

Abstract
A strategy employing gene-trap mutagenesis and site-specific recombination (Cre/loxP) has been developed to isolate genes that are transcriptionally activated during programmed cell death. Interleukin-3 (IL-3)-dependent hematopoietic precursor cells (FDCP1) expressing a reporter plasmid that codes for herpes simplex virus–thymidine kinase, neomycin phosphotransferase, and murine IL-3 were transduced with a retroviral gene-trap vector carrying coding sequences for Cre-recombinase (Cre) in the U3 region. Activation of Cre expression from integrations into active genes resulted in a permanent switching between the selectable marker genes that converted the FDCP1 cells to factor independence. Selection for autonomous growth yielded recombinants in which Cre sequences in the U3 region were expressed from upstream cellular promoters. Because the expression of the marker genes is independent of the trapped cellular promoter, genes could be identified that were transiently induced by IL-3 withdrawal.
Keywords: insertional mutagenesis
Apoptosis or programmed cell death (PCD) is a genetically controlled program of cellular self-destruction, which is of central importance for the development and homeostasis of virtually all animals. It is a highly regulated process involving an increasing number of genes that are conserved in all metazoans (reviewed in ref. 1).
Some of the initial evidence for an active cell–suicide program was derived from experiments in which cell death could be repressed by inhibitors of RNA and protein synthesis, suggesting that apoptosis is controlled at the transcriptional level. However, it was subsequently shown that these inhibitors fail to block and may even induce apoptosis in many other situations, suggesting that apoptotic effector molecules are always present in the cell. Indeed, several protein families have been identified whose members are products of constitutively expressed genes that either stimulate or inhibit apoptosis (reviewed in refs. 2 and 3). A disturbed balance between these proteins is thought to impair development and cause diseases such as degenerative disorders or cancer (reviewed in ref. 4).
Compelling evidence for PCD genes that are transcriptionally regulated has been obtained only recently. Two related genes referred to as reaper and head involution defective have been isolated from Drosophila melanogaster and shown to induce apoptosis by activating pre-existing suicide molecules (5–7). Thus, RNA and protein synthesis appears necessary to generate molecules capable of modifying the balance between the antagonistic components of the execution machinery.
To identify such molecules in mammalian cells, where the types of genetic analyses used with Drosophila are impractical, we have developed a strategy that is based on the induction of gene fusions between a promoterless reporter gene and the controlling elements of a cellular gene by means of special vectors referred to as “gene traps” (reviewed in ref. 4). We and others have shown that by inserting a gene-trap vector into a large collection of chromosomal sites and selecting for gene expression, recombinants are obtained in which the transduced reporter gene is fused to an upstream cellular promoter. Transcripts generated by such fusions faithfully reflect the activity of a disrupted cellular gene and serve as a molecular tag to clone any gene linked to a specific function (8–15).
However, to recover genes that are induced in cells undergoing apoptosis and might be only transiently expressed, an additional procedure was required to uncouple the trapped cellular promoter from the transduced reporter gene. To achieve this, the site-specific recombination system Cre/loxP (16, 17) was used to permit a permanent switching between two selectable marker genes by means of a retroviral gene-trap expressing Cre recombinase (Cre). This rescues interleukin 3 (IL-3) growth factor-dependent hematopoietic cells (FDCP1) (18, 19) from apoptotic cell death induced by factor deprivation and, thus, allowed selection for gene-trap integrations into potential suicide genes.
MATERIALS AND METHODS
The sequences for Cre and loxP were derived from pMCCre and pGEM30, respectively (17). The mouse phosphoglycerate kinase (pgk) promoter was obtained from ppgkCAT (20) and the thymidine kinase (tk)neo fusion gene was constructed as described (21). The mouse cDNA-encoding IL-3 was derived from pcMulti-CSF (22) and the bovine growth hormone (bpa) and simian virus 40 polyadenylation sequences were obtained from pRC/CMV (Invitrogen) and pSBC2 (23), respectively. Most components of the reporter plasmid ppgklxtkneoIL3 were assembled in pBluescript(IIKS) as follows: A loxP site was first inserted into the Bluescript polylinker as an EcoRI/PstI fragment. Subsequently, pgk was ligated into the XbaI/BamHI sites of the polylinker and tkneo was cloned blunt-ended into the downstream EcoRV site. The IL-3 gene was initially subcloned into the EcoRI site of the loxP flanking polylinker of pGEM30 after first cleaving the plasmid with ClaI/SalI and religating the filled-in ends to remove an additional EcoRI site. IL-3–loxP was then recovered from pGEM30 as an SalI/XhoI fragment and cloned into the Bluescript polylinker. One copy of bpa obtained as a XbaI/AvaI fragment was cloned blunt-ended into the ClaI site downstream of tkneo. To obtain ppgklxtkneoIL3, an XhoI fragment containing the assembled sequences was cloned into pSBC2, upstream of a simian virus 40 polyadenylation site. Because with this initial construct expression of IL-3 in transfected FDCP1 cells was leaky, a second copy of bpa was cloned blunt-ended into the SalI site of pSBC2 to obtain the final ppgklxtkneoIL3.
pGgU3Creen(−) was derived from pGgU3Neoen(−) (10) by replacing the neomycin gene with Cre-coding sequences. The Cre sequences were amplified by PCR from pMCCre.
Cells and Viruses.
U3Cre virus-containing supernatants were obtained from high titer psi2 producer cells transfected with pGgU3Creen(−) as described (24). FDCP1 cells were infected in suspension cultures by incubating for 2 hr with virus-containing supernatant. Following incubation for 24 hr in fresh medium, cells were placed in selection using 0.6 μg/ml of active G418 (GIBCO).
FDCP1 cells were propagated at concentrations of 2 × 105 cells/ml in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO), supplemented with 10% (vol/vol) fetal bovine serum (HyClone) and 10 ng/ml recombinant mouse IL-3 (Sandoz Pharmaceutical). Agar cultures were an equal volume mixture of double-strength DMEM supplemented with 40% (vol/vol) fetal bovine serum (HyClone) and 0.6% (wt/vol) Bacto-agar (Difco) in double-distilled water as described (25). Where indicated, cultures contained 5 μM ganciclovir (Syntex, Palo Alto, CA).
Electroporation of ppgklxtkneoIL3 into FDCP1 cells was performed using a Bio-Rad GenePulser according to the manufacturer’s instructions. Recombinants were isolated in agar cultures containing 0.6 mg/ml G418. Developing clones were isolated after 10 days and expanded in suspension cultures as described above.
Amplification and Cloning of Upstream Sequences.
Genomic DNAs from autonomous FLOXIL3 cells were digested with MseI and ligated at concentrations of 5 μg/ml to obtain circular molecules. After cleavage with PvuII, 1 μg of each sample was used for PCR as described (20). Reactions were performed using an Expand high fidelity PCR system (Boehringer Mannheim) according to the manufacturer’s instructions. The DNA was amplified for 35 cycles in a Perkin–Elmer thermocycler (denaturation at 94°C for 30 sec, annealing, and elongation at 68°C for 3 min) using the Cre-specific primers 5′-GCATTGCTGTCACTTGGTCGCGGCAGCC-3′ and 5′-CGGTGAACGTGCAAAACAGGCTCTAGCGTT-3′. Where required, nested PCRs were performed for an additional 35 cycles (denaturation at 94°C for 1 min, annealing at 68°C for 1 min, elongation at 72°C for 2 min) using the primers 5′-ATTCAACTTGCACCATGCCGCCCACGAC-3′ and 5′-CTGTTACGTATAGCCGAAATTGCCAGGATC-3′. Amplification products were cloned into the T vector (Invitrogen) and sequenced with an automatic sequencer (Applied Biosystems).
Nucleic Acid Hybridization Analyses.
Genomic DNA and polyadenylated RNA obtained by oligo(dT)-cellulose chromatography (OligoTex; Qiagen, Chatsworth, CA) were hybridized to 32P-labeled probes as described (26). Flanking sequences were end-labeled using [32P-γ]-ATP and T4 polynucleotide kinase (New England Biolabs). Blots were scanned with a PhosphorImager (Molecular Dynamics) and analyzed with imagequant nt software (Molecular Dynamics).
RESULTS
Construction of an FDCP1 Reporter Cell Line Expressing Two Selectable Marker Genes Flanked by loxP Recombination Targets.
The FDCP1 hematopoietic precursor cells are strictly dependent on IL-3 for growth and promptly initiate apoptosis upon growth factor withdrawal (18, 19).
To obtain a cell line convertible to factor independence by Cre, FDCP1 cells were electroporated with the reporter plasmid ppgklxtkneoIL3 (Fig. 1A). ppgklxtkneoIL3 consists of two tandemly arrayed selectable marker genes that are transcribed from a pgk promoter. The 5′ gene encodes for a herpes simplex virus 2 thymidine kinase (tk)-neomycin phosphotransferase (neo) fusion protein (21) and is flanked by two loxP recombination targets. The 3′ gene encodes for murine IL-3 and terminates in a simian virus 40 polyadenylation sequence. To suppress IL-3 translation from dicistronic transcripts, two tandem copies of bovine growth hormone polyadenylation sequence (bpa) were inserted downstream of tkneo. Thus, cells expressing ppgklxtkneoIL3 were expected to still be IL-3 dependent. Because Cre typically excises the sequences flanked by direct loxP site repeats, Cre expression was expected to delete the tkneo gene, thereby placing the IL-3 gene just downstream of the pgk promoter. As a result, cells would lose neomycin resistance and acquire factor independence by synthesizing IL-3 (Fig. 1A).
Figure 1.
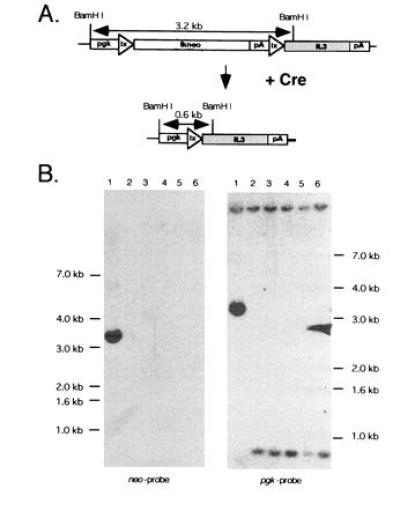
Cre-loxP-mediated recombination of ppgklxtkneoIL3 in FLOXIL3 cells with U3Cre gene-trap integrations into expressed genes. (A) Predicted structure of ppgklxtkneoIL3 before and after recombination. (B) Southern blot analysis of autonomous and parental FLOXIL3 cells. Genomic DNAs were cleaved with BamHI, fractionated on agarose gels, blotted onto nylon filters, and hybridized to a 32P-labeled neo (Left) or pgk (Right) probe as follows. Lanes: 1, parental FLOXIL3 cells; 2–6, factor-independent FLOXIL3 clones 1–5. Molecular weights were estimated with a 1-kb ladder (BRL).
Stable FDCP1 transformants were selected in G418 and five clonal cell lines were isolated from agar cultures. One cell line expressing a single copy plasmid, referred to as “FLOXIL3,” was selected for further analysis.
We first determined whether FLOXIL3 cells were still dependent on IL-3 for growth by plating 5 × 107 cells at densities of 2 × 105/ml into semi-solid cultures without IL-3. Because no colonies developed within 10 days (data not shown), we concluded that neither leaky IL-3 translation nor spontaneous recombination occurs within these cells.
To address this further, FLOXIL3 and parental FDCP1 cells were preincubated in agar cultures without IL-3 for up to 24 hr and subsequently rescued by adding IL-3. As seen in Fig. 2, both cell types initiated apoptosis with similar kinetics, indicating that the expression of ppgklxtkneoIL3 did not alter the cellular response to factor deprivation. Moreover, most cells survived for more than 12 hr without IL-3, thus leaving sufficient time for the gene-trap transduced sequences to be expressed.
Figure 2.
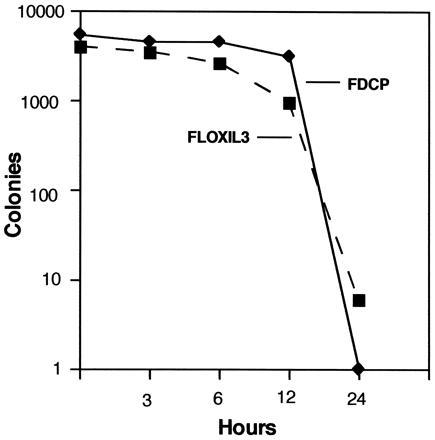
Survival of FLOXIL3 and FDCP1 cells in cultures without IL-3. Cells were plated into agar cultures at concentrations of 5 × 103 per ml. After 3, 6, 12, and 24 hr of preincubation, IL-3 was added to duplicate plates to obtain a final concentration of 10 ng/ml. Colonies were counted after incubating for 10 days.
A Gene-Trap Expressing Cre (U3Cre) Converts FLOXIL3 Cells to Factor Independence.
A U3Cre gene-trap vector was derived from the Moloney murine leukemia virus-based pGgU3Neo(en−) vector (26) by replacing the neo gene in the U3 region with the Cre coding sequence derived from pMCCre (17). The plasmid was transfected into helper cells and supernatants from lines producing high titers of recombinant virus were used to infect FLOXIL3 cells. As has been shown in previous studies, virus replication and long terminal repeat (LTR)-mediated duplication places the sequences inserted in U3 just 30 nt downstream of the flanking cellular DNA. This enables their expression from integrations into transcribed genes (24, 26). Thus, the prediction was that FLOXIL3 cells with U3Cre integrations in expressed genes would convert to factor independence. To select for such events, virus infected FLOXIL3 cells were plated into agar cultures without IL-3. Several autonomously growing colonies were obtained and expanded in suspension cultures. As was expected from the expression of a recombined reporter plasmid, all clones lost G418 resistance and proliferated extensively without IL-3 (data not shown).
To confirm recombination, genomic DNAs from five autonomous clones were digested with BamHI and analyzed by Southern blotting. Because BamHI cleaves within the 5′ ends of pgk and IL-3, respectively, non-recombined parental cells generate an internal fragment of 3.2 kb that hybridizes to both pgk- and neo-specific probes (Fig. 1). Since this fragment accommodates all sequences flanked by loxP, including tkneo, its deletion should be associated with a size reduction of 2.6 kb. Indeed, all clones produced a 0.6 kb restriction fragment that did not hybridize to neo, indicating that recombination has taken place (Fig. 1B).
Moreover, when analyzed by Northern blotting, all clones expressed cell–provirus fusion transcripts, suggesting that Cre was expressed from active cellular promoters (see next section and data not shown).
A U3Cre/FLOXIL3 Integration Library Preselected in G418 Yields Autonomous Clones with Recombined Reporter Plasmids.
A library consisting of approximately 2 × 106 independent proviral integrations was constructed by incubating FLOXIL3 cells with supernatant from U3Cre producer cells at an approximate multiplicity of infection of 0.5. Because the U3Cre vector lacks a constitutively expressed drug resistance marker, virus titers were derived by multiplying the number of factor independent colonies with the average frequency of integrations that enable U3 gene activation of other gene-trap vectors (26).
To enrich for integrations that are not expressed in FLOXIL3 cells, the library was initially selected in G418. Since U3Cre activation results in loss of tkneo, G418 was expected to eliminate all integrations into genes that enable Cre expression to levels high enough to cause recombination. Selection was monitored by plating aliquots of the library into agar cultures without IL-3 and counting colonies after 10 days (Fig. 3). Because preliminary experiments have shown that autonomous cells secrete IL-3 and thus support the proliferation of non-recombined parental cells, all cultures contained ganciclovir, which was found to reduce the number of colonies derived from parental cells by one order of magnitude (data not shown).
Figure 3.
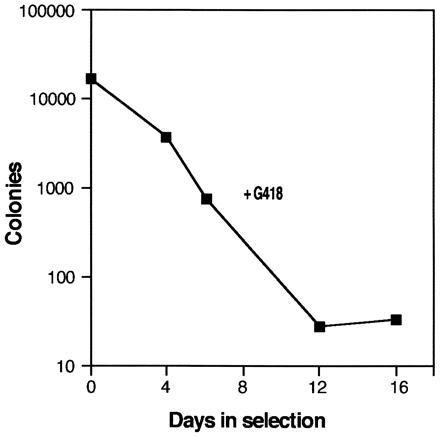
Selection of a U3Cre/FLOXIL3 cell integration library in G418. FLOXIL3 cells (4 × 106) were infected with U3Cre virus and incubated for 16 days in suspension cultures containing 10 ng/ml IL-3 and 0.6 μg/ml active G418. At various time points aliquots were plated in agar cultures containing 5 μM ganciclovir, but no IL-3, using 1 × 105 cells per ml. Colonies were counted after incubating for 10 days. Colony numbers represent the total number of colonies obtained from 106 seeded cells.
As shown in Fig. 3, colony numbers decreased progressively in G418 reaching a plateau by day 12, suggesting that most Cre-expressing clones had been eliminated by this time. Accordingly, surviving cells were likely to have provirus integrations in transcriptionally inactive chromosomal regions (including silent genes) or in genes expressed too weakly to cause recombination. Therefore, a simple representation of the preselected library was plated into agar cultures in presence of ganciclovir.
A total of 120 autonomous colonies were recovered, and 110 were successfully expanded. Southern blot analysis of genomic DNA digested by BamHI showed that each clone had single and unique proviral integrations consistent with a simple genomic representation and a multiplicity of infection of 0.5. Since BamHI cleaves within the 5′ third of Cre (Fig. 4 Left), each provirus generates three Cre-hybridizing fragments. Variable bands represent sequences extending from the BamHI sites in the LTRs to BamHI sites in the 5′ and 3′ flanking cellular DNA, respectively. The constant band derives from the provirus and was of expected size in 108 of 110 clones, indicating that most proviruses have not rearranged (Fig. 4 Left). When the same blots were hybridized to a pgk-specific probe, all clones harbored recombined reporter plasmids, suggesting that functional Cre was induced after withdrawing IL-3 (Fig. 4 Right).
Figure 4.
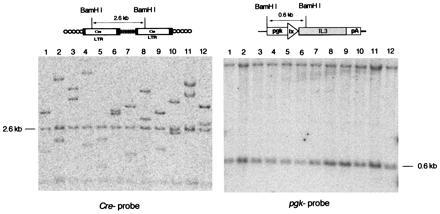
U3Cre provirus integrations and site-specific recombination in autonomous clones isolated after IL-3 withdrawal. (Upper) Structure of U3Cre proviruses (Left) and recombined reporter plasmids (Right). (Lower) Southern blot analysis of clones recovered from the U3Cre/FLOXIL3 integration library following IL-3 withdrawal. Genomic DNAs from individual clones were cleaved with BamHI, processed as described in the legend to Fig. 1, and hybridized to 32P-labeled Cre- (Left) or pgk- (Right) specific probes as follows. Lanes: 1, 26–11-1; 2, 26–11-3; 3, 26–11-4; 4, 26–11-5; 5, 26–11-6; 6, 26–11-7; 7, 26–11-8; 8, 26–12-1; 9, 26–12-2; 10, 26–12-3; 11, 26–12-4; 12, 26–12-5. The samples shown are representative for all clones derived from the library.
To investigate this further, Northern blots containing mRNAs from 11 independent clones were hybridized to a Cre-specific probe. Constitutively expressed U3Cre integrations typically generate a fusion transcript of variable size initiating in a nearby cellular promoter and terminating in the polyadenylation site of the 5′ LTR, and a “genomic” transcript of constant size initiating in the 5′ proviral LTR and terminating in the polyadenylation site of the 3′ LTR (Fig. 5, lanes 5 and 6). Because U3Cre proviruses lack the viral enhancer, genomic transcripts are only seen following the acquisition of a cellular enhancer (24, 26). This suggests that Cre activation could in principle occur from integrations nearby a genomic enhancer even in absence of a cellular promoter. However, in most cases we and others have examined, gene-trap activation was always associated with cell–provirus fusion transcripts from which reporter genes are translated (this study, refs. 12, 20, 24, 27, and 28, S. Nigro and H.v.M., unpublished work, and G. Hicks and H. E. Ruley, unpublished work). Moreover, reporter genes within 3′ LTRs are unlikely to be translated due to multiple short open reading frames positioned between the LTRs.
Figure 5.
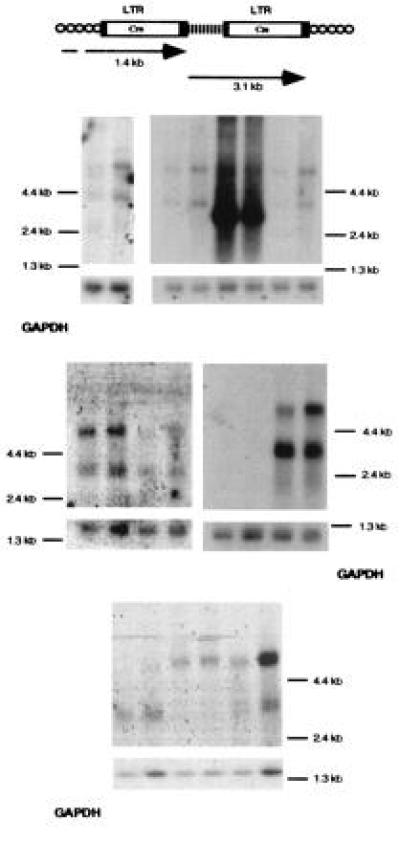
Analysis of cell–provirus fusion transcripts in clones recovered after IL-3 withdrawal. (Upper) Predicted transcripts from U3Cre integrations into expressed genes. (Lower) Northern blot analysis of cell–provirus fusion transcripts before and after serum deprivation. Polyadenylated RNAs (5 μg per lane) were fractionated on formaldehyde–agarose gels, blotted onto nylon filters, and hybridized to 32P-labeled Cre- or glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-specific probes before (odd number lanes) and after (even number lanes) 16 hr of serum deprivation. Lanes 5 and 6, clone with a constitutively expressed U3Cre integration. Note that exposure times for visualizing Cre-specific transcripts were 145 times longer than those required for GAPDH.
In 9 of 11 clones fusion transcripts were undetectable or weak (Table 1, Fig. 5, lanes 1, 3, 7, 9, 11, 13, 17, 19, and 21), indicating that Cre was only transiently expressed to levels high enough to cause recombination. The result is consistent with a mechanism of U3Cre repression mediated by intracellular IL-3.
Table 1.
Summary of results obtained with FLOXIL3 cell lines and cloned flanking sequences
| FLOXIL3 clone | Fusion transcripts | Provirus flanking DNA, nt | Hybridization to cellular transcripts | Transcript size, kb |
|---|---|---|---|---|
| 26-12-1 | + | 150 | − | |
| 26-12-2 | +iS | 50 | ND | |
| 26-12-3 | +iS | 50 | +iIL-3 | 4.4 |
| 26-12-4 | − | 230 | − | |
| 26-12-5 | − | 9 | ND | |
| 26-32-1 | + | 322 | − | |
| 26-32-2 | − | 54 | −iIL-3 | 8.4 |
| 26-32-3 | +iS | 104 | +iIL-3 | 4.2 |
| 26-32-4 | +iS | 19 | ND | |
| 26-41-2 | +iS | 153 | ND | |
| 26-41-4 | +iS | 353 | +iIL-3 | 7.5 |
Fusion transcripts in FLOXIL3 cell lines were analyzed on Northern blots as described in the legend to Fig. 5. Genomic DNAs from FLOXIL3 cell lines were digested with MseI and religated to obtain circular molecules. After cleavage with PvuII, 1 μg of DNA from each sample was used for PCR as described in Materials and Methods. Amplification products were sequenced using an automatic sequencer and hybridized to polyadenylated RNA from wild-type FDCP1 cells as described in the legend to Fig. 6. Transcripts are labeled (+) when present and (−) when absent. iS, inducible by serum starvation; iIL-3, inducible by IL-3 deprivation; ND, not done.
Cell–Provirus Fusion Transcripts Are Inducible by Serum Starvation.
Because all clones expressed IL-3 constitutively, attempts to reinduce fusion transcripts by IL-3 starvation were impractical. Instead, we reasoned that if the fusion transcripts were initially induced as part of an apoptotic program, they should be reinducible by an alternative apoptotic stimulus. To test this hypothesis the cells were serum-starved for 16 hr and then analyzed by Northern blotting. As shown in Fig. 5 (lanes 2, 4, 8, 12, 16, and 22), fusion transcripts were induced by serum starvation in 6 of 11 clones, suggesting that the disrupted genes are directly or indirectly associated with PCD.
Sequences Flanking U3Cre Proviruses Hybridize to Inducible Cellular Transcripts.
Upstream proviral sequences were isolated by inverse PCR and sequenced. All sequences showed typical cell DNA–provirus junctions (29) and varied in size from 9 to 353 nt (Table 1). Extensive data base searches (EMBL, GenBank, expressed sequence tag data base) revealed no homologies to known genes or expressed sequence tags.
Four of seven flanking sequences hybridized to unique cellular transcripts that were induced in wild-type FDCP1 cells by IL-3 starvation (Fig. 6). The result is consistent with previous observations, showing that only 50–60% of the provirus flanking regions contain sufficient exon sequence to detect cellular transcripts (20, 26). Nevertheless, the presence of hybridizing transcripts in some clones provides direct evidence for U3Cre integrations into genes responsive to IL-3.
Figure 6.
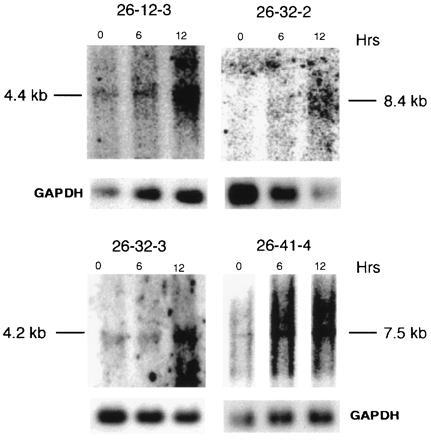
Analysis of cellular transcripts induced by IL-3 withdrawal. Polyadenylated RNAs (5 μg/lane) extracted from FDCP1 cells after 0, 6, and 12 hr of IL-3 starvation, were processed as described in the legend to Fig. 5 and hybridized to 32P-labeled flanking sequence or GAPDH probes.
DISCUSSION
This study exploited the combined features of gene-trap mutagenesis and site-specific recombination to identify genes induced in apoptotic cells. Factor-dependent hematopoietic cells expressing specific reporter constructs were infected with retrovirus gene-trap vectors containing coding sequences for Cre in the U3 region. Selection for factor independence following factor withdrawal produced cell clones in which Cre sequences in the 5′ LTR of the integrated provirus were expressed on inducible transcripts initiating in the flanking cellular DNA. Moreover, 4 of 7 flanking sequences tested hybridized to inducible cellular transcripts of so far unknown genes. This was not unexpected since we and others have shown that only 10% of retroviral gene-trap integrations occur into previously characterized genes (reviewed in ref. 27). Although this percentage may have increased with the size of the data bases that double every 18–24 months, three considerations suggest that the present strategy does not favor recovery of integrations into known genes. First, selection was applied for genes that are hardly active in exponentially growing cells and therefore unlikely to be recovered by conventional cDNA cloning. Second, most genes identified in this study were expressed very weakly and therefore were likely to be missing from cDNA libraries, which are strongly biased toward highly expressed genes (30, 31). Third, the vectors integrate in or near 5′ exons that are often absent from all but full-length cDNA clones.
Several features of the strategy seem ideally suited for isolating sequence tags from genes that are transcriptionally regulated during PCD. First, permanent selectable marker switching between two selectable marker genes by means of a gene-trap expressing Cre essentially converts dying cells into proliferating cells by activating endogenous cytokine production. This permits selection for integrations in genes that are active during cell death. Second, by uncoupling the expression of the trapped cellular gene from the expression of the selectable marker gene, the strategy allows identification of genes that are only transiently expressed. This is important, because cells are unlikely to express suicide genes constitutively. Third, unlike cDNA cloning, the method does not appear biased toward highly expressed genes, and gene representation is more uniform (28). Fourth, unlike differential reverse transcriptase-PCR amplification (32, 33), gene trapping seems fairly reproducible, avoids redundancy, and allows quantification. Finally, gene-trap proviruses are consistently located in or near 5′ exons and have the same transcriptional orientation relative to the gene, features which greatly facilitate cloning.
One hundred and twenty autonomous clones were isolated from a library of approximately 2 × 106 independent integrations, indicating that 1 in 17,000 proviruses adopted an inducible cellular promoter. Although this frequency seems low, it is consistent with the prediction that following G418 selection most remaining U3Cre integrations are in non-transcribed chromosomal regions and only a small fraction of these are in transcriptionally silent genes. Assuming that 107 proviral integrations are required to saturate the genome (one integration every 300 nt to cover 3 × 109 nt, the size of the haploid genome), a maximum of 600 genes may be induced by IL-3 withdrawal or during apoptosis. This represents 0.6% of the estimated total number of genes (1 × 105) and 1% of all genes expressed at any one time (2–4 × 104) (34). This calculation assumes that retroviruses integrate randomly throughout most of the genome (35–37) and possible sites of integration (38) neither preferentially permit nor exclude U3Cre expression. Moreover, the recorded frequency of induced events is based on the use of exon traps and may be significantly higher with 3′ splice site-activated intron traps (reviewed in ref. 27).
Fifty percent of the analyzed fusion transcripts were also inducible by serum starvation, suggesting that the disrupted genes are associated with an apoptotic program or with growth arrest. Whether these genes directly control the suicide machinery or are part of other mechanisms that are activated as a consequence of PCD (such as DNA repair or cell cycle progression), remains to be established. In any case, the characterization of the cellular transcripts induced by IL-3 starvation is likely to provide further insight into the complex molecular changes taking place during apoptosis.
Finally, although the described strategy seems unique in terms of allowing selection of cells that are predisposed to die, it is not restricted to the analysis of PCD and may also be useful for recovering transcriptional targets of other signal transduction pathways.
Acknowledgments
We thank H. Gu and K. Rajewsky for providing the plasmids pMCCre and pGEM30 and C. Stocking for pMultiCSF. We also thank Drs. M. Grez and M. Nehls for carefully reviewing the manuscript, and Dr. R. Bruyns and Ms. S. Reindel for helping with the mRNA purification. Finally, we thank Ms. Uta Matysiak-Scholze for assistance during DNA sequencing. This work was supported by grants from Deutsche Forschungsgemeinschaft and Deutsche Krebshilfe to H.v.M.
Footnotes
Abbreviations: Cre, Cre recombinase; FDCP, factor-dependent cell line; IL-3, interleukin 3; LTR, long terminal repeat; PCD, programmed cell death; tk, thymidine kinase; pgk, phosphoglycerate kinase.
References
- 1.Steller H. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 2.White E. Genes Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Oltvai Z, Korsmeyer S. Cell. 1994;79:189–192. doi: 10.1016/0092-8674(94)90188-0. [DOI] [PubMed] [Google Scholar]
- 4.Thompson C. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 5.White K, Tahaoglu E, Steller H. Science. 1996;271:805–807. doi: 10.1126/science.271.5250.805. [DOI] [PubMed] [Google Scholar]
- 6.White K, Grether M, Abrams J, Young L, Farrell K, Steller H. Science. 1994;264:677–683. doi: 10.1126/science.8171319. [DOI] [PubMed] [Google Scholar]
- 7.Grether M, Abrams J, Agapite J, White K, Steller H. Genes Dev. 1995;9:1694–1708. doi: 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z, Friedrich G, Soriano P. Genes Dev. 1994;8:2293–2301. doi: 10.1101/gad.8.19.2293. [DOI] [PubMed] [Google Scholar]
- 9.Forrester L, Nagy A, Sam M, Watt A, Stevenson L, Bernstein A, Joyner A, Wurst W. Proc Natl Acad Sci USA. 1996;93:1677–1682. doi: 10.1073/pnas.93.4.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeGregori J, Russ A, von Melchner H, Rayburn H, Priyaranjan P, Jenkins N A, Copeland N G, Ruley H E. Genes Dev. 1994;8:265–276. doi: 10.1101/gad.8.3.265. [DOI] [PubMed] [Google Scholar]
- 11.Skarnes W, Moss J, Hurtley S, Beddington R. Proc Natl Acad Sci USA. 1995;92:6592–6596. doi: 10.1073/pnas.92.14.6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reddy S, DeGregori J V, von Melchner H, Ruley H E. J Virol. 1991;65:1507–1515. doi: 10.1128/jvi.65.3.1507-1515.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li L, Cohen S. Cell. 1996;85:319–329. doi: 10.1016/s0092-8674(00)81111-3. [DOI] [PubMed] [Google Scholar]
- 14.Lih C J, Cohen S N, Wang C, Chao S L. Proc Natl Acad Sci USA. 1996;93:4617–4622. doi: 10.1073/pnas.93.10.4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerr W G, Heller M, Herzenberg L A. Proc Natl Acad Sci USA. 1996;93:3947–3952. doi: 10.1073/pnas.93.9.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sauer B, Henderson N. Proc Natl Acad Sci USA. 1988;85:5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu H, Zou Y, Rajewsky K. Cell. 1993;73:1155–1164. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- 18.Vaux D, Cory S, Adams J M. Nature (London) 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 19.Dexter T, Garland J, Scott D, Scolnick E, Metcalf D. J Exp Med. 1980;152:1036–1047. doi: 10.1084/jem.152.4.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Melchner H, Reddy S, Ruley H E. Proc Natl Acad Sci USA. 1990;87:3733–3737. doi: 10.1073/pnas.87.10.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russ A P, Friedel C, Grez M, von Melchner H. J Virol. 1996;70:4927–4932. doi: 10.1128/jvi.70.8.4927-4932.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haeuptle M T, Flint N, Gough N M, Dobberstein B. J Cell Biol. 1989;108:1227–1236. doi: 10.1083/jcb.108.4.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dirks W, Wirth M, Hauser H. Gene. 1993;128:247–249. doi: 10.1016/0378-1119(93)90569-o. [DOI] [PubMed] [Google Scholar]
- 24.von Melchner H, Ruley H E. J Virol. 1989;63:3227–3233. doi: 10.1128/jvi.63.8.3227-3233.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metcalf D. Recent Results in Cancer Research. Vol. 61. Berlin: Springer; 1977. [PubMed] [Google Scholar]
- 26.von Melchner H, DeGregori J V, Rayburn H, Reddy S, Friedel C, Ruley H E. Genes Dev. 1992;6:919–927. doi: 10.1101/gad.6.6.919. [DOI] [PubMed] [Google Scholar]
- 27.von Melchner H, Ruley H E. In: Gene Entrapment: In Functional Analysis of the Human Genome. Farzaneh F, Cooper D N, editors. Oxford: Bios Scientific; 1995. pp. 109–129. [Google Scholar]
- 28.Chang W, Hubbard C, Friedel C, Ruley H. Virology. 1993;193:737–747. doi: 10.1006/viro.1993.1182. [DOI] [PubMed] [Google Scholar]
- 29.Varmus H. Science. 1988;240:1427–1435. doi: 10.1126/science.3287617. [DOI] [PubMed] [Google Scholar]
- 30.Adams M D, Kelley J M, Gocayne J D, Dubnick M, Polymeropoulos M H, Xiao H, Merril C R, Wu A, Olde B, Moreno R F, Kerlavage A R, McCombie W R, Venter J C. Science. 1991;252:1643–1651. doi: 10.1126/science.2047873. [DOI] [PubMed] [Google Scholar]
- 31.Adams M D, Kerlavage A R, Fields C, Venter J C. Nat Genet. 1993;4:256–267. doi: 10.1038/ng0793-256. [DOI] [PubMed] [Google Scholar]
- 32.Liang P, Zhu W, Zhang X, Guo Z, O’Connell R P, Averboukh L, Wang F, Pardee A B. Nucleic Acids Res. 1994;22:5763–5764. doi: 10.1093/nar/22.25.5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCleland M, Mathieu-Daude F, Welsh J. Trends Genet. 1995;11:242–246. doi: 10.1016/s0168-9525(00)89058-7. [DOI] [PubMed] [Google Scholar]
- 34.Lewin B. Cell. 1975;4:77–93. doi: 10.1016/0092-8674(75)90113-0. [DOI] [PubMed] [Google Scholar]
- 35.Goff S P. Cancer Cells. 1990;2:172–178. [PubMed] [Google Scholar]
- 36.Sandmeyer S B, Hansen L J, Chalker D L. Annu Rev Genet. 1990;24:491–518. doi: 10.1146/annurev.ge.24.120190.002423. [DOI] [PubMed] [Google Scholar]
- 37.Withers-Ward E S, Kitamura Y, Barnes J P, Coffin J M. Genes Dev. 1994;8:1473–1487. doi: 10.1101/gad.8.12.1473. [DOI] [PubMed] [Google Scholar]
- 38.Shih C C, Stoye J P, Coffin J M. Cell. 1988;53:531–537. doi: 10.1016/0092-8674(88)90569-7. [DOI] [PubMed] [Google Scholar]