

Abstract
The α7 subtype of the nicotinic acetylcholine receptor (nAChR) is the target of studies aimed at identifying features that will lead to the development of selective therapeutics. Five arylidine anabaseines, three with pyridine rings and two with the pyrrole rings, were synthesized in 35 –65% yield via aldol condensation. The compounds are homologs of benzylidine anabaseine and were chosen for synthesis because they provide either a hydrogen bond acceptor (pyridines) or hydrogen bond donor (pyrroles) that may interact with the receptor within the benzylidine selectivity motif. Initial analysis of the new compounds at 100 μM concentration reveal that the two pyrrole anabaseines are good partial agonists of the α7 nAChR, having 40% of the efficacy of ACh, efficacy comparable to 4OH-GTS-21, and dramatically enhanced efficacy relative to the 2- and 4- pyridinyl compounds. The pyrrole compounds were confirmed to be α7 selective, displaying preference for this receptor over muscle and heteromeric neuronal receptor subtypes.
Nicotinic acetylcholine receptors (nAChRs) are pentameric ligand-gated ion channels locating in the cell membrane, which allow cations to flow through upon activation.1,2 Although primarily expressed in muscle cells and neurons they are also expressed in glia and non neuronal tissues.3 In the brain, heteromeric α4β2 and homomeric α7 subtype are the two major nAChRs: the first one has high binding affinity with acetylcholine and nicotine; the latter one can bind with α-bungarotoxin tightly. Though α4β2 receptors may be the most prevalent nAChRs subtype in the brain,4,5 α7 nAChRs have been implicated as influential in neuroprotection, attentional and cognitive enhancement, and inflammatory signaling inhibition. Therefore, selective α7 nAChR agonists are of interest for treatment of Alzheimer’s disease, schizophrenia and inflammatory disorders.6–8
Targeting α7 receptors for either inflammation or CNS disorders relies on the development of selective agents so that other nicotinic receptor subtypes are not affected, possibly alleviating serious side effects such as autonomic dysfunction, seizures and drug dependency.9–11 Previous studies have shown that functionalization of non selective nAChR agonists can confer α7 selectivity.1,13,14 For example, the nAChR agonist anabaseine binds with nAChRs tightly, yet nonselectively with regard to subtype. With an extended hydrophobic group, benzylidene anabaseine (BA, 1, Fig 1) exhibits α7 selectivity.13 We have attributed this selectivity to the interaction of the hydrophobic benzylidine ring with a complementary recognition site in the α7 receptor, termed the benzylidine motif.1 Superimposed on this effect, is the interaction between aryl substituents and residues within the benzylidine motif. For example, 3-(4-hydroxy,2-methoxybenzylidene) anabaseine (4-OH GTS-21), 2 (Fig 1), is a good nAChR partial agonist with high α7 selectivity in both human and rat.15 Varying the ring substituents of BA compounds will result in changes of the agonists potency and efficacy.13,16 A general trend appears to be that polar substituents such as hydroxyl, amino, or methoxy groups tend to give more efficacious agonists.13,17 We considered the possibility that these polar groups undergo favorable hydrogen bonding interactions with the receptor, but dissecting the interaction is problematic because hydroxyl and amino groups are both hydrogen bond acceptors and donors. To simplify the analysis, we sought to characterize agonists where the substituents hydrogen bonding interactions could only be H-bond donating or only H-bond accepting, but not both at the same time. Figure 1 presents three novel pyridinyl methylene anabaseines (H-bond acceptor only), 3a–c, and two pyrrol-3-yl methylene anabaseines (H-bond donor only), 4a,b, that we synthesized and screened as agonists for different types of nAChRs (α7, α4β2, α3β4, α1β1εδ).
Figure 1.

Arylidine anabaseines. Benzylidene anabaseine (BA, 1); 4-OH GTS-21, 2; pyridinyl methylene anabaseines, 3a–c; pyrrolyl methylene anabaseine, 4a,b. Whereas 4OH-GTS-21 presents a complex pattern of H-bond donation and acceptor function, compounds 3 and 4 are either H bond donors or acceptors, respectively.
Most starting materials and reagents for the synthesis were purchased from Sigma-Aldrich or Fischer Scientific. However, we synthesized pyrrole-3-carboxyldehyde and anabaseine by modifications of the reported methods.18,19 Thus, 3-pyrrole carboxaldehyde (5b) was synthesized in 22 % yield from the triflic acid catalyzed equilibration of 2-pyrrole carboxaldehyde (5a). We omitted the tedious continuous extraction step, and found that two consecutive chromatographic steps, a dry silica column followed by a normal silica gel column, were required to purify the crude product(both eluted with CH2Cl2/EtOAc at 12:1 ratio).18 Anabaseine was synthesized as shown in Scheme 1. The gram scale synthesis of anabaseine utilized a mixed Claisen type condensation between N-protected piperidinone 8 and ethyl nicotinate.20 The synthesis of 8 proceeded in low yields, so we sought to optimize the Mannich reaction of valerolactam 7 leading to N-protected 8. Substitution of paraformaldehyde for aqueous formaldehyde and use of a Dean-Stark trap led to protected 8 in 37% yield after chromatography (silica, CH2Cl2/CH3OH, 15:1).
Scheme 1.
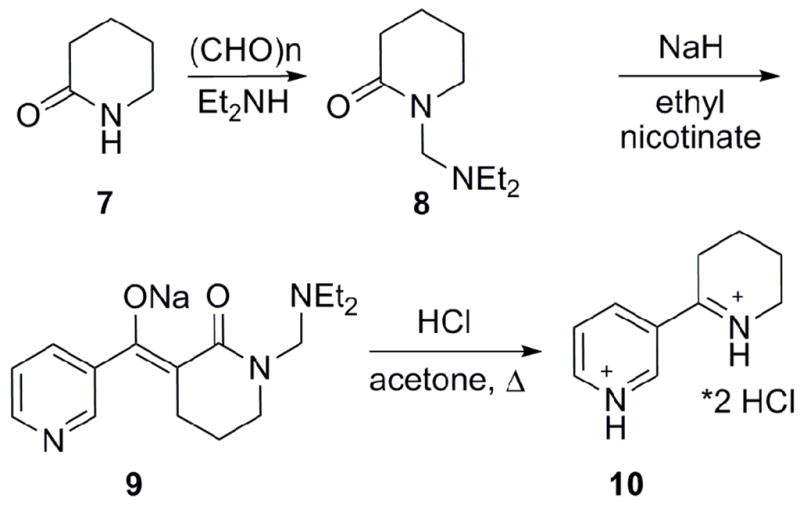
Synthesis of anabaseine.
Mixed Claisen condensation of 1-(diethylaminomethyl)-2-piperidone 8 and ethyl nicotinate afforded the sodium salt, 9, in 37% yield. Conversion of 9 to anabaseine dihydrochloride was achieved by refluxing in a 5:1 concentrated HCl/acetone mixture.18 After recrystallization from absolute ethanol, we obtained anabaseine dihydrochloride salt 10 as a white solid in 49% yield.
The general method for synthesis of benzylidine anabaseines and aryl analogs involves aldol-type condensation between an aromatic aldehyde and anabaseine, presumably via its enamine form. Though the reaction may be catalyzed by acid, e.g. HCl, conjugate acid/base systems such as acetic acid/acetate can be utilized for aromatic carboxaldehydes with strong electron withdrawing groups.21 Thus, condensation of anabaseine dihydrochloride (or anabaseine dihydrobromide) and pyridine carboxaldehydes or pyrrole carboxaldehydes in methanolic solution optimally produced 3a–c and 4a,b in the presence of acetic acid and sodium acetate (mole ratio: 3 to 1) at room temperature (Scheme 2). It was important to conduct these reactions in inert atmosphere (N2 or Ar) to minimize side product formation. Reactions for generating 3a–c were finished within twelve hours. The syntheses of 4a,b required longer reaction times with the less reactive pyrrole carboxaldehydes; 24 h for compound 4a and 96 h for compound 4b. Although some aldol condensation reactions can require heat to effect dehydration,22 double bond formation can often be readily achieved at room temperature. In the present system with pyridine and pyrrole carboxaldehydes, higher temperature tends to adversely affect the yields for condensation. For example, in the synthesis of 3a, running the reaction at 60 °C, 40 °C and 25 °C gave 17%, 34% and 58% yields, respectively. Compounds 3a–c and 4a,b were carefully purified by silica column chromatography, using CH2Cl2/CH3OH or CH2Cl2/iPrOH as eluent. It was found that CH2Cl2/iPrOH was the superior choice for purification of 3c. The yields for the five compounds were as follows: 3a, 58%; 3b, 50%; 3c, 65%; 4a, 39 %; 4b, 35 %. The E-double bond geometry for 3a–c and 4a,b were confirmed by NOESY.(Figure 2) For 3a–c, irradiation of H6 resulted in enhancement of both H2 and H4, which would only arise from the E-isomer of 3. Moreover, interaction of the pyridine ring’s hydrogen with H5 can also be seen in all of the three 3 isomers. In the case of the pyrrole compounds 4a,b, irradiating H5 produced a positive NOE on H8 of the pyrrole ring, leading to confirmation of the E-olefin geometry for compounds 4a and 4b. MOPAC semi-empirical calculations reveal that the two aromatic rings found in compounds 3 or 4 can π-stack face to face in the Z-isomer. This still doesn’t compensate for other destabilizing effects: the Z-isomers for compounds 3 and 4 are all higher in energy than the E-isomers by more than 4 kcal/mol.
Scheme 2.

Synthesis of pyridinyl methylene anabaseine and pyrrolyl methylene anabaseine: 6a, 6b and 6c refer to ortho, meta, para pyridine carboxylaldehyde respectively; 5a and 5b refer to 2- and 3- pyrrole carboxylaldehyde respectively
Figure 2.

NOE enhancement used for assigning the olefin geometry of 3a–c and 4a, 4b.
The agonist activity of compounds 3a–c and 4a,b was studied in Xenopus oocytes expressing mRNAs corresponding to α1β1εδ, α3β4, α4β2 or α7 subunits of nAChRs. Compounds were screened at a concentration of 100 μM. Non-α7 receptors were not activated by any of the compounds, with the possible exception of 4b/α3β4 which showed very weak (5%) activation relative to control ACh. Interestingly, the pyrrole H-bond donor compounds 4a,b proved to be good partial agonists with 40% activation relative to ACh with potencies only 2–3 fold lower than benzylidene anabaseines such as 2MeO4OHBA (4OH-GTS-21) with comparable efficacy.24 It is also noteworthy that compared to the “parent” unsubstituted benzylidine anabaseine, 1, compounds 4a,b are almost four times more efficacious.13 Pyridine 3b produced comparable activation (~ 35% ACh maximum) but was approximately 10-fold less potent (ED50 ≈ 100 μM), while 3a and 3c were very weak, producing less than 20% of the ACh response at a concentration of 1 mM. In summary, the results suggest that H-bond donation by substituents on the extended hydrophobic group of BA analogs leads to preferential activation of the α7 nAChR subtype. Further studies aimed at the detailed characterization of the electrophysiology of these compounds will be reported elsewhere.
Acknowledgments
This work was supported by grant R01 GM57481. We wish to thank Dr. I. Ghiviriga for help with the NOESY experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Dani JA, Bertrand D. Ann Rev Pharmacol Toxicol. 2007;47:699. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- 2.Romanelli MN, Gratteri P, Guandalini L, Martini E, Bonaccini C, Gualtieri F. ChemMedChem. 2007;2:746. doi: 10.1002/cmdc.200600207. [DOI] [PubMed] [Google Scholar]
- 3.Sharma G, Vijayaraghavan S. J Neurobio. 2002;53:524. doi: 10.1002/neu.10114. [DOI] [PubMed] [Google Scholar]
- 4.Whiting PJ, Lindstrom JM. J Neurosci. 1988;8:3395. doi: 10.1523/JNEUROSCI.08-09-03395.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. Mol Pharm. 1992;41:31. [PubMed] [Google Scholar]
- 6.Carson R, Craig D, McGuinness B, Johnston JA, O’Neil FA, Passmore AP, Ritchie CW. J Med Genet. 2008;45:244. doi: 10.1136/jmg.2007.052704. [DOI] [PubMed] [Google Scholar]
- 7.Waldo MC, Adler LE, Leonard S, Olincy A, Ross RG, Harris JG, Freedman R. Biol Psychiatry. 2000;47:231. doi: 10.1016/s0006-3223(99)00272-3. [DOI] [PubMed] [Google Scholar]
- 8.De Jonge WJ, Ulloa L. Brit J Pharm. 2007;151:915. doi: 10.1038/sj.bjp.0707264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franceschini D, Orr-Urtreger A, Yu W, Mackey LY, Bond RA, Armstrong D, Patrick JW, Beaudet AL, De Biasi M. Behav Brain Res. 2000;113:3. doi: 10.1016/s0166-4328(00)00195-9. [DOI] [PubMed] [Google Scholar]
- 10.Damaj MI, Glassco W, Dukat M, Martin BR. J Pharmacol Exp Ther. 1999;291:1284. [PubMed] [Google Scholar]
- 11.Damaj MI, Kao W, Martin BR. J Pharm Exp Ther. 2003;307:526. doi: 10.1124/jpet.103.054908. [DOI] [PubMed] [Google Scholar]
- 12.Horenstein NA, Leonik FM, Papke RL. Molecular Pharmacology. in press. [Google Scholar]
- 13.Papke RL, Meyer EM, Lavieri S, Bollampally SR, Papke TAS, Horenstein NA, Itoh Y, Papke JKP. Neuropharm. 2004;46:1023. doi: 10.1016/j.neuropharm.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 14.Leonik FM, Papke RL, Horenstein NA. Bioorg Med Chem Lett. 2007;17:1520. doi: 10.1016/j.bmcl.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Meyer EM, Tay ET, Papke RL, Meyers C, Huang GL, DeFiebre CM. Brain Res. 1997;768:49. doi: 10.1016/s0006-8993(97)00536-2. [DOI] [PubMed] [Google Scholar]
- 16.Kem WR, Mahnir VM, Prokai L, Papke RL, Cao X, LeFrancois S, Wildeboer K, Prokai-Tatrai K. doi: 10.1124/mol.65.1.56. [DOI] [PubMed] [Google Scholar]
- 17.Porter-Papke J, Soti F. Mol Pharm. 2004;65:56. doi: 10.1124/mol.65.1.56. [DOI] [PubMed] [Google Scholar]
- 18.Papke, R. L.; Kem, W. R.; Horenstein, N. A. unpublished results.
- 19.Carson JR, Davis NM. J Org Chem. 1981;46:839. [Google Scholar]
- 20.Zoltewicz John A, Cruskie Michael P., Jr OPPI BRIEFS. 1995;27:510. [Google Scholar]
- 21.Katritzky AR, Suzuki K, He H. J Org Chem. 2002;67:8224. doi: 10.1021/jo0203241. [DOI] [PubMed] [Google Scholar]
- 22.Zoltewicz JA, Prokai-Tatrai K, Bloom L, Kem WR. Heterocycles. 1993;35:171. [Google Scholar]
- 23.Nielsen AT, Houlihan WJ. Org React. 1968;16:1–438. [Google Scholar]
- 24.Papke RL, Porter-Papke JK. Br J Pharmacol. 2002;137:49–61. doi: 10.1038/sj.bjp.0704833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.As pointed out by a reviewer, the impact of pyrrole ring size on the activity of 4a,b relative to 6-membered ring compounds remains an area for investigation.