

Abstract
The use of Moloney murine leukemia virus (Mo-MLV)-based vectors to deliver therapeutic genes into target cells is limited by their inability to transduce nondividing cells. To test the capacity of HIV-based vectors to deliver genes into nondividing cells, we have generated replication-defective HIV type 1 (HIV-1) reporter vectors carrying neomycin phosphotransferase or mouse heat stable antigen, replacing the HIV-1 sequences encoding gp160. These vectors also harbor inactive vpr, vpu, and nef coding regions. Pseudotyped HIV-1 particles carrying either the ecotropic or the amphotropic Mo-MLV envelope proteins or the vesicular stomatitis virus G protein were released after single or double transfections of either human 293T or monkey COS-7 cells with titers of up to 107 colony-forming units per milliliter. A simple ultrafiltration procedure resulted in an additional 10- to 20-fold concentration of the pseudotyped particles. These vectors along with Mo-MLV-based vectors were used to transduce primary human skin fibroblasts and human peripheral blood CD34+ cells. The HIV-1 vector system was significantly more efficient than its Mo-MLV-based counterpart in transducing human skin fibroblasts arrested at the G0/G1 stage of the cell cycle by density-dependent inhibition of growth. Human CD34+ cells were transduced efficiently using HIV-1 pseudotype particles without prior stimulation with cytokines.
Keywords: gene therapy, quiescent cells, human skin fibroblasts, CD34+ cells, cell cycle
Vectors based on onco-retroviruses, including Moloney murine leukemia virus (Mo-MLV), are used routinely to deliver therapeutic genes into primary cells in vitro (1). These viruses depend on cell proliferation for completion of their life cycle (2, 3). Breakdown of the nuclear envelope that accompanies mitosis appears to be essential for the nuclear import of the viral preintegration complex and to allow its integration into the genome of the host cell (4, 5). In contrast, lentiviruses, including HIV type 1 (HIV-1), differ fundamentally from onco-retroviruses in that they are relatively independent of cell division for completion of their replicative cycle. For instance, HIV-1 has been shown to infect terminally differentiated macrophages (6, 7), T lymphocytes (8, 9), brain microglial cells (10), and cells that are artificially arrested in the G1/S or G2 phases of the cell cycle (2, 5, 11, 12). However, the state of the viral genome and the extent of viral gene expression in quiescent, primary cells remain unclear. HIV-1, which is present in CD4+ T lymphocytes of the peripheral circulation and lymph nodes, is maintained predominantly either as extrachromosomal viral DNA (13, 14) or in a state of restricted transcription (15), and the demonstration of HIV-1 infection of primary, quiescent T cells in vitro has remained controversial. Several studies have shown that HIV-1 can readily enter quiescent T cells in culture, resulting in partially reverse-transcribed viral DNA (16, 17) or in full-length, but transcriptionally inactive, viral DNA (8), and that productive infection depends on subsequent cell activation. However, a recent report has provided evidence that highly purified CD4+ T lymphocytes in the G0/G1 state of the cell cycle are infectible by HIV-1, although such cells could not subsequently be stimulated to produce virus (9). Studies concerning the productive HIV-1 infection of monocytes or macrophages are equally contradictory. A number of reports have indicated that nonproliferating monocytes can be infected with HIV-1 independent of cellular DNA synthesis (6, 7, 18, 19), whereas Schuitemaker and coworkers (20) have shown that productive infection of monocyte-derived macrophages is restricted to the cell fraction with proliferating capacity and that the activation state that coincides with the G1/S phase of the cell cycle is essential, indicating that neither DNA synthesis nor mitosis is sufficient for establishing a productive HIV-1 infection in such cells. Taken together, these results show that HIV-1 is able to infect quiescent cells in vitro and that cell activation and/or differentiation, rather than cell division, are required, in some instances, for optimal replication of this virus.
We have been interested for some time in designing retrovirus-based vectors that are capable of infecting nondividing cells, and we have explored the possibility of using HIV-1-based vectors to deliver genes into primary, nondividing cells, including hematopoietic progenitor and stem cells and post-mitotic neurons. Page and coworkers (21) and Landau and coworkers (22) were the first to describe an HIV-1-based vector system consisting of a replication-defective HIV-1 vector with the sequences encoding the viral envelope (Env) protein (gp120) deleted. Cotransfection of the env-deficient genomes with expression vectors encoding heterologous Env proteins, including ecotropic and amphotropic Mo-MLV Env or human T-cell leukemia virus type I Env, led to the efficient formation of HIV-1 pseudotypes. In this paper, we describe an improved high-titer, two-plasmid HIV-1 expression system and show its applicability for the efficient transduction of nondividing, primary human cells.
MATERIALS AND METHODS
Plasmid Constructions.
HIV-1 BH10 sequences (23) from EcoRI (nucleotide 5101) to BamHI (nucleotide 7829) were subcloned into EcoRI- and BamHI-cut pBluescript II SK(−) (Stratagene). A 1.2-kb deletion in the env gene from the NdeI site at nucleotide 5758 to the BglII site at nucleotide 6976 was introduced. A 1169-bp PCR fragment from pBK-CMV (Stratagene) spanning the simian virus 40 (SV40) early promoter and neomycin phosphotransferase (neo) encoding regions was subsequently inserted at this site. The mouse heat stable antigen (HSA) coding region (24) was PCR-amplified from a cDNA plasmid kindly provided by R. Keith Humphries (Terry Fox Laboratory, Vancouver). A 168-bp fragment carrying the SV40 origin of replication was derived from pBK-CMV, as was the human cytomegalovirus (CMV) promoter (nucleotides 1300–1600). The plasmids were cut with SalI and BamHI, and the insert was subcloned into pHIVgpt (21), previously cut with SalI and BamHI to yield pHIV-neo and pHIV-HSA, respectively. Plasmid pSV-GL (25) was kindly provided by John Rose (Yale University, New Haven, CT). Plasmid pLTR-G was constructed by subcloning a 720-bp XhoI–HindIII fragment derived from pNL4-3 (26), spanning the 3′ HIV-1 long terminal repeat (LTR) into pGFP-C1 (CLONTECH) with the CMV promoter deleted. A 1.6-kb fragment encoding vesicular stomatitis virus G protein (VSV-G) was subsequently cloned downstream of the LTR. The following plasmids were obtained through the AIDS Research and Reference Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health (Bethesda): pHIVgpt from Kathleen Page and Dan Littman (21), pSV-A-MLV-env from Nathaniel Landau and Dan Littman (22), and pNL4-3 from Malcom Martin (26).
Cells.
293T, BOSC-23, and BING cells (27) were kindly provided by Warren Pear (Rockefeller University, New York). Human osteosarcoma (HOS) cells (CRL 1543), TE671 cells (CRL 8805), COS-7 cells (CRL 1651), and primary human skin fibroblasts (HSF; CRL 2072) were obtained from the American Type Culture Collection. The cells were grown in DMEM (GIBCO/BRL) containing 10% heat-inactivated fetal bovine serum (FBS). The human A3.01 T-cell line was obtained from Thomas Folks (28) through the AIDS Research and Reference Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health. The cells were grown in RPMI 1640 medium (GIBCO/BRL) containing 10% FBS. Quiescent HSF were obtained by growing passage 5 or 9 cells to confluency for 20–30 days in DMEM containing 10% FBS (29). Dividing HSF were obtained by treatment with trypsin and plating of the the cells 1 day before infection. G-CSF-mobilized human peripheral blood CD34+ cells previously enriched using a Ceprate Stem Cell Concentrator (CellPro, Bothell, WA) (30) were kindly provided by Susan Leitman (Department of Transfusion Medicine, National Institutes of Health). The cells were incubated in 6-well plates in DMEM containing 15% FBS and 20 ng/ml interleukin 3 (PreproTech, Inc., Rocky Hill, NJ), 50 ng/ml interleukin 6 (PreproTech, Inc.), and 100 ng/ml stem cell factor (R & D Systems).
Virus Production.
Pseudotyped viruses were generated by transfection of plasmid DNA into 293T or COS-7 cells by calcium-phosphate precipitation (27). Transfections were done in 6-well dishes using 5 μg of vector DNA and 5 μg of the env plasmid DNA per well containing 2 ml of DMEM including 10% FBS. Chloroquine was added to the 293T cells before the DNA precipitate was added to yield a final concentration of 25 μM. The medium was changed 12 hr later, and 48–72 hr after the start of transfection, the medium was removed and filtered through a 0.45-μm Millex-HA filter (Millipore). Viruses were concentrated by centrifugation through a Centriprep 100 concentrator (Amicon) for 30 min at 1500 rpm (5°C) in a Beckman GPR centrifuge. Virus stocks were aliquoted and stored at −80°C. Extracellular p24 levels were measured using a RETRO-TEK ELISA kit (Cellular Products).
Transduction of Cells.
A3.01 cells were infected in the presence of 10 μg/ml DEAE-dextran. All other cells were infected in DMEM/FBS containing 4–8 μg/ml polybrene for 4–24 hr.
PCR.
High molecular weight DNA was extracted using the Hirt procedure (31). Semiquantitative PCR amplification with neo gene-specific primers (32) was performed for 28 cycles. PCR products were transferred to a Hybond-N+ membrane (Amersham) and subsequently probed using an oligonucleotide (5′-CCGGCTGTCAGCGCAGGGGCGCCC) applying an Amersham enhanced chemiluminescence kit.
Flow Cytometry.
Cells for fluorescence-activated cell sorter (FACS) analysis were stained with a fluorescein isothiocyanate-labeled anti-HSA monoclonal antibody (Caltag, South San Francisco, CA) for 30 min on ice in Hanks’ balanced salt solution (GIBCO/BRL) containing 2% FBS. The cells were washed twice with PBS and resuspended in 4% paraformaldehyde. For cell cycle analysis, cells were pelleted and resuspended in 1 ml of cold PBS. Subsequently, 2 ml of ice-cold methanol were added. The cells were processed using a CycleTEST PLUS DNA reagent kit (Becton Dickinson).
RESULTS
Construction of HIV-1-Based Vectors.
The design of the prototype HIV-1 vector is outlined in Fig. 1A. The vector backbone was derived from the HXB2 molecular clone (33) that encodes truncated forms of Vpr, Vpu, and Nef. A 1.2-kb deletion in the env coding region was introduced, leaving the rev responsive element and the tat and rev exons intact. An expression cassette consisting of the SV40 early promoter driving the bacterial neo gene was subsequently introduced into the deleted env coding region. This vector is referred to as HIV-neo. An additional vector was constructed carrying the mouse HSA coding region as a reporter gene under the control of the human CMV early promoter. With a view toward boosting the vector copy number in transiently transfected cells producing SV40 T antigen, such as 293T and COS-7 cells, a 168-bp fragment spanning the SV40 origin of replication was added to the vector upstream of the CMV promoter. This vector is referred to here as HIV-HSA.
Figure 1.
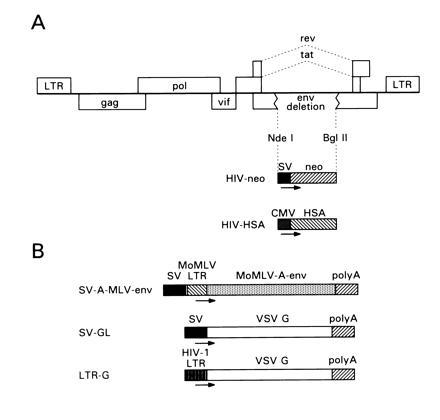
Recombinant plasmids for production of HIV-1 pseudotypes. (A) Recombinant HIV vectors. (B) Structure of Env expression vectors.
Pseudotyping of HIV-1 Vectors Using Heterologous Envelopes.
To test the functionality of the HIV-neo vector construct, high-titer virus stocks were generated by transient cotransfection of human 293T, BING, or BOSC-23 cells or monkey COS-7 cells using the pHIV-neo plasmid together with an Env-encoding plasmid (Fig. 1B). The efficiency of transfection was assessed indirectly by measuring the levels of extracellular p24 in the supernatants of the cells, and the yield of infectious virus particles was determined by measuring the efficiency of transduction of HOS cells, human medulloblastoma (TE671) cells, and mouse 3T3 cells and scored through the formation of G418-resistant colonies. The results presented in Table 1 show that transduction was strictly dependent on the presence of an Env-encoding plasmid and that a number of heterologous envelopes including the amphotropic and ecotropic Env proteins of Mo-MLV and VSV-G led to the formation of pseudotyped virus. Pseudotype formation was very efficient with the amphotropic Mo-MLV Env and with VSV-G in 293T cells resulting in titers of up to 107 colony-forming units (cfu)/ml or greater. The viral stocks contained up to 600 cfu/pg of p24. These values are up to 200-fold higher than the ones previously described for HIV-1 particles (34), indicating that HIV-1 pseudotypes are more infectious than their natural counterparts.
Table 1.
Generation of HIV-neo pseudotypes
| Cells transfected | Virus envelope | p24, ng/ml | Virus
titer on cells,* cfu/ml
|
||
|---|---|---|---|---|---|
| 3T3 | HOS | TE671 | |||
| 293T | None | 242 | <101 | <101 | <101 |
| Mo-MLV-A | 278 | 6.1 × 106 | 6.0 × 106 | 7.8 × 106 | |
| VSV-G (SV-GL) | 44 | 1.2 × 105 | 1.1 × 106 | 8.0 × 105 | |
| VSV-G (LTR-G) | 95 | 2.3 × 107 | 3.1 × 107 | 3.1 × 107 | |
| BOSC-23 | Mo-MLV-E | 278 | 5.3 × 106 | NA | NA |
| BING | Mo-MLV-A | 266 | 1.7 × 105 | NA | NA |
| COS-7 | Mo-MLV-A | 27.5 | 5.8 × 106 | 4.2 × 106 | 3.7 × 106 |
| VSV-G (SV-GL) | 7.9 | 2.0 × 106 | 3.0 × 105 | 6.0 × 105 | |
| VSV-G (LTR-G) | 6.7 | 2.2 × 106 | 2.0 × 106 | 4.0 × 106 | |
NA, not available; cfu, colony-forming units.
Infected cells were trypsinized 3 days after transduction and cultured for 10–14 days in DMEM/FBS containing G418 (0.35 mg/ml active drug). Colonies were stained using 0.2% crystal violet in 20% ethanol. Data from one representative experiment of several are shown.
The results in Table 2 show that pseudotyped virus stocks harboring amphotropic Mo-MLV Env or VSV-G can be concentrated by ultrafiltration. The recovery of infectious virus particles harboring Mo-MLV amphotropic Env varied between 66% and 100%. A 12- to 14-fold reduction in volume resulted in an 8.5- to 10-fold increase in titer with a 60–83% recovery of infectious virus particles harboring VSV-G.
Table 2.
Concentration of virus stocks by ultrafiltration
| Virus envelope | Initial titer, cfu/ml | Final titer, cfu/ml | Volume change | Titer increase | % recovery |
|---|---|---|---|---|---|
| Experiment 1 | |||||
| Mo-MLV-A | 2.0 × 106 | 2.8 × 107 | 14-fold | 14.2-fold | 100 |
| VSV-G (SV-GL) | 1.8 × 105 | 1.5 × 106 | 14-fold | 8.5-fold | 60 |
| Experiment 2 | |||||
| Mo-MLV-A | 1.0 × 107 | 6.7 × 107 | 10-fold | 6.7-fold | 66 |
| VSV-G (LTR-G) | 1.0 × 107 | 1.0 × 108 | 12-fold | 10-fold | 83 |
In experiment 1, 293T cells in 82-mm dishes were transfected with 22 μg of pHIV-neo along with 21 μg of pSV-A-MLV env or 10.5 μg of pSV-GL. Sixty-five hours after transfection, the culture supernatants were collected and filtered through a 0.45-μm filter. In experiment 2, transfection was carried out in 6-well dishes with 5 μg of pHIV-neo and 5 μg of the Env-encoding plasmid per well. Ten-milliliter aliquots of the supernatants were concentrated by using a Centriprep 100 concentrator. Virus titers were determined by measuring the formation of G418-resistant colonies using TE671 cells (experiment 1) or HOS cells (experiment 2).
The functionality of the HIV-HSA vector construct was assessed by FACS analysis. HOS cells and the A3.01 T-cell line were infected with G-pseudotyped particles, and the cells were stained 72–96 hr later with fluorescein isothiocyanate-labeled anti-HSA. Conservatively, at least 65% of the infected HOS cells expressed HSA (Fig. 2A), and 28% of the transduced A3.01 cells were positive (Fig. 2B), indicating that the infection and expression of the reporter cell surface antigen were very efficient.
Figure 2.
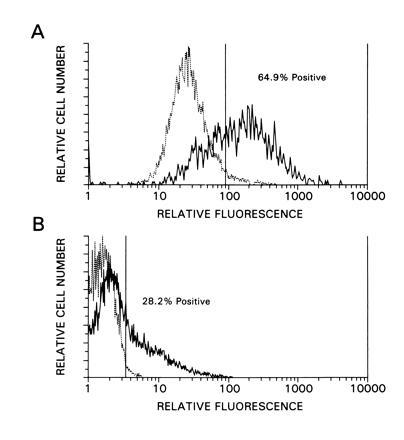
Infection efficiency of HIV-HSA. (A) HOS cells were infected and tested for the expression of the HSA reporter gene by FACS analysis 96 hr after infection. The broken line represents uninfected cells, whereas the thick line corresponds to infected cells. (B) Human A3.01 cells were subjected to FACS analysis 72 hr after infection. The broken line in each panel represents uninfected cells, whereas the thick line corresponds to infected cells.
Transduction of Quiescent and Proliferating HSF.
With a view toward testing the performance of the pseudotyped HIV-neo vectors in nondividing cells, quiescent primary HSF were used as a model. Contact-inhibited HSF were prepared by incubation in DMEM containing 10% FBS for up to 4 weeks, resulting in a cell population with about 90% of the cells in G0/G1 (Fig. 3A). These cells were subsequently infected with amphotropic HIV-neo pseudotyped viral constructs or an amphotropic Mo-MLV-derived virus stock (LGSN). Dividing cells were prepared by trypsin treatment and subsequent subcultivation 1 day before infection. The results in Fig. 3B show that between 60% and 70% of the dividing HSF were transduced efficiently by both HIV-neo and LGSN as measured by the formation of G418-resistant cell clones. In contrast, stationary HSF were preferentially transduced by HIV-neo as were HSF cultures that were split 3 days after transduction. This shows that the HIV-neo virus in contrast to the Mo-MLV-based virus infected stationary HSF cultures efficiently under the conditions used. To analyze the fate of the neo reporter gene, high molecular weight DNA from stationary and dividing HSF previously infected with HIV-neo pseudotypes was extracted 11 days after infection and analyzed by semiquantitative PCR using neo-specific primers. Between 25% and 50% of the DNA was positive for the neo fragment (data not shown). This is consistent with the view that proviral DNA has integrated into the genome of a large fraction of the cells.
Figure 3.
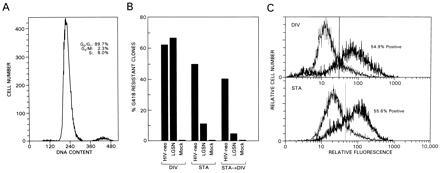
Transduction of stationary and dividing primary HSF cultures. (A) Cell cycle analysis of stationary HSF cultures. The percentage of cells in a particular stage of the cell cycle is indicated. (B) Relative transduction efficiency of dividing (DIV) and stationary (STA) fibroblasts. Dividing cultures were prepared by treatment with trypsin and replating 24 hr before infection or 72 hr after infection (STA→DIV). Transduction efficiency was assessed after trypsin treatment by plating the cells into medium with and without G418 (0.35 mg/ml of active drug after 14 days) and counting the clones 2 weeks later. The percentage of G418-resistant clones reflects the number of surviving clones in DMEM/10% FBS containing G418 relative to the number of clones present in DMEM/10% FBS alone. (C) FACS analysis of dividing and stationary HSF infected with HIV-HSA. The cells were subjected to FACS analysis 4 days after infection. Thick line, infected cells; thin line, uninfected cells.
Stationary and dividing HSF were infected using HIV-HSA pseudotyped with VSV-G and analyzed by FACS analysis to assess the impact of cell division on the expression of the reporter gene. At least 54% of the infected HSF expressed HSA (Fig. 3C) whether or not the cells were dividing or stationary, indicating that efficient gene expression was independent of cell division.
Transduction of Nondividing Human Peripheral Blood CD34+ Cells.
Hematopoietic progenitor cells, including CD34+ cells, require prestimulation with cytokines to make them accessible for efficient infection with Mo-MLV-based vectors. To test the performance of HIV-HSA pseudotypes in unstimulated human CD34+ cells, peripheral blood human CD34+ cells, highly enriched for G0/G1 cells (Fig. 4A) were infected with HIV-HSA pseudotyped with VSV-G and cultivated in media supplemented with IL-3, IL-6, and stem cell factor for 71 hr and subsequently subjected to FACS analysis. Well over 80% of the infected cells expressed the HSA reporter gene after only one cycle of infection. This contrasts favorably to results obtained using a Mo-MLV-based HSA vector (35). In these experiments, only 10% of the cells expressed the HSA reporter gene despite the fact that the cells had gone through four cycles of infection in the presence of stromal cells.
Figure 4.
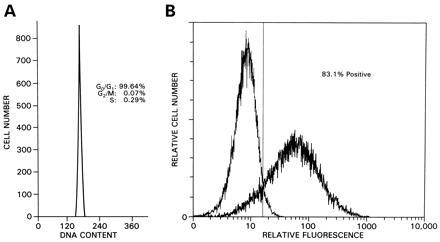
Transduction of nondividing human CD34+ cells. (A) Cell cycle analysis of peripheral blood CD34+ cells. (B) FACS analysis. CD34+ cells (4 × 105) were infected and processed for FACS analysis 71 hr later. Dead cells were gated away after propidium iodide staining. Thick line: infected cells; thin line: mock infected cells.
DISCUSSION
The mixing of Env glycoproteins in doubly infected cells to form pseudotyped HIV virions has been reported in the past, and a number of studies have demonstrated that HIV produced in cells infected with xenotropic murine leukemia virus (36, 37), amphotropic murine leukemia virus (38, 39), or herpes simplex virus (40) gave rise to phenotypically mixed virions with an expanded host range, suggesting that pseudotyped virions had formed. Also, phenotypic mixing of viral envelopes was shown to occur between HIV-1 and VSV in cells coinfected with these viruses (40). Page and coworkers (21) have shown that expression of amphotropic or ecotropic Mo-MLV Env glycoproteins in cells transfected with a HIV-1 vector construct resulted in the production of virus capable of infecting both human and murine cells, and Landau and coworkers (22) have demonstrated that HIV-1 efficiently incorporated the human T-cell leukemia virus type I Env. The results presented in this manuscript confirm and extend these earlier observations in that the VSV-G was found to be incorporated very efficiently into HIV-1 virions with pseudotyped viral titers reaching 107 cfu/ml or higher. Moreover, we have developed a facile ultrafiltration procedure that has allowed us to concentrate such HIV-1 pseudotypes 10-fold or more, thus raising the titers to 108 cfu/ml or higher. Also, good recoveries were obtained with VSV-G as well as Mo-MLV amphotropic Env containing particles. This finding is in contrast to the concentration attempts using ultracentrifugation in which retrovirus particles with Mo-MLV-based envelopes suffer dramatic losses in infectivity (41).
A number of HIV-1 vector systems have been described in the past. With the original two-plasmid expression system of Page and coworkers (21) and Landau and coworkers (22), titers of up to 2 × 105 cfu/ml were obtained using HOS cells. A HIV-1 reporter vector that encodes human placental alkaline phosphatase incorporated within the nef coding region with pseudotyped virus titers close to 2 × 105/ml was recently described by He and Landau (42). Several groups have designed three-component HIV expression systems consisting of a packaging construct, a plasmid encoding HIV Env, and an expression vector carrying a reporter gene (43–50). In general, these systems proved to be quite inefficient with titers around 104 cfu/ml or below.
While this work was in progress, two papers describing HIV-1 vector systems pseudotyped with VSV-G were published. Naldini and coworkers (51) described a three-plasmid expression system to generate HIV-1 based vectors by transient transfection of 293T cells. The titers obtained in this system were in the order of 105 transducing units/ml. The pseudotyped vectors transduced Escherichia coli β-galactosidase and firefly luciferase reporter genes into proliferating rat 208F fibroblasts, but cells arrested in G0 by density-dependent inhibition of growth in the presence of dexamethasone were not transduced efficiently. The vectors were shown to be capable of transducing human monocyte-derived macrophages, and highly concentrated stocks could mediate stable in vivo gene transfer into terminally differentiated neurons. In a recent study, Akkina and coworkers (52) described the use of VSV-G-pseudotyped vectors to transduce CD34+ cells. Although the authors have claimed that their vector system is efficient, the exact viral titers are difficult to judge from the data presented. Also, the culture conditions used for the CD34+ cells were not reported, and it is not clear from the data whether the cells were cycling before infection. Bartz and coworkers (53) also used the VSV-G protein to pseudotype env-deficient HIV-1 backbones. Titers of 107 infectious units/ml were obtained from cell-free supernatants after transient cotransfection of 293T cells.
The improved first-generation HIV-1 vector system described here was readily capable of infecting cell populations highly enriched for G0/G1 cells under conditions where Mo-MLV vectors performed much less efficiently, making the HIV-1 vector system potentially more attractive for the delivery of therapeutic genes under conditions where Mo-MLV vectors are not useful. A number of recent reports (54–56) have clearly demonstrated that gene expression from the HIV LTR and from heterologous promoters in HIV-1 vectors is strictly dependent on integration. Thus, we are presenting indirect evidence for the fact that the proviral DNA had integrated. Our data also show that the Vpr, Vpu, and Nef functions are not required for the production of high-titer HIV-1 vector stocks in the cells used.
Acknowledgments
This project was supported, in part, by a grant from the Office of AIDS Research, National Institutes of Health. We thank Dr. Cecilia Fahlmann, Dr. Makoto Migita, and Dr. Jeff Medin for their help and suggestions. Ann Brun and Mike Amiri provided competent technical assistance. We are grateful to Dr. Jeff Medin and Dr. A. Dusty Miller for critical reading of the manuscript.
Footnotes
Abbreviations: cfu, colony-forming units; CMV, cytomegalovirus; env, envelope; FACS, fluorescence-activated cell sorter; FBS, fetal bovine serum; HIV-1, HIV type 1; HOS, human osteosarcoma; HSA, heat stable antigen; HSF, human skin fibroblasts; LTR, long terminal repeat; Mo-MLV, Moloney murine leukemia virus; neo, neomycin phosphotransferase; RT, reverse transcription; SV40, simian virus 40; VSV-G, vesicular stomatitis virus G protein.
References
- 1.Crystal R G. Science. 1995;270:404–410. doi: 10.1126/science.270.5235.404. [DOI] [PubMed] [Google Scholar]
- 2.Springett G M, Moen R C, Anderson S, Blaese R M, Anderson W F. J Virol. 1989;63:3865–3869. doi: 10.1128/jvi.63.9.3865-3869.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller D G, Adam M A, Miller A D. Mol Cell Biol. 1990;10:4239–4242. doi: 10.1128/mcb.10.8.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roe T, Reynolds T C, Yu G, Brown P O. EMBO J. 1993;12:2099–2108. doi: 10.1002/j.1460-2075.1993.tb05858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis P F, Emerman M. J Virol. 1994;68:510–516. doi: 10.1128/jvi.68.1.510-516.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinberg J B, Matthews T J, Cullen B R, Malim M H. J Exp Med. 1991;174:1477–1482. doi: 10.1084/jem.174.6.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freed E O, Englund G, Martin M A. J Virol. 1995;69:3949–3954. doi: 10.1128/jvi.69.6.3949-3954.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spina C A, Guatelli J C, Richman D R. J Virol. 1995;69:2977–2988. doi: 10.1128/jvi.69.5.2977-2988.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang S, Patterson B, Levy J A. J Virol. 1995;69:5659–5665. doi: 10.1128/jvi.69.9.5659-5665.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watkins B A, Dorn H H, Kelly W B, Armstrong R C, Potts B J, Michaels F, Kufta C B, Dubois-Dalcq M. Science. 1990;249:549–553. doi: 10.1126/science.2200125. [DOI] [PubMed] [Google Scholar]
- 11.Lewis P, Hensel M, Emerman M. EMBO J. 1992;11:3053–3058. doi: 10.1002/j.1460-2075.1992.tb05376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li G, Simm M, Potash M J, Volsky D J. J Virol. 1993;67:3969–3977. doi: 10.1128/jvi.67.7.3969-3977.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevenson M, Stanwick T L, Dempsey M P, Lamonica C A. EMBO J. 1990;9:1551–1560. doi: 10.1002/j.1460-2075.1990.tb08274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bukrinsky M I, Stanwick T L, Dempsey M P, Stevenson M. Science. 1991;254:423–427. doi: 10.1126/science.1925601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCune J M. Cell. 1995;82:183–188. doi: 10.1016/0092-8674(95)90305-4. [DOI] [PubMed] [Google Scholar]
- 16.Zack J A, Arrigo S J, Weitsman S R, Go A S, Haislip A, Chen I S Y. Cell. 1990;61:213–222. doi: 10.1016/0092-8674(90)90802-l. [DOI] [PubMed] [Google Scholar]
- 17.Zack J A, Haislip A M, Krogstad P, Chen I S Y. J Virol. 1992;66:1717–1725. doi: 10.1128/jvi.66.3.1717-1725.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinzinger N K, Bukrinsky M L, Haggerty S A, Ragland A M, Kewalramani V, Lee M-A, Gendelman H E, Ratner L, Stevenson M, Emerman M. Proc Natl Acad Sci USA. 1994;91:7311–7315. doi: 10.1073/pnas.91.15.7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Schwendler U, Kornbluth R S, Trono D. Proc Natl Acad Sci USA. 1994;91:6992–6996. doi: 10.1073/pnas.91.15.6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuitemaker H, Koostra N A, Fouchier R A M, Hooibrink B, Miedema F. EMBO J. 1994;13:5929–5936. doi: 10.1002/j.1460-2075.1994.tb06938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Page K A, Landau N R, Littman D R. J Virol. 1990;64:5270–5276. doi: 10.1128/jvi.64.11.5270-5276.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Landau N R, Page K A, Littman D R. J Virol. 1991;65:162–169. doi: 10.1128/jvi.65.1.162-169.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ratner L, Haseltine W, Patarca R, Livak K J, Starcich B, Josephs S F, Doran E R, Rafalski J A, Whitehorn E A, Baumeister K, Ivanoff L, Petteway S R, Jr, Pearson M L, Lautenberger J A, Papas T S, Ghrayeb J, Chang N T, Gallo R C, Wong-Staal F. Nature (London) 1985;313:277–284. doi: 10.1038/313277a0. [DOI] [PubMed] [Google Scholar]
- 24.Kay R, Takei F, Humphries R K. J Immunol. 1990;145:1952–1959. [PubMed] [Google Scholar]
- 25.Rose J K, Bergman J E. Cell. 1982;30:753–782. doi: 10.1016/0092-8674(82)90280-x. [DOI] [PubMed] [Google Scholar]
- 26.Adachi A, Gendelman H E, Loenig S, Folks T, Willey R, Rabson A, Martin M A. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pear W, Nolan G, Scott M, Baltimore D. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Folks T, Benn S, Rabson A, Theodore T, Hoggan M D, Martin M, Lightfoote M, Sell K. Proc Natl Acad Sci USA. 1985;82:4539–4545. doi: 10.1073/pnas.82.13.4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tobey R A, Valdez J G, Crissman H A. Exp Cell Res. 1988;179:400–416. doi: 10.1016/0014-4827(88)90279-0. [DOI] [PubMed] [Google Scholar]
- 30.Dunbar C E, Cottler-Fox M, O’Shaughnessy J A, Doren S, Carter C, Berenson R, Brown S, Moen R C, Greenblatt J, Stewart F M, Leitman S F, Wilson W H, Cowan K, Young N S, Nienhuis A W. Blood. 1995;85:3048–3057. [PubMed] [Google Scholar]
- 31.Hirt B. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 32.Lam J S, Reeves M E, Cowherd R, Rosenberg S A, Hwu P. Hum Gene Ther. 1996;7:1415–1422. doi: 10.1089/hum.1996.7.12-1415. [DOI] [PubMed] [Google Scholar]
- 33.Ratner L, Fisher A, Jagodzinski L L, Mitsuya H, Liou R-S, Gallo R, Wong-Staal F. AIDS Res Hum Retroviruses. 1987;3:57–69. doi: 10.1089/aid.1987.3.57. [DOI] [PubMed] [Google Scholar]
- 34.Kimpton J, Emerman M. J Virol. 1992;66:2232–2239. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Medin J A, Migita M, Pawliuk R, Jacobson S, Amiri M, Kluepfel-Stahl S, Brady R O, Humphries R K, Karlsson S. Blood. 1996;87:1754–1762. [PubMed] [Google Scholar]
- 36.Lusso P, di Marzo Veronese F, Ensoli B, Franchini G, Jemma C, de Rocco S E, Kalyanaraman V S, Gallo R C. Science. 1990;247:848–852. doi: 10.1126/science.2305256. [DOI] [PubMed] [Google Scholar]
- 37.Canivet M, Hoffman A D, Hardy D, Sernatinger J, Levy J A. Virology. 1990;178:543–551. doi: 10.1016/0042-6822(90)90352-r. [DOI] [PubMed] [Google Scholar]
- 38.Spector D, Wade E, Wright D A, Koval V, Clark C, Jaquish D, Spector S A. J Virol. 1990;64:2298–2308. doi: 10.1128/jvi.64.5.2298-2308.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chesebro B, Wehrly K, Maury W. J Virol. 1990;64:4553–4557. doi: 10.1128/jvi.64.9.4553-4557.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu Z, Chen S S L, Huang A S. J Acquired Immune Defic Syndr. 1990;3:215–219. [PubMed] [Google Scholar]
- 41.Burns J C, Friedmann T, Driever W, Burrascano M, Yee J-K. Proc Natl Acad Sci USA. 1993;90:8033–8037. doi: 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He J, Landau N R. J Virol. 1995;69:4587–4592. doi: 10.1128/jvi.69.7.4587-4592.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poznansky M, Lever A, Bergeron L, Haseltine W, Sodroski J. J Virol. 1991;65:532–536. doi: 10.1128/jvi.65.1.532-536.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimada T, Fujii H, Mitsuya H, Nienhuis A W. J Clin Invest. 1991;88:1043–1047. doi: 10.1172/JCI115365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buchschacher G L, Jr, Panganiban A T. J Virol. 1992;66:2731–2739. doi: 10.1128/jvi.66.5.2731-2739.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strair R K, Medina D J, Nelson C J, Graubert T A, Mellors J W. Nucleic Acids Res. 1993;21:4836–4842. doi: 10.1093/nar/21.20.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parolin C, Dorfman T, Palú G, Göttlinger H, Sodroski J. J Virol. 1994;68:3888–3895. doi: 10.1128/jvi.68.6.3888-3895.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carroll R, Lin J-T, Joy Dacquel E, Mosca J D, Burke D S, St. Louis D C. J Virol. 1994;68:6047–6051. doi: 10.1128/jvi.68.9.6047-6051.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richardson J H, Kaye J F, Child L A, Lever A M L. J Gen Virol. 1995;76:691–696. doi: 10.1099/0022-1317-76-3-691. [DOI] [PubMed] [Google Scholar]
- 50.Yu H, Rabson A B, Kaul M, Ron Y, Dougherty J P. J Virol. 1996;70:4530–4537. doi: 10.1128/jvi.70.7.4530-4537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage F, Verma I M, Trono D. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 52.Akkina R K, Walton R M, Chen M L, Li Q-X, Planelles V, Chen I S Y. J Virol. 1996;70:2581–2585. doi: 10.1128/jvi.70.4.2581-2585.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bartz S R, Rogel M E, Emerman M. J Virol. 1996;70:2324–23331. doi: 10.1128/jvi.70.4.2324-2331.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wiskerchen M, Muesing M A. J Virol. 1995;69:376–386. doi: 10.1128/jvi.69.1.376-386.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Englund G, Theodore T S, Freed E O, Engelman A, Martin M A. J Virol. 1995;69:3216–3219. doi: 10.1128/jvi.69.5.3216-3219.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leavitt A D, Robles G, Alesandro N, Varmus H E. J Virol. 1996;70:721–728. doi: 10.1128/jvi.70.2.721-728.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]