stromer H, Dienesch C, Neubauer S. Chronic phosphocreatine depletion by the creatine analogue beta-guanidinopropionate is associated with increased mortality and loss of ATP in rats after myocardial infarction. Circulation. 2001;104:1844–1849. doi: 10.1161/hc3901.095933. [DOI] [PubMed] [Google Scholar]
- 69.Abraham MR, Selivanov VA, Hodgson DM, Pucar D, Zingman LV, Wieringa B, et al. Coupling of cell energetics with membrane metabolic sensing. Integrative signaling through creatine kinase phosphotransfer disrupted by M-CK gene knock-out. J Biol Chem. 2002;277:24427–24434. doi: 10.1074/jbc.M201777200. [DOI] [PubMed] [Google Scholar]
- 70.Janssen E, Dzeja PP, Oerlemans F, Simonetti AW, Heerschap A, de Haan A, et al. Adenylate kinase 1 gene deletion disrupts muscle energetic economy despite metabolic rearrangement. EMBO J. 2000;19:6371–6381. doi: 10.1093/emboj/19.23.6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wallis J, Lygate CA, Fischer A, ten Hove M, Schneider JE, Sebag-Montefiore L, et al. Supranormal myocardial creatine and phosphocreatine concentrations lead to cardiac hypertrophy and heart failure: insights from creatine transporter-overexpressing transgenic mice. Circulation. 2005;112:3131–3139. doi: 10.1161/CIRCULATIONAHA.105.572990. [DOI] [PubMed] [Google Scholar]
- 72.Bak MI, Ingwall JS. Acidosis during ischemia promotes adenosine triphosphate resynthesis in postischemic rat heart. In vivo regulation of 5′-nucleotidase. J Clin Invest. 1994;93:40–49. doi: 10.1172/JCI116974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tian R, Musi N, D'Agostino J, Hirshman MF, Goodyear LJ. Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation. 2001;104:1664–1669. doi: 10.1161/hc4001.097183. [DOI] [PubMed] [Google Scholar]
- 74.Allard MF, Parsons HL, Saeedi R, Wamboldt RB, Brownsey R. AMPK and metabolic adaptation by the heart to pressure overload. Am J Physiol Heart Circ Physiol. 2007;292:140–148. doi: 10.1152/ajpheart.00424.2006. [DOI] [PubMed] [Google Scholar]
- 75.Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994;267:H742–H750. doi: 10.1152/ajpheart.1994.267.2.H742. [DOI] [PubMed] [Google Scholar]
- 76.Liao R, Jain M, Cui L, D'Agostino J, Aiello F, Luptak I, et al. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure due to pressure-overload in mice. Circulation. 2002;106:2125–2131. doi: 10.1161/01.cir.0000034049.61181.f3. [DOI] [PubMed] [Google Scholar]
- 77.Gong G, Liu J, Liang P, Guo T, Hu Q, Ochiai K, et al. Oxidative capacity in failing hearts. Am J Physiol Heart Circ Physiol. 2003;285:541–548. doi: 10.1152/ajpheart.01142.2002. [DOI] [PubMed] [Google Scholar]
- 78.Murakami Y, Zhang Y, Cho YK, Mansoor AM, Chung JK, Chu C, et al. Myocardial oxygenation during high work states in hearts with postinfarction remodeling. Circulation. 1999;99:942–948. doi: 10.1161/01.cir.99.7.942. [DOI] [PubMed] [Google Scholar]
- 79.Kong SW, Bodyak N, Yue P, Liu Z, Brown J, Izumo S, et al. Genetic expression profiles during physiological and pathological cardiac hypertrophy and heart failure in rats. Physiol Genomics. 2005;21:31–42. doi: 10.1152/physiolgenomics.00226.2004. [DOI] [PubMed] [Google Scholar]
- 80.Arany Z, Wagner BK, Ma Y, Chinsomboon J, Laznik D, Spiegelman BM. Gene expression-based screening identifies microtubule inhibitors as inducers of PGC-1alpha and oxidative phosphorylation. Proc Natl Acad Sci USA. 2008;105:4721–4726. doi: 10.1073/pnas.0800979105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Murray AJ, Cole MA, Lygate CA, Carr CA, Stuckey DJ, Little SE, et al. Increased mitochondrial uncoupling proteins, respiratory uncoupling and decreased efficiency in the chronically infarcted rat heart. J Mol Cell Cardiol. 2008;44:694–700. doi: 10.1016/j.yjmcc.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 82.Sheeran FL, Pepe S. Energy deficiency in the failing heart: linking increased reactive oxygen species and disruption of oxidative phosphorylation rate. Biochem Biophys Acta. 2006;1757:543–552. doi: 10.1016/j.bbabio.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 83.Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura-Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol. 2003;551:491–501. doi: 10.1113/jphysiol.2003.045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, et al. Transcriptional coactivator PGC-1α controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 85.Lehman JJ, Boudina S, Banke NH, Sambandam N, Han X, Young DM, et al. The transcriptional coactivator PGC-1α is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am J Physiol Heart Circ Physiol. 2008;295:H185–H196. doi: 10.1152/ajpheart.00081.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPARγ coactivator 1α. Proc Natl Acad Sci USA. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, et al. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1α promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- 89.Hansson A, Hance N, Dufour E, Rantanen A, Hultenby K, Clayton DA, et al. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc Natl Acad Sci USA. 2004;101:3136–3141. doi: 10.1073/pnas.0308710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van den Bosch BJ, van den Burg CM, Schoonderwoerd K, Lindsey PJ, Scholte HR, de Coo RF, et al. Regional absence of mitochondria causing energy depletion in the myocardium of muscle LIM protein knockout mice. Cardiovasc Res. 2005;65:411–418. doi: 10.1016/j.cardiores.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 91.Huss JM, Imahashi K-i, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, et al. The nuclear receptor ERRα is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007;6:25–37. doi: 10.1016/j.cmet.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 92.Sano M, Wang SC, Shirai M, Scaglia F, Xie M, Sakai S, et al. Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure. EMBO J. 2004;23:3559–3569. doi: 10.1038/sj.emboj.7600351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brixius K, Lu R, Boelck B, Grafweg S, Hoyer F, Pott C, et al. Chronic treatment with carvedilol improves Ca(2+)-dependent ATP consumption in triton X-skinned fiber preparations of human myocardium. J Pharmacol Exp Ther. 2007;322:222–227. doi: 10.1124/jpet.106.116798. [DOI] [PubMed] [Google Scholar]
- 94.Lamberts RR, Caldenhoven E, Lansink M, Witte G, Vaessen RJ, St Cyr JA, et al. Preservation of diastolic function in monocrotaline-induced right ventricular hypertrophy in rats. Am J Physiol Heart Circ Physiol. 2007;293:H1869–H1876. doi: 10.1152/ajpheart.00294.2007. [DOI] [PubMed] [Google Scholar]
- 95.Tuunanen H, Engblom E, Naum A, Nagren K, Hesse B, Airaksinen KE, et al. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation. 2006;114:2130–2137. doi: 10.1161/CIRCULATIONAHA.106.645184. [DOI] [PubMed] [Google Scholar]
- 96.Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, et al. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96:2190–2196. doi: 10.1161/01.cir.96.7.2190. [DOI] [PubMed] [Google Scholar]
- 97.Tian R, Nascimben L, Kaddurah-Daouk R, Ingwall JS. Depletion of energy reserve via the creatine kinase reaction during the evolution of heart failure in cardiomyopathic hamsters. J Mol Cell Cardiol. 1996;28:755–765. doi: 10.1006/jmcc.1996.0070. [DOI] [PubMed] [Google Scholar]