

Abstract
Aims
The role of nitric oxide (NO) in heart failure (HF) is complex and remains controversial. We tested the hypothesis that the role of NO in isolated atria and cardiomyocytes is altered in isoproterenol-induced HF.
Methods and results
Rats received isoproterenol (ISO, 5 mg/kg/day, intraperitoneally) or vehicle for 1 week. Haemodynamic parameters were obtained by left ventricular catheterization. Effects of NOS inhibition on isolated atria and on electrically paced left ventricular myocytes were determined. Additionally, expressions of nitric oxide synthases and their allosteric modulators hsp90, caveolin-1, and caveolin-3 proteins in the left ventricles were measured. ISO increased left ventricular mass by 33% and decreased indices of left ventricular systolic and diastolic function dp/dtmin and dp/dtmax (both P < 0.05). Isolated atria from HF rats had a lower spontaneous beating rate (P < 0.05). NOS inhibition by L-NAME increased basal frequency and attenuated the positive chronotropic effect of beta-adrenergic stimulation in the HF group (P < 0.05). Ventricular myocytes from failing hearts had impaired cell shortening. L-NAME decreased contractility of control, but not failing myocytes. Left ventricular expressions of eNOS, hsp90, iNOS, but not nNOS or caveolins, were increased.
Conclusion
Despite the increased capacity for NO synthesis in isoproterenol-induced HF, NO does not sustain contractility of failing myocytes. NO may contribute to the decreased basal heart rate and it may accelerate beta-adrenergic stimulation of chronotropy.
Keywords: Isoproterenol-induced heart failure, Nitric oxide, Nitric oxide synthases, Heart rate, Ventricular function
Introduction
In the heart, nitric oxide is produced by all three isoforms of nitric oxide synthases, endothelial (eNOS), inducible (iNOS), and neuronal (nNOS) nitric oxide synthase. Although eNOS is expressed mainly in the endothelial cells, cardiac myocytes can express all three isoforms of the enzyme and may regulate several aspects of cardiac function including contractility and heart rate.1
In a healthy heart, physiological amounts of NO may help sustain cardiac inotropy.2 In heart failure, both upregulation3 and downregulation4 of eNOS have been observed. Overexpression of eNOS in cardiomyocytes was found to improve cardiac function and reduce hypertrophy in heart failure after myocardial infarction.5 eNOS-derived nitric oxide can decrease the extent of myocardial hypertrophy and fibrosis6 and decrease myocyte loss.7 It can also increase the perfusion of the myocardium through vasodilation and increase in angiogenesis.8 This suggests a protective role of eNOS in heart failure.
On the other hand, iNOS-derived NO may depress cardiac contractility in heart failure,9 where overexpression of this enzyme has been reported.10 Yet, data from transgenic mice have shown little effect of iNOS on cardiac contractility,11 but increased occurrence of sudden cardiac death.12 Similarly, Bendall et al.13 reported a role for overexpressed nNOS in the blunted response to a beta-agonist in post-infarction heart failure in the rat and nNOS has been found to be overexpressed in human heart failure.14
Isoproterenol-induced cardiac hypertrophy is a reliable, reproducible, and well-characterized model of cardiac hypertrophy15 associated with arrhythmias, myocyte loss, and fibrosis, with progression to heart failure. As opposed to pressure overload-induced heart failure, no surgical intervention is needed and, specifically, isoproterenol treatment is associated with marked myocardial ischaemia. Transgenic mice overexpressing eNOS were partly protected against hypertrophy and fibrosis evoked by isoproterenol administration.6 We have previously reported overexpression of eNOS in the vessels of rats with isoproterenol-induced heart failure.16 Caveolin-1 expressed mainly in the endothelial cells and the muscle-specific isoform caveolin-3 are negative regulators of eNOS activity, while heat shock protein 90 (hsp90) enhances eNOS activation.17 The status of the NO system in the heart of rats with isoproterenol-induced cardiac hypertrophy and failure has not been determined. Therefore, the aim of this work was to describe effects of nitric oxide on the regulation of heart rate and myocardial contractility and expression of nitric oxide synthases with their allosteric regulators in isoproterenol-induced heart failure.
Methods
We used male Wistar rats (16 weeks old). Rats were kept under standard conditions and received food and water ad libitum. Isoproterenol (5 mg/kg) was administered for one week, once daily by intraperitoneal injection. Control animals received the vehicle of isoproterenol (0.05% ascorbic acid in 0.9% NaCl). Separate groups of rats were used for left ventricular catheterization, spontaneously beating atria, left ventricular myocytes, and finally for western blot analyses. All studies were performed 24 h after the last isoproterenol administration.
To detect myocardial ischaemia, 12-lead electrocardiography was performed in rats anaesthetized with tribromoethanol (15 µl of 2.5% solution per g of body weight),18 which has good analgesic properties and has been demonstrated to have a less depressive influence on cardiac function than some other commonly used anaesthetic procedures.19 ST segment depression was taken as a sign of ischaemic injury. Left ventricular catheterization was then performed in closed-chest rats using modifications of the method described previously.20 Briefly, the left jugular vein was cannulated with a custom-fashioned polyethylene tube connected to a microinfusion pump for drug administration. A polyethylene tube (PE50, 0.58 mm ID × 0.96 mm OD, Portex, England) was inserted into the aorta via the right carotid artery and advanced into the left ventricle under continuous monitoring of the pressure waveform. Pressure signals were digitized and recorded with a S.P.E.L. Advanced Haemosys system (Experimetria Ltd, Hungary). Heart rate, left ventricular pressure (LVP), and the first derivatives of left intraventricular pressure (rate of pressure development +dP/dt and rate of pressure decrease −dP/dt) were monitored continuously and recorded and analysed after 10 min of stabilization. Additionally, the effect of acute beta-adrenergic stimulation (isoproterenol, 60 ng min−1, intravenously) on the above parameters was determined. For determination of fibrosis, paraffin-embedded left ventricular samples were processed by standard Sirius-red staining.
Function of spontaneously beating isolated right atria was evaluated at 37°C in Tyrode’s solution. Atria were stabilized for 30 min with tension of 10 mN. Basal beating rate was measured. Isoproterenol was then added cumulatively into the organ bath at a concentration of 10−10 to 10−6 M. Basal and isoproterenol-stimulated heart rate was measured in the presence or absence of the NOS inhibitor L-NAME (3 × 10−4 M).
Left ventricular cardiac myocytes were isolated by collagenase digestion. The cells were electrically paced at 0.5 Hz frequency. Relative cell shortening was measured by videomicroscopy. The method has been described previously.20 The effects of isoproterenol (10−7 M) and nitric oxide synthase inhibitor L-NAME (3 × 10−4 M) on cell shortening were determined.
For western blot analysis, tissue samples from rats sacrificed by CO2 asphyxiation were frozen in liquid nitrogen and stored at –20°C until further processing. Expressions of eNOS, iNOS, nNOS, hsp90, caveolin-1, and caveolin-3 were determined by western blot analysis with chemiluminescent detection (ECL Plus, Amersham) and compared with the expression of actin. In preliminary experiments, we verified that specific anti-eNOS, iNOS, nNOS, hsp90, caveolin-1, caveolin-3 (BD Pharmingen, Franklin Lakes, USA), and anti-actin (Sigma-Aldrich) antibodies detected a single band of expected molecular weight in positive controls or ventricular homogenates, and that expression of actin was not changed by isoproterenol administration. As a positive control for iNOS, we used homogenates from lungs of rats exposed to bacterial lipopolysacharide (LPS, 3 mg/kg, Sigma-Aldrich) for 4 h to increase expression of iNOS. For nNOS, brain homogenates were used as a positive control. Quantification was performed as described previously.16
All procedures involving the use of experimental animals were approved by the State Veterinary and Food Administration of the Slovak Republic. The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1985).
Results are expressed as average ± standard error of the mean. Means were compared using unpaired Student’s t-test in GraphPad Prism 4.0 (GraphPad Software, Inc.).
Results
Biometric data and histology
Overall mortality associated with 1 week of isoproterenol administration was 30%. Biometric parameters in the surviving rats are presented in Table 1. Body weight was slightly decreased by the treatment. The atria and ventricles were hypertrophied significantly (Table 1) and fibrosis was observed in the ventricles (Figure 1).
Table 1.
Heart and body weights after 1 week of isoproterenol administration (n = 10–11 per group)
| Control | Heart failure | % difference | |
|---|---|---|---|
| Body weight (g) | 327 ± 19 | 293 ± 26* | −10 |
| Left ventricle (g) | 0.67 ± 0.05 | 0.89 ± 0.09* | +33 |
| Right ventricle (g) | 0.15 ± 0.03 | 0.21 ± 0.04* | +40 |
| Left atrium (g) | 0.032 ± 0.007 | 0.043 ± 0.010* | +34 |
| Right atrium (g) | 0.036 ± 0.01 | 0.048 ± 0.011* | +34 |
*P < 0.05 vs. Control.
Figure 1.
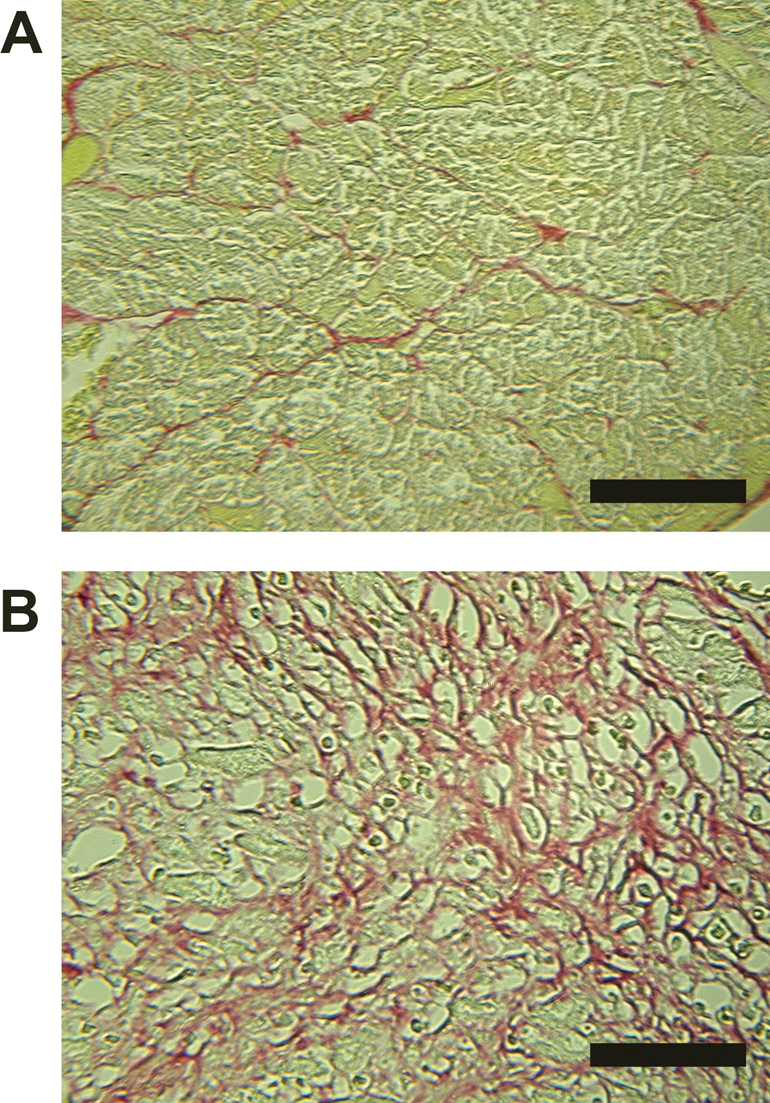
Representative Sirius-red-stained sections of the left ventricles of control rats (A) and rats with isoproterenol-induced heart failure (B). Red areas represent fibrosis. Black bar represents 50 µm.
Haemodynamic data and electrocardiography
Heart rate was lower in the heart failure group than in the control group (336 ± 10 vs. 411 ± 14 min−1, n = 7; P < 0.05). A. carotis systolic blood pressures were decreased in the heart failure group (106 ± 4.2 vs. 118 ± 2.9 mmHg, n = 7; P < 0.05), while diastolic blood pressure only tended to be decreased (70 ± 7 vs. 77 ± 3.5 mmHg; n = 7; P = NS). Systolic LVPs were lower in the heart failure group. The rate of pressure development dp/dtmax in the left ventricle was markedly lower in heart failure, as was the rate of pressure decrease dp/dtmin (Table 2). Acute beta-adrenergic stimulation using an infusion of isoproterenol (60 ng/min) was not able to increase the function of the failing hearts with the exception of heart rate increase (Table 2). ECG showed a depression of ST segment (negative T wave) in the heart failure group (Figure 2), indicating myocardial ischaemia.
Table 2.
Functional parameters of the heart in vivo under basal conditions and under beta adrenergic stimulation (isoproterenol infusion, 60 ng/min, ‘Beta Stimulation’) in control rats and rats with heart failure (n = 7 per group)
| Left ventricle | Control |
Heart failure |
||
|---|---|---|---|---|
| Basal | Beta stimulation | Basal | Beta stimulation | |
| sLVP (mmHg) | 139 ± 4.8 | 166 ± 3.6* | 119 ± 4.1# | 134 ± 7.3§ |
| dp/dtmax (mmHg s−1) | 6182 ± 436 | 8779 ± 747* | 4454 ± 377# | 5037 ± 631§ |
| dp/dtmin (mmHg s−1) | 5009 ± 358 | 6554 ± 590* | 3573 ± 256# | 4547 ± 799 |
| Heart rate (min−1) | 411 ± 14.4 | 467 ± 2.6* | 336 ± 9.5# | 407 ± 6.8*,§ |
sLVP, systolic left ventricular pressure.
*P < 0.05 vs. respective basal.
#P < 0.05 vs. Control basal.
§P < 0.05 vs. Control stimulated.
Figure 2.

Representative ECG recordings from control rats and rats with isoproterenol-induced heart failure. Asterisk indicates ST segment depression with a negative T-wave. Similar results were observed in at least 8/10 rats.
Effect of NOS inhibition on the function of isolated atria
Isolated atria from failing hearts had a lower spontaneous beating rate than controls (Figure 3A). Addition of NOS inhibitor L-NAME increased spontaneous beating rate in atria from failing hearts, but not in controls (Figure 3B). In control atria, in vitro beta-adrenergic stimulation with isoproterenol had a positive chronotropic effect which was not affected by NOS inhibition (Figure 3C). However, in atria from failing hearts, effect of NOS inhibition on the effect of beta-adrenergic stimulation was biphasic—L-NAME increased beating rate at low concentrations of isoproterenol (10−10 M and 3 × 10−10 M), but decreased the beating rate at higher concentrations of isoproterenol (10−8 M, 3 × 10−8 M and 10−7 M) (Figure 3D).
Figure 3.

Function of atria isolated from control rats and rats with heart failure. (A) Spontaneous beating rate. *P < 0.05 vs. Control. (B) Effect of L-NAME on basal beating rate in control and failing hearts. *P < 0.05 vs. vehicle. (C and D) Effect of L-NAME on isoproterenol-stimulated beating rate of isolated atria from control and failing hearts. *P < 0.05 vs. −L-NAME; n = 5 per group.
Effect of NOS inhibition on the function of isolated left ventricular myocytes
Isolated paced ventricular myocytes under basal conditions had a relative cell shortening of 10.4 ± 0.6%, which was lower in myocytes isolated from failing hearts (8.7 ± 0.5%; P < 0.05). Beta-adrenergic stimulation by isoproterenol (10−7 M) added in vitro increased cell shortening in myocytes from both groups, however, the increase in cell shortening was significantly smaller in failing cells (P < 0.05; Figure 4). Addition of L-NAME (10−4 M) to the cell medium decreased cell shortening in control (P < 0.05; Figure 4), but not in failing myocytes, indicating a different role for NO in healthy and failing hearts.
Figure 4.

Effect of NO synthase inhibition with L-NAME (10−4 M) on cell shortening of left ventricular myocytes isolated from control (A) and failing hearts (B). Cells were electrically paced at 0.5 Hz and cell shortening was measured by videomicroscopy under basal conditions (‘Basal’) and after addition of isoproterenol (10−7 M) into the cell media (‘Beta-adrenergic stimulation’). n = 5/27–40 per group (rats/cells), *P < 0.05 vs. Vehicle; #P < 0.05 vs. Basal.
Expression of NOS isoforms and their allosteric modulators in the left ventricles
Results of immunoblotting analysis of the left ventricles are shown in Figure 5 with quantitative data in Table 3. In the failing left ventricle expression of eNOS and of its positive allosteric activator hsp90 were increased (both P < 0.05). Furthermore, expression of caveolin-1 showed a trend towards a decrease (−10 ± 3%, P = 0.065) and that of muscle-specific caveolin-3 was unchanged in the failing ventricle. Expression of inducible nitric oxide synthase was elevated (P < 0.05). The neuronal isoform of NOS in the left ventricle was not detected, despite a successful detection of nNOS in brain homogenates (Figure 5).
Figure 5.

Expression of endothelial nitric oxide synthase (eNOS), inducible nitric oxide synthase (iNOS), neuronal nitric oxide synthase (nNOS), eNOS positive allosteric modulator hsp90, and negative allosteric modulators caveolin-1 (cav-1), caveolin-3 (cav-3) after 1 week of isoproterenol administration. Actin was used as a loading control. Representative autoluminograms. HF, heart failure; C, control.
Table 3.
Expression of nitric oxide synthases and their allosteric modulators caveolin-1, caveolin-3, and hsp90 at protein level in the left ventricle of rats with isoproterenol-induced heart failure (n = 8–11 per group)
| Control | Heart failure | |
|---|---|---|
| eNOS | 100.0 ± 5.1 | 122.2 ± 7.3* |
| iNOS | 100.0 ± 7.0 | 123.1 ± 7.0* |
| nNOS | ND | ND |
| cav-1 | 100.0 ± 3.8 | 90.6 ± 3.0 |
| cav-3 | 100.0 ± 4.3 | 93.8 ± 5.1 |
| Hsp90 | 100.0 ± 8.7 | 121.2 ± 5.4* |
ND, not detected.
*P < 0.05 vs. Control.
Discussion
In the present study, we provide novel observations showing that capacity for nitric oxide synthesis was increased in isoproterenol-induced heart failure and was associated with an altered nitric oxide-related cardiac function, namely regulation of heart rate and a loss of positive inotropic action of nitric oxide in the left ventricular myocytes.
We observed a decreased heart rate and a selective positive chronotropic effect of NOS inhibition by L-NAME under basal conditions in isolated atria from heart failure rats, but not from controls. Under beta-adrenergic stimulation, isolated atria from failing hearts responded to L-NAME in a biphasic manner, with an increased frequency at low concentrations of isoproterenol, but with a reduction of the maximal response to the beta-agonist. This has not been reported previously and could be related to increased eNOS expression and/or activation in the atria. In line with this observation, eNOS did not regulate basal chronotropy in control mice, but the positive chronotropic effect of beta-adrenergic stimulation was attenuated when eNOS was upregulated.21 This contrasts with a report of no effect of myocyte-specific eNOS overexpression on the regulation of heart rate in mice.22 The positive chronotropic effect of L-NAME in atria from failing hearts in our experiment using low concentrations of the beta-agonist, could in theory also be related to beta3 receptors coupled to eNOS activation. This is supported by reports that NOS inhibition increased the positive chronotropic effect of a beta3-selective agonist.23 Increased expression of beta3 receptors in our model remains to be demonstrated. It is nevertheless likely, that in isoproterenol-induced heart failure, NO participates on the regulation of heart rate.
We found an increased expression of eNOS and its positive allosteric modulator hsp90 in the failing hearts, while the negative modulator caveolin-1 tended to be decreased. This could increase output of eNOS-derived NO and may be a compensatory mechanism against excessive beta-adrenergic stimulation. Ozaki et al.6 observed that transgenic mice overexpressing eNOS are partly protected against isoproterenol-induced cardiac hypertrophy. In the rats with heart failure, we found a depression of the ST segment of the ECG, which is indicative of ischaemia. Nitric oxide increases coronary flow and increases oxygen availability for the myocardium under catecholamine stimulation24 and could thus have a protective role. Incorporation of the human eNOS gene into the rat myocardium prior to myocardial infarction led to an inhibition of apoptotic and necrotic changes25 and of myocardial hypertrophy and fibrosis.5 Moreover, eNOS-derived NO stimulates angiogenesis.8 Development of hypertrophy after isoproterenol administration is rapid, it is maximal as early as 1 day after the start of isoproterenol administration and then stabilizes.26 The slow process of angiogenesis may not be sufficient to compensate for the high oxygen demand of the hypertrophied myocytes. Increased eNOS expression after 1 week of isoproterenol administration could, in theory, be linked to the activation of angiogenesis, but confirmation of this hypothesis is beyond the intended scope of our study.
We observed increased iNOS expression in the failing left ventricles. Inducible nitric oxide synthase is physiologically present at very low levels, but its expression can increase in heart failure.27 This can considerably increase NO levels in the heart because iNOS produces large quantities of NO independently of calcium dynamics in the cell. Our data show that upregulation of iNOS is an early event in isoproterenol-induced heart failure. Increased iNOS expression has been observed in a model of isoproterenol-induced heart failure, however with a different experimental setup (6 weeks after two high doses of isoproterenol).28 A 2 week infusion of isoproterenol in mice led to an induction of iNOS in the heart which was associated with increased apoptosis, which was partially prevented by a selective iNOS inhibitor and almost normalized in iNOS-deficient mice.29 Apoptosis of cardiomyocytes is increased ∼9-fold in isoproterenol-induced cardiac hypertrophy,30 which supports our finding of increased iNOS expression. It is likely that neuronal nitric oxide synthase did not contribute to NO formation in the left ventricle as it was undetectable in our rat left ventricular homogenates, this is in contrast to the findings of some,31 but not all studies.23
Left ventricular function in vivo was markedly impaired after 1 week of treatment with isoproterenol. To circumvent the sequelae of myocardial fibrosis on the mechanical properties of the heart, we also measured the function of isolated left ventricular myocytes. We found that they had impaired cell shortening both under basal condition and in response to beta-adrenergic stimulation, indicating a functional defect intrinsic to the cardiac muscle. Inhibition of NO synthesis resulted in decreased cell shortening in the control, but not in the failing myocytes. This indicates that in control rats, endogenous NO consistently stimulated myocyte contractility, whereas this effect was absent in myocytes from failing hearts, both under basal conditions and in vitro beta-adrenergic stimulation, despite eNOS and iNOS upregulation. The difference in L-NAME’s effect between healthy and failing myocytes in our study could be reconciled by an increased capacity for NO synthesis in the failing cells. NO was reported to have a bimodal effect on the contractility of isolated myocytes—low concentrations of NO can increase contractility of cardiac myocytes while high concentrations can attenuate it.2 Alternatively, with NO synthesis increased, we can hypothesize that bioavailability of NO in hypertrophied myocytes may be lower due to oxidative stress in this model32 or that signalling pathways downstream of NO are altered in the hypertrophied and failing heart. In line with this premise, there are reports that non-specific NOS inhibitors have little effect on basal function in heart failure33 or they increase contractility in failing myocytes.34 Furthermore, in three other models of cardiac hypertrophy, NO was devoid of effect on myocardial contractility.35
Although isoproterenol-induced heart failure has similarities to end-stage human heart failure, the findings of our study are limited to the model we used, in which the emphasis rests on a single, but important pathophysiological feature of end-stage heart failure—catecholamine overload.
We conclude that in rats 1 week of treatment with isoproterenol resulted in cardiac hypertrophy, and dysfunction of the left ventricle and isolated left ventricular myocytes. Increased capacity for NO synthesis evidenced by an upregulation of eNOS, its positive allosteric modulator hsp90, and iNOS in the failing left ventricles was associated with a lower heart rate, and reversal of the positively inotropic effect of nitric oxide in isolated left ventricular myocytes, indicating a possible role for nitric oxide in cardiac dysfunction and heart rate regulation in this model.
Funding
This work was supported by grants of the Comenius University UK/26/2006, UK/418/2006, VEGA 1/2286/05, VEGA 1/0109/08 and a grant of the Slovak Society of Cardiology (J.K.).
Acknowledgements
The authors thank Eva Hasonova and Simona Kolembusova for their skilful technical assistance.
Conflict of interest: none declared.
References
- 1.Pelat M, Massion PB, Balligand JL. Nitric oxide ‘at heart’: emerging paradigms after a decade. Arch Mal Coeur. 2005;98:242–248. [PubMed] [Google Scholar]
- 2.Kojda G, Kottenberg K, Nix P, Schlüter KD, Piper HM, Noack E. Low increase in cGMP induced by organic nitrates and nitrovasodilators improves contractile response of rat ventricular myocytes. Circ Res. 1996;78:91–101. doi: 10.1161/01.res.78.1.91. [DOI] [PubMed] [Google Scholar]
- 3.Stein B, Eschenhagen T, Rüdiger J, Scholz H, Förstermann U, Gath I. Increased expression of constitutive nitric oxide synthase III, but not inducible nitric oxide synthase II, in human heart failure. J Am Coll Cardiol. 1998;32:1179–1186. doi: 10.1016/s0735-1097(98)00399-4. [DOI] [PubMed] [Google Scholar]
- 4.Smith CJ, Sun D, Hoegler C, Roth BS, Zhang X, Zhao G, Xu XB, Kobari Y, Pritchard K, Jr, Sessa WC, Hintze TH. Reduced gene expression of vascular endothelial NO synthase and cyclooxygenase-1 in heart failure. Circ Res. 1996;78:58–64. doi: 10.1161/01.res.78.1.58. [DOI] [PubMed] [Google Scholar]
- 5.Janssens S, Pokreisz P, Schoonjans L, Pellens M, Vermeersch P, Tjwa M, Jans P, Crosbie SM, Picard MH, Szelid Z, Gillijns H, Werf VF, Collen D, Bloch D. Cardiomyocyte-specific overexpression of nitric oxide synthase 3 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circ Res. 2004;94:1256–1262. doi: 10.1161/01.RES.0000126497.38281.23. [DOI] [PubMed] [Google Scholar]
- 6.Ozaki M, Kawashima S, Yamashita T, Hirase T, Ohashi Y, Inoue N, Hirata K, Yokoyama M. Overexpression of endothelial nitric oxide synthase attenauates cardiac hypertrophy induced by chronic isoproterenol infusuon. Circ J. 2002;66:851–856. doi: 10.1253/circj.66.851. [DOI] [PubMed] [Google Scholar]
- 7.Prabhu SD. Nitric oxide protects against pathological ventricular remodeling: reconsideration of the role of NO in the failing heart. Circ Res. 2004;94:1256–1262. doi: 10.1161/01.RES.0000129569.07667.89. [DOI] [PubMed] [Google Scholar]
- 8.Duda DG, Fukumura D, Jain RK. Role of eNOS in neovascularization: NO for endothelial progenitor cells. Trends Mol Med. 2004;10:143–145. doi: 10.1016/j.molmed.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Funakoshi H, Kubota T, Kawamura N, Machida Y, Feldman AM, Tsutsui H, Shimokawa H, Takeshita A. Disruption of inducible nitric oxide synthase improves beta-adrenergic inotropic responsiveness but not the survival of mice with cytokine-induced cardiomyopathy. Circ Res. 2002;90:959–965. doi: 10.1161/01.res.0000017632.83720.68. [DOI] [PubMed] [Google Scholar]
- 10.Thoenes M, Förstermann U, Tracey WR, Bleese NM, Nüssler AK, Scholz H, Stein B. Expression of inducible nitric oxide synthase in failing and non-failing human heart. J Mol Cell Cardiol. 1996;28:165–169. doi: 10.1006/jmcc.1996.0016. [DOI] [PubMed] [Google Scholar]
- 11.Heger J, Gödecke A, Flögel U, Merx MW, Molojavyi A, Kühn-Velten WN, Schrader J. Cardiac-specific overexpression of inducible nitric oxide synthase does not result in severe cardiac dysfunction. Circ Res. 2002;90:93–99. doi: 10.1161/hh0102.102757. [DOI] [PubMed] [Google Scholar]
- 12.Mungrue IN, Gros R, You X, Pirani A, Azad A, Csont T, Schulz R, Butany J, Stewart DJ, Husain M. Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J Clin Invest. 2002;109:735–743. doi: 10.1172/JCI13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bendall JK, Damy T, Ratajczak P, Loyer X, Monceau V, Marty I, Milliez P, Robidel E, Marotte F, Samuel JL, Heymes C. Role of myocardial neuronal nitric oxide synthase-derived nitric oxide in beta-adrenergic hyporesponsiveness after myocardial infarction-induced heart failure in rat. Circulation. 2004;110:2368–2375. doi: 10.1161/01.CIR.0000145160.04084.AC. [DOI] [PubMed] [Google Scholar]
- 14.Damy T, Ratajczak P, Shah AM, Camors E, Marty I, Hasenfuss G, Marotte F, Samuel JL, Heymes C. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet. 2004;363:1365–1367. doi: 10.1016/S0140-6736(04)16048-0. [DOI] [PubMed] [Google Scholar]
- 15.Szabo J, Csaky L, Szegi J. Experimental cardiac hypertrophy induced by isoproterenol in the rat. Acta Physiol Acad Sci Hung. 1975;46:281–285. [PubMed] [Google Scholar]
- 16.Krenek P, Klimas J, Kroslakova M, Gazova A, Plandorova J, Kucerova D, Fecenkova A, Svec P, Kyselovic J. Increased expression of endothelial nitric oxide synthase and caveolin-1 in the aorta of rats with isoproterenol-induced cardiac hypertrophy. Can J Physiol Pharmacol. 2006;84:1245–1250. doi: 10.1139/y06-073. [DOI] [PubMed] [Google Scholar]
- 17.Feron O, Balligand JL. Caveolins and the regulation of endothelial nitric oxide synthase in the heart. Cardiovasc Res. 2006;69:788–797. doi: 10.1016/j.cardiores.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 18.Bacharova L, Kyselovic J, Klimas J. The initial stage of left ventricular hypertrophy in spontaneously hypertensive rats is manifested by a decrease in the QRS amplitude/left ventricular mass ratio. Clin Exp Hypertens. 2004;26:557–567. doi: 10.1081/ceh-200031835. [DOI] [PubMed] [Google Scholar]
- 19.Hart CYT, Burnett JC, Redfield M. Effects of avertin versus xylazine-ketamine anesthesia on cardiac function in normal mice. Am J Physiol Heart Circ Physiol. 2001:H1938–H1945. doi: 10.1152/ajpheart.2001.281.5.H1938. [DOI] [PubMed] [Google Scholar]
- 20.Kirchhefer U, Klimas J, Baba HA, Buchwalow IB, Fabritz L, Huls M, Matus M, Muller FU, Schmitz W, Neumann J. Triadin is a critical determinant of cellular Ca cycling and contractility in the heart. Am J Physiol Heart Circ Physiol. 2007;293:H3165–H3174. doi: 10.1152/ajpheart.00799.2007. [DOI] [PubMed] [Google Scholar]
- 21.Danson EJ, Zhang YH, Sears CE, Edwards AR, Casadei B, Paterson DJ. Disruption of inhibitory G-proteins mediates a reduction in atrial beta-adrenergic signaling by enhancing eNOS expression. Cardiovasc Res. 2005;67:575–577. doi: 10.1016/j.cardiores.2005.04.034. [DOI] [PubMed] [Google Scholar]
- 22.Brunner F, Andrew P, Wölkart G, Zechner R, Mayer B. Myocardial contractile function and heart rate in mice with myocyte-specific overexpression of endothelial nitric oxide synthase. Circulation. 2001;104:3097–3102. doi: 10.1161/hc5001.101966. [DOI] [PubMed] [Google Scholar]
- 23.Sterin-Borda L, Bernabeo G, Ganzinelli S, Joensen L, Borda E. Role of nitric oxide/cyclic GMP and cyclic AMP in beta3 adrenoceptor-chronotropic response. J Mol Cell Cardiol. 2006;40:580–588. doi: 10.1016/j.yjmcc.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 24.Setty S, Tune JD, Downey HF. Nitric oxide modulates right ventricular flow and oxygen consumption during norepinephrine infusion. Am J Physiol Heart Circ Physiol. 2002;282:H696–H703. doi: 10.1152/ajpheart.00398.2001. [DOI] [PubMed] [Google Scholar]
- 25.Smith RS, Jr, Agata J, Xia CF, Chao L, Chao J. Human endothelial nitric oxide synthase gene delivery protects against cardiac remodeling and reduces oxidative stress after myocardial infarction. Life Sci. 2005;76:2457–2471. doi: 10.1016/j.lfs.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 26.Linck B, Boknik P, Baba HA, Eschenhagen T, Haverkamp U, Jackel E, Jones LR, Kirchhefer U, Knapp J, Laer S, Muller FU, Schmitz W, Scholz H, Syska A, Vahlensieck U, Neumann J. Long-term beta adrenoceptor-mediated alteration in contractility and expression of phospholamban and sarcoplasmic reticulum Ca(++)-ATPase in mammalian ventricle. J Pharmacol Exp Ther. 1998;286:531–538. [PubMed] [Google Scholar]
- 27.Haywood GA, Tsao PS, von der Leyen HE, Mann MJ, Keeling PJ, Trindade PT, Lewis NP, Byrne CD, Rickenbacher PR, Bishopric NH, Cooke JP, McKenna WJ, Fowler MB. Expression of inducible nitric oxide synthase in human heart failure. Circulation. 1996;93:1087–1094. doi: 10.1161/01.cir.93.6.1087. [DOI] [PubMed] [Google Scholar]
- 28.Grosjean SA, Arstall MA, Mitchell RN, Klappacher GW, Kelly RA, Pfeffer MA, Pfeffer JM. Inducible nitric oxide synthase and tumor necrosis factor in animal models of myocardial necrosis induced by coronary artery ligation or isoproterenol injection. J Card Fail. 1999;5:236–245. doi: 10.1016/s1071-9164(99)90008-8. [DOI] [PubMed] [Google Scholar]
- 29.Hu A, Jiao X, Gao E, Koch WJ, Sharifi-Azad S, Grunwald Z, Ma XL, Sun JZ. Chronic beta-adrenergic receptor stimulation induces cardiac apoptosis and aggravates myocardial ischemia/reperfusion injury by provoking inducible nitric-oxide synthase-mediated nitrative stress. J Pharmacol Exp Ther. 2006;318:469–475. doi: 10.1124/jpet.106.102160. [DOI] [PubMed] [Google Scholar]
- 30.Shizukuda Y, Buttrick PM, Geenen DL, Borczuk AC, Kitsis RN, Sonnenblick EH. Beta-adrenergic stimulation causes cardiocyte apoptosis: influence of tachycardia and hypertrophy. Am J Physiol. 1998;275:H961–H968. doi: 10.1152/ajpheart.1998.275.3.H961. [DOI] [PubMed] [Google Scholar]
- 31.Piech A, Dessy C, Havaux X, Feron O, Balligand JL. Differential regulation of nitric oxide synthases and their allosteric regulators in heart and vessels of hypertensive rats. Cardiovasc Res. 2003;57:456–467. doi: 10.1016/s0008-6363(02)00676-4. [DOI] [PubMed] [Google Scholar]
- 32.Zhang GX, Kimura S, Nishiyama A, Shokoji T, Rahman M, Yao L, Nagai Y, Fujisawa Y, Miyatake A, Abe Y. Cardiac oxidative stress in acute and chronic isoproterenol-infused rats. Cardiovasc Res. 2005;65:230–238. doi: 10.1016/j.cardiores.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 33.Massion PB, Feron O, Dessy C, Balligand JL. Nitric oxide and cardiac function: ten years after and continuing. Circ Res. 2003;93:388–398. doi: 10.1161/01.RES.0000088351.58510.21. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto S, Tsutsui H, Tagawa H, Saito K, Takahashi M, Tada H, Yamamoto M, Katoh M, Egashira K, Takeshita A. Role of myocyte nitric oxide in beta-adrenergic hyporesponsiveness in heart failure. Circulation. 1997;95:1111–1114. doi: 10.1161/01.cir.95.5.1111. [DOI] [PubMed] [Google Scholar]
- 35.Kotchi KE, Weisselberg T, Röhnert P, Preiss M, Heinroth-Hoffmann I, Osten B, Brodde OE. Nitric oxide inhibits isoprenaline-induced positive inotropic effects in normal, but not in hypertrophied rat heart. Naunyn Schmiedebergs Arch Pharmacol. 1998;357:579–583. doi: 10.1007/pl00005211. [DOI] [PubMed] [Google Scholar]