

Abstract
The understanding of the mutational mechanism that generates high levels of variation at microsatellite loci lags far behind the application of these genetic markers. A phylogenetic approach was developed to study the pattern and rate of mutations at a dinucleotide microsatellite locus tightly linked to HLA-DQB1 (DQCAR). A random Japanese population (n = 129) and a collection of multiethnic samples (n = 941) were typed at the DQB1 and DQCAR loci. The phylogeny of DQB1 alleles was then reconstructed and DQCAR alleles were superimposed onto the phylogeny. This approach allowed us to group DQCAR alleles that share a common ancestor. The results indicated that the DQCAR mutation rate varies drastically among alleles within this single microsatellite locus. Some DQCAR alleles never mutated during a long period of evolutionary time. Sequencing of representative DQCAR alleles showed that these alleles lost their ability to mutate because of nucleotide substitutions that shorten the length of uninterrupted CA repeat arrays; in contrast, all mutating alleles had relatively longer perfect CA repeat sequences.
Microsatellites, which are abundant in the human genome, are highly polymorphic due to allelic variation in the number of repeat units of 2–5 base pairs. These genetic markers are widely used in human genetics, although an understanding of the mutational mechanism that generates such a high level of variation lags far behind their applications. The high similarity in allele sizes at each locus inspired the hypothesis that stepwise mutation mechanisms through replication slippage might be involved (1–4). The direct knowledge of spontaneous mutation and the estimation of mutation rates in microsatellites are largely due to the contribution of large scale pedigree studies in the search for disease genes (5–9). Such studies have shown that more than 90% of mutations result in the expansion or contraction of the alleles by a single repeat unit (2 to 4 bp). These studies also estimated that the mutation rates of microsatellites studied range from 1.2 × 10−4 to 1.5 × 10−2. Comparison of allele sequences revealed very complex mutation patterns (10–12). However, the laborious pedigree studies prevent one from observing enough mutations for each locus and consequently all the conclusions drawn are based on a large collection of loci.
Population genetic studies of microsatellite loci have been fruitful in revealing possible pattern of mutations. It has been shown that a simple stepwise mutation model (SMM; ref. 13), which is intuitively compatible with replication slippage mechanism, can describe the behaviors of many microsatellite loci in the populations (14–16). An extended SMM that allows a few big changes in repeat number (multistep SMM) may be more suitable for microsatellites (16, 17). However, since only simple summary statistics were used in the above studies, characterization of individual locus was not possible. Furthermore, the pattern and rate of mutations at microsatellites may vary among loci (14, 18). The understanding of the pattern and rate of mutations is very relevant to the applications of those genetic markers in evolutionary studies as well as in gene mapping studies (19–22).
Phylogenetic approaches reconstruct evolutionary relationships not only among taxa at various levels but also among genes as well as alleles from a single locus. A phylogeny usually spans over a much larger number of generations or meiotic events than a pedigree. Consequently, phylogenetic approaches allow the study of low frequency genetic events such as mutations or recombinations using a relatively smaller sample size. The objective of this work is to demonstrate the utility of this approach in studying the tempo and mode of mutations at a microsatellite locus while the principles used have much broader applications for problems of similar nature.
A CA-repeat microsatellite locus tightly linked to HLA DQB1 locus (DQCAR; ref. 23) was recently characterized in several populations (24). Since DQB1 can be typed relatively easily, it provides a unique opportunity to reconstruct the evolutionary history of DQCAR and consequently to study the pattern and rate of DQCAR mutations by identifying common ancestors of DQCAR alleles. By typing random Japanese samples and a collection of multiethnic samples at both DQB1 and DQCAR, we could demonstrate that mutation rates vary drastically among alleles of a single microsatellite locus. The mechanism of such variation is discussed in this report.
MATERIALS AND METHODS
A Japanese population of 129 unrelated individuals was collected and typed at DRB1, DQA1, and DQB1. A collection of 941 samples from various ethnic backgrounds (Japanese, Papua New Guinean, African-American, and Caucasians including cell line and patients with various autoimmune diseases) were also typed at DRB1, DQA1, and DQB1 in several clinical laboratories and in our lab. All the samples selected for this study were oligotyped by using PCR–sequence specific oligonucleotide probe or related method at DQB1 (see refs. 24 and 25 for detailed descriptions). Typing at DQCAR locus was performed in our lab (24).
DQB1-DQCAR haplotypes were inferred using three different methods. First, haplotypes were inferred based on known associations established from homozygotes and previously published papers (24). An expectation maximization algorithm developed by Excoffier and Slatkin (26) was used to estimate haplotype frequencies. A new haplotyping algorithm based on parsimony principle by minimizing the number of conflicting inferences with exhaustive search was also used (L.J., unpublished work).
The phylogeny of DQB1 alleles was reconstructed based upon the aligned complete coding sequences obtained from the European Molecular Biology Laboratory Data Library. Several measures of genetic distance including Kimura’s two-parameter model (27) were used in the reconstruction of distance matrix phylogenies (see refs. 28 and 29 for the definition of genetic distance measures and the detailed description of phylogeny reconstruction). The maximum parsimony method was also used (30). The tree was rooted by including pig and horse DQB sequences. mega (29) was used in this analysis.
RESULTS
Two Major Groups of DQB1 Alleles Were Found.
A phylogeny of 14 DQB1 alleles presented in our samples was reconstructed based upon the aligned complete coding sequences. Fig. 1 presents a Neighbor-joining tree (31) using Kimura’s two-parameter model (27). Several other measures of genetic distance were also used, and all gave identical results in terms of the topology of phylogeny using the Neighbor-joining method. The maximum parsimony tree displayed a slightly different topology which became identical with the Neighbor-joining tree shown in Fig. 1 once peptide-binding sites (PBSs) were removed from the analysis. The discrepancy of the phylogeny with and without PBS is probably due to either the variation of substitution rate among nucleotide sites driven by selection variation (32) or the very frequent gene conversion (33). Interestingly, DQB1 alleles were grouped into two major clusters: DQB1*02 (0201, 0202), DQB1*03 (0301, 0302, 03032), and DQB1*04 (0401, 0402) formed the non-DQ1 group; and DQB1*05 (0501, 0502, 05031) and DQB1*06 (06011, 0602, 0603, 0604) formed another cluster (DQ1). The high bootstrapping values shown on internal branches indicate the reliability of the phylogeny (see ref. 29 for detailed explanation of bootstrapping procedure).
Figure 1.
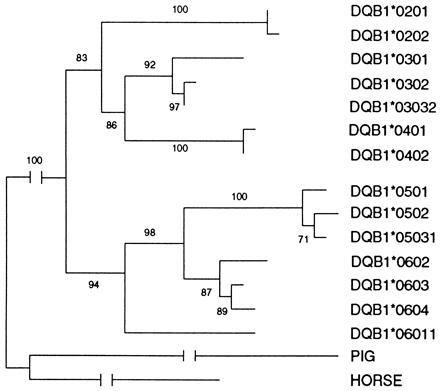
Neighbor-joining tree of DQB1 based on Kimura’s two parameter distance. The numbers are bootstraping values (in percentage) with 500 replications.
The Level of DQCAR Variation Differs Greatly Between the Two Groups of DQB1 Alleles.
DQB1-DQCAR haplotypes were inferred using three different methods. The haplotypes were first inferred manually based on (i) the haplotypes of homozygote cell lines, (ii) known associations established from homozygotes at one of the two loci (DQB1 or DQCAR), and (iii) pedigree information for some individuals. Two independent inferrences generated identical results. A maximum likelihood algorithm developed by Excoffier and Slatkin (23) was used to estimate haplotype frequencies. Only those haplotypes with non-zero frequencies were accepted. A new haplotyping algorithm based on parsimony principle minimizing the number of conflicting inferences with exhaustive search was also used (L.J., unpublished work). Identical haplotypes were obtained using all three different methods.
The Japanese DQB1-DQCAR haplotypes (number of chromosomes in parenthesis) are presented in Table 1. The haplotype data for the mixed population including both 941 multiethnic samples and 129 Japanese samples are shown in Fig. 2. The alleles observed only once in all 2140 chromosomes are indicated by parentheses (Table 2). The number of each DQB1 allele is listed in the last column of Fig. 2. For both the Japanese and the mixed population samples, the combined observed frequencies for DQ1 and non-DQ1 alleles were similar. However, the level of DQCAR variation differs greatly between the two groups of DQB1 alleles. In both populations, the numbers of DQCAR alleles in non-DQ1 lineages were larger than those found in the DQ1 lineages, thus suggesting that DQCAR alleles associated with non-DQ1 alleles might have much higher mutation rates than those associated with DQ1 alleles. Furthermore, the DQCAR alleles found in non-DQ1 lineages tended to have larger fragment sizes (109–125 bp) than those observed in DQ1 lineages with the exception of DQB1*0201. In contrast, most of the DQCAR alleles in the DQ1 group were monomorphic with a 103-bp size (24).
Table 1.
Haplotype frequencies of DQB1-DQCAR in Japanese samples
| DQB1 alleles | DQCAR allele size | Total | |||
|---|---|---|---|---|---|
| DQB1*0201 | 99(1) | 1 | |||
| DQB1*0202 | 113(1) | 1 | |||
| DQB1*0301 | 113(3) | 117(14) | 121(12) | 123(4) | 33 |
| DQB1*0302 | 109(1) | 111(11) | 113(10) | 22 | |
| DQB1*03032 | 111(1) | 115(30) | 117(2) | 33 | |
| DQB1*0401 | 113(16) | 115(9) | 117(4) | 29 | |
| DQB1*0402 | 113(3) | 115(2) | 117(4) | 9 | |
| DQB1*0501 | 103(22) | 22 | |||
| DQB1*0502 | 103(3) | 3 | |||
| DQB1*05031 | 107(5) | 5 | |||
| DQB1*0602 | 103(17) | 17 | |||
| DQB1*0603 | 103(2) | 2 | |||
| DQB1*0604 | 103(21) | 21 | |||
| DQB1*06011 | 107(60) | 60 | |||
Numbers in parentheses are numbers of chromosomes.
Figure 2.
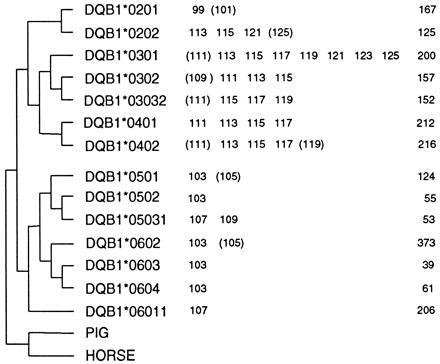
DQCAR alleles in a mixed population (2140 chromosomes).
The number of DQCAR alleles observed in our population for a given DQB1 lineage should be the result of several factors: (i) The mutation rate of each individual DQCAR allele, (ii) the age of the DQB1 lineage studied, and (iii) the effective number of individuals in the population bearing the DQB1 allele. The relative importance of these factors can be estimated by studying the properties of the phylogeny and the frequency distribution of DQB1 allele in various populations.
Do DQCAR Alleles Associated with Specific DQB1 Lineage Share a Common Ancestor?
The DQCAR alleles associated with a given DQB1 allele share a unique common ancestral microsatellite allele right after the DQB1 lineage emerged in the population. This becomes only evident if it can be shown that the “new” DQB1 allele contains specific mutation(s) unique to this lineage. A maximum parsimony principle (30) was used to infer the mutations for each external lineages of the DQB1 phylogeny based upon the sequences of oligonucleotide (24) used in genotyping. A DQB1 lineage was considered as a new derivative if it had a unique mutation that was not present in other lineages. Sites that have to be explained by multiple and/or recurrent mutations (most probably due to gene conversion) were excluded from the analysis. An asterisk is added to each of the newly derived DQB1 lineage in Fig. 3. Note that the phylogeny was reconstructed (see Fig. 1) based on the sequences of the complete coding region while the samples were typed by PCR-SSOP oligotyping which includes only a few sites within the DQB1 gene (mostly in the second exon).
Figure 3.

Estimated branch length for each DQB1 lineage. Number, size range, and the number of uninterrupted CA repeats of DQCAR alleles carried by each DQB1 lineage.
The age of DQB1 lineage-specific mutations can be estimated by the length of external lineages. The branch lengths of these lineages were estimated using synonymous substitution rate (34) based on the full-length sequences but not those of oligonucleotide. These data are presented in Fig. 3 along with the number and the range of DQCAR allele sizes for each DQB1 lineage observed in the multi-ethnic population. The number of allele is the total number of alleles associated with each DQB1 lineage. The range of allele is the number of possible CA repeat units between the minimum and maximum observed alleles (including both). For example, if the sizes of allele associated with DQB1*0202 range from 113 bp to 125 bp, the range of allele observed in this DQB1 lineage is (125)/2 + 1 = 7. Both the number and the range of alleles associated with a DQB1 lineage reflect the level of variation at the DQCAR locus (17).
A careful observation of the phylogeny displayed in Fig. 3 clearly demonstrate that the number of DQCAR allele observed in each individual DQB1 branch does not correlate with the estimated branch length. In all cases, the number and the range of DQCAR alleles observed in the non-DQ1 lineages are much larger than those associated with DQ1 lineages independent of branch length and of the number of chromosomes tested with each DQB1 subtype. This was evident not only for alleles that were found frequently within a specific population (e.g., DQB1*06011 in Japanese, DQB1*02 in Caucasians) but also for alleles with high frequency across various ethnic groups (DQB1*03, DQB1*04, DQB1*0602, and DQB1*0604; see ref. 35 for DQB1 allele frequencies across worldwide populations). For example, DQB1*06011 is featured with an extremely long lineage and a very large number of individuals compared with others but it is still monomorphic at the DQCAR locus. Similarly, DQB1*0602 is generally monomorphic in a very large number of individuals across all populations. In contrast, several DQB1 lineages such as DQB1*0202, DQB1*0302, and DQB1*0401 have almost negligible branch lengths, yet they have much larger numbers of DQCAR alleles. Therefore, the variation of effective numbers of chromosomes bearing DQB1 lineages does not necessarily contribute to the much larger variation of DQCAR alleles in non-DQ lineages.
Mutation Rates Vary Among DQCAR Alleles.
The above observation suggests that DQCAR alleles with larger fragment sizes in non-DQ1 lineages (109–125 bp) have higher mutation rates than DQ1 DQCAR alleles with small fragment sizes (103 bp). The only exception is DQB1*0201, a non-DQ1 allele, which shows low DQCAR variation in a large number of chromosomes in spite of its position in the tree. In this lineage, however, the allele size of DQCAR (99 bp) is the smallest amongst all samples. This is thus in fact consistent with the hypothesis that microsatellite alleles with a larger number of repeats tend to have higher mutation rates (4).
At least one DQCAR allele for each DQB1 lineage was subcloned and sequenced. All non-DQ1 DQCAR alleles were found to share almost identical flanking sequences while those for DQ1 were also identical but different from non-DQ1 DQCAR sequences (C.M., unpublished work). Several nucleotide substitutions were observed in DQCAR allele sequences associated with DQ1 lineages, and one of them occurred in the middle of the CA repeat array disrupting the CA repeat structure. The number of uninterrupted CA repeats for each DQB1 lineage is presented in Fig. 3. Non-DQ1 DQCAR alleles do have a larger number of CA repeats than those bearing DQ1 haplotypes. Interestingly, the number of CA repeats at DQB1*0201 is 9, similar to DQ1 DQCAR alleles, although its length (99 bp) is shorter than the latter (103 bp).
DISCUSSION
In this report, we demonstrate that DQB1 alleles can be classified into two major groups: DQ1 and non-DQ1. The number of DQCAR alleles associated with these two DQB1 groups was also found to vary drastically, with non-DQ1 lineages being very variable and DQ1 lineages being almost monomorphic. Further analysis indicated that this difference correlates mostly with the number of uninterrupted CA repeat sequences within the microsatellite rather than other population genetic factors such as the age of DQB1 lineages, effective population sizes, or recent population expansion.
The DQCAR allele sizes observed within individual DQB1 lineages were very close to each other and often differed by increments of 2 bp, indicating that replication slippage is a reasonable hypothetical mechanism underlying size polymorphism at the level of the DQCAR locus. This was especially obvious for the low mutating lineages such as DQB1*0201, DQB1*0501, DQB1*05031, and DQB1*0602 where only two neighboring DQCAR alleles (differing by 2 bp) were observed suggesting single step replication slippage events.
Major histocompatibility complex alleles are well known to be subject to balance selection (30), thus tending to have much longer lineages than neutral markers. It is expected therefore to be an ideal system to study low frequency genetic events. In spite of this, three out of four informative lineages in non-DQ1 show negligible numbers of synonymous substitutions, indicating one might have to study longer sequences. The presence of natural selection could be a problem since it would make it difficult to compare traits across DQB1 lineages if the intensity of the selection among lineages are different. However, there is no reason to believe that DQCAR alleles associated with a single DQB1 lineage are subject to different level of natural selection. Therefore, the conclusions in this report that were mostly based upon the number of alleles among lineages would not be affected by such variation of selection.
Recombination between DQCAR and the DQB1 gene could be a problem. However, the recombination rate within the HLA class II region is extremely low as indicated by strong linkage disequilibrium (36). Furthermore, we have never observed the most frequent DQ1 specific DQCAR allele (103 bp), which has an extremely high frequency in natural populations in any non-DQ1 sample. We therefore conclude that recombination between DQCAR and DQB1 is negligible.
Many approaches in human genetics rely solely on pedigree data. A large number of individuals and pedigrees have to be typed to study low frequency events such as mutations and recombinations since pedigrees only span over a few generations. In contrast, a phylogenetic approach includes events of interest that have accumulated over hundreds or thousands of generations. In this report, we demonstrated the utility of the approach in studying the mutational mechanisms of a microsatellite locus. The principle employed, we believe, could have much broader applications for problems of similar nature.
Acknowledgments
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS33797 to E.M. and by National Institutes of Health Training Grant T32-GS08404 to L.J. We thank Drs. P. Underhill and P. Oefner for the suggestions regarding cloning and sequencing strategies, Dr. L. Excoffier for providing program exhapfre, and Drs. N. Takahata and T. Ota for technical suggestions on phylogeny reconstruction of DQB1 alleles.
References
- 1.Tautz D, Renz M. Nucleic Acids Res. 1984;12:4127–4138. doi: 10.1093/nar/12.10.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levinson G, Gutman G. Mol Biol Evol. 1987;4:203–221. doi: 10.1093/oxfordjournals.molbev.a040442. [DOI] [PubMed] [Google Scholar]
- 3.Stephen W. Mol Biol Evol. 1989;6:198–212. doi: 10.1093/oxfordjournals.molbev.a040542. [DOI] [PubMed] [Google Scholar]
- 4.Weber J. Genomics. 1990;7:524–530. doi: 10.1016/0888-7543(90)90195-z. [DOI] [PubMed] [Google Scholar]
- 5.Kwiatkowski D, Henske E, Weimer K, Ozelius L, Gusella J, Haines J. Genomics. 1992;12:229–240. doi: 10.1016/0888-7543(92)90370-8. [DOI] [PubMed] [Google Scholar]
- 6.Petrukhin K, Speer M, Cayanis E, Bonaldo M, Tantravali U, Soares M, Fischer S, Warburton D, Gilliam T, Ott J. Genomics. 1993;15:76–85. doi: 10.1006/geno.1993.1012. [DOI] [PubMed] [Google Scholar]
- 7.Bowcock A, Osborne-Lawrence S, Barnes R, Chakravarti A, Washington S, Dunn C. Genomics. 1993;15:376–386. doi: 10.1006/geno.1993.1071. [DOI] [PubMed] [Google Scholar]
- 8.Mahtani M, Willard H. Hum Mol Genet. 1993;2:431–437. doi: 10.1093/hmg/2.4.431. [DOI] [PubMed] [Google Scholar]
- 9.Weber J, Wong C. Hum Mol Genet. 1993;8:1123–1128. doi: 10.1093/hmg/2.8.1123. [DOI] [PubMed] [Google Scholar]
- 10.Puers C, Hammond H A, Jin L, Caskey T, Schumm J W. Am J Hum Genet. 1993;53:953–958. [PMC free article] [PubMed] [Google Scholar]
- 11.Garza J C, Freimer N B. Genome Res. 1996;6:211–217. doi: 10.1101/gr.6.3.211. [DOI] [PubMed] [Google Scholar]
- 12.Garza J C, Slatkin M, Freimer N B. Mol Biol Evol. 1995;4:594–603. doi: 10.1093/oxfordjournals.molbev.a040239. [DOI] [PubMed] [Google Scholar]
- 13.Ohta T, Kimura M. Genet Res. 1973;22:201–204. doi: 10.1017/s0016672300012994. [DOI] [PubMed] [Google Scholar]
- 14.Edwards A, Hammond H, Jin L, Caskey C, Chakraborty R. Genomics. 1992;12:241–253. doi: 10.1016/0888-7543(92)90371-x. [DOI] [PubMed] [Google Scholar]
- 15.Valdes A, Slatkin M, Freimer N. Genetics. 1993;133:737–749. doi: 10.1093/genetics/133.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shriver M, Jin L, Chakraborty R, Boerwinkle E. Genetics. 1993;134:983–993. doi: 10.1093/genetics/134.3.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Rienzo A, Peterson A, Garza J, Valdes A, Slatkin M. Proc Natl Acad Sci USA. 1994;91:3166–3170. doi: 10.1073/pnas.91.8.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin, L., Zhong, Y., Shriver, M., Deka, R. & Chakraborty, R. (1994) Am. J. Hum. Genet. 55, Suppl., A39.
- 19.Shriver, M., Jin, L., Chakraborty, R. & Boerwinkle, R. (1993) Am. J. Hum. Genet. 53, Suppl., 858.
- 20.Slatkin M. Genetics. 1995;139:457–462. doi: 10.1093/genetics/139.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldstein D, Linares A, Cavalli-Sforza L, Feldman M. Genetics. 1995;139:463–471. doi: 10.1093/genetics/139.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shriver M, Jin L, Boerwinkle E, Deka R, Ferrell E, Chakraborty R. Mol Biol Evol. 1995;12:914–920. doi: 10.1093/oxfordjournals.molbev.a040268. [DOI] [PubMed] [Google Scholar]
- 23.Satyanarayana K, Strominger J. Immnogenetics. 1992;35:235–240. doi: 10.1007/BF00166828. [DOI] [PubMed] [Google Scholar]
- 24.Macaubas C, Hallmayer J, Kalil J, Kimura A, Yasunaga S, Grumet F, Mignot E. Hum Immnol. 1995;42:209–220. doi: 10.1016/0198-8859(94)00101-u. [DOI] [PubMed] [Google Scholar]
- 25.Kimura A, Dong R, Harada H, Sasazuki T. Tissue Antigens. 1992;40:5–12. doi: 10.1111/j.1399-0039.1992.tb01951.x. [DOI] [PubMed] [Google Scholar]
- 26.Excoffier L, Slatkin M. Mol Biol Evol. 1995;12:921–927. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- 27.Kimura M. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 28.Nei M. Molecular Evolutionary Genetics. New York: Columbia Univ. Press; 1987. [Google Scholar]
- 29.Kumar, S., Tamura, R. & Nei, M. (1993) mega, Molecular Evolutionary Genetics Analysis (Pennsylvania State Univ., University Park, PA), Version 1.0.
- 30.Fitch W. J Mol Evol. 1981;18:30–37. doi: 10.1007/BF01733209. [DOI] [PubMed] [Google Scholar]
- 31.Saitou N, Nei M. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 32.Hughes A, Nei M. Proc Natl Acad Sci USA. 1989;86:958–962. doi: 10.1073/pnas.86.3.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zangenberg G, Huang M-M, Arnheim N, Erlich H. Nat Genet. 1995;10:407–414. doi: 10.1038/ng0895-407. [DOI] [PubMed] [Google Scholar]
- 34.Nei M, Gojobori T. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 35.Tsuji K, Aizawa M, Sasazuki T. Proceedings of the Eleventh International Histocompatibility Workshop and Conference. Oxford: Oxford Univ. Press; 1992. [Google Scholar]
- 36.Martin M, Mann D, Carrington M. Hum Mol Genet. 1995;4:423–428. doi: 10.1093/hmg/4.3.423. [DOI] [PubMed] [Google Scholar]