

Abstract
Epigenetic modification in the nuclear genome plays a key role in human tumorigenesis. In this paper, we investigated whether changes in the mtDNA copy number frequently reported to vary in a number of human tumors induce methylation changes in the nucleus. We utilized the Restriction Landmark Genomic Scanning (RLGS) to identify genes that undergo changes in their methylation status in response to the depletion and repletion of mtDNA. Our study demonstrates that depletion of mtDNA results in significant changes in methylation pattern of a number of genes. Furthermore, our study suggests that methylation changes are reversed by the restoration of mtDNA in cells otherwise lacking the entire mitochondrial genome. These studies provide the first direct evidence that mitochondria regulate epigenetic modification in the nucleus that may contribute to tumorigenesis.
Keywords: mitochondria, epigenetic, retrograde, mitochondrial DNA, mtDNA depletion, RLGS, OXPHOS, rho0
Introduction
One of the most common and consistent phenotypes of cancers is their defective mitochondria.1-15 Mitochondria have been implicated in carcinogenesis because of their vital role in energy production, nuclear-mitochondrial and mitochondria-to-nucleus signal integration, control of apoptosis and various metabolic pathways. Mitochondrial dysfunction leads to resistance to apoptosis16 and to the well-known Warburg Effect described in 1956.1 Since Warburg's hypothesis, a number of mitochondrial abnormalities in cancers both at the genetic and metabolic levels have been reported.6-8
Mitochondria contain their own genome. The mitochondrial genome is a 16.6 kb closed-circular, double-helical molecule and is inherited only through the mother. The mtDNA encodes two rRNAs, 22 tRNAs and 13 proteins.6-8 Each of these proteins is a subunit of one of four respiratory enzyme complexes localized in the mitochondria. They include seven subunits of respiratory enzyme complex I, one subunit of complex III, three subunits of complex IV and two subunits of complex V. All other (∼1,500) mitochondrial proteins, including those involved in the replication, transcription and translation of mtDNA, are encoded by nuclear genes and are targeted to the mitochondrion by a specific transport system.17 Although mtDNA represents less than 1% of total cellular DNA, its gene products are essential for normal cell function. Replication of mtDNA is error prone and, unlike nuclear DNA, mammalian mtDNA contains no introns, has no protective histones and is exposed to deleterious reactive oxygen species generated by oxidative phosphorylation (OXPHOS).4 These factors contribute to the accumulation of mutations in mtDNA at an approximately tenfold greater rate than in nuclear DNA.18,19 Thus, somatic mutations in mtDNA have been reported in tumor cells.6 The majority of these mutations are homoplasmic in nature, indicating that the mutant mtDNA becomes dominant in tumor cells.
Human mtDNA contains one single control region called D-loop that controls mtDNA replication and the transcription of mtDNA-encoded OXPHOS genes. Mutations in the D-loop region have been reported in all tumors examined to date.6-8 Mutations in the D-loop region result in altered binding affinities of the nuclear proteins involved in mtDNA replication and transcription, leading to the depletion of mtDNA content.20,21 Our laboratory recently reported the absence of mtDNA-encoded cytchrome c-oxidase subunit II expression in more than 40% of breast and ovarian tumors.22 Other laboratories have also measured mtDNA content in tumors and report a decrease in mtDNA content in breast,23,24 renal,25 hepatocellular24,26 and gastric tumors.27 The depletion of mtDNA is also supported by a decrease in oxidative phosphorylation (OXPHOS) levels in renal tumors.28 It is also noteworthy that drugs used for the treatment of HIV inhibit not only their target HIV reverse transcriptase but also the human POLG responsible for the replication of mtDNA. Thus, the treatment of HIV induces depletion of mtDNA.29 Tamoxifen, a commonly used drug for the treatment of breast cancer, also depletes mtDNA.30 A recent study also demonstrates that the depletion of mtDNA correlates with tumor progression and prognosis in breast cancer patients.31
Recent studies suggest cross-talk between the mitochondrial and the nuclear genome.6-8,22,32,33 Our studies indicate that the depletion of mtDNA results in genetic changes in the nuclear genome due to the impaired ability of the cell to repair oxidative damage.34,35 This effect was related to an imbalance in nucleotide pools with a five- to six-fold reduction in dTTP and dCTP pools in human cells that have been depleted of mtDNA.36 We found that chromosomal instability (CIN) was increased in rho0 cells compared to the parental lines.35 We also demonstrated that depletion of mtDNA activates an evolutionary-conserved error-prone DNA repair pathway involving Rev1, Rev3 and Rev7 proteins.34 In addition to changes in nucleotide metabolism and DNA repair, we have also demonstrated gene-expression changes linked to inter-genomic cross-talk between the nucleus and mitochondria.37 The apurinic/apyrimidinic endo-nuclease (APE1) is downregulated in rho0 cells. Importantly, this downregulation can be reversed if wild-type mitochondria are reintroduced to the rho0 cells to generate a cybrid. Furthermore, we have demonstrated that tumorigenicity was also reversed by the transfer of wild-type mitochondria to rho0 cells.35 Together, these studies provide evidence for the role of mtDNA levels in tumorigenesis.
While it is quite clear that the mitochondrial impairment commonly seen in cancer cells leads to increased damage to the nuclear genome and to changes in nuclear gene-expression, the question of epigenetic changes related to mitochondrial dysfunction has not been addressed. DNA methylation is an epigenetic modification of the DNA that is frequently disrupted in nearly all types of cancer. Hypomethylation of the repetitive elements associated with increased genomic instability is frequently seen38-40 and the hypermethylation of specific CpG islands in promoter regions of several tumor-suppressor genes is commonly observed to be associated with transcriptional silencing of the gene.41-44 To date, it is unclear whether the mtDNA affects epigenetic changes in the nuclear genome. In this study, we took a genomic-scanning approach to identifying CpG island methylation changes associated with the depletion and repletion of mtDNA. Our study suggests that mitochondrial impairment induces DNA methylation in the nuclear genome, and that some, but not all, of the changes induced by the depletion of the mitochondrial genome can be reversed by reintroduction of wild-type mitochondria.
Results
DNA methylation changes associated with the depletion of mtDNA
The human oxidative phosphorylation system consists of five multi-subunit complexes, of which the individual subunits, with the exception of complex II, are encoded either by mitochondrial or nuclear DNA. The generation of a cell-line devoid of mtDNA and, as a consequence, lacking all mtDNA-encoded subunits is possible by culturing cells in the presence of a low concentration of ethidium bromide 37. This chemical generation of an mtDNA depletion cell-line occurred by the preferential inhibition of mtDNA replication. In this study, we have used the MDA-MB-435 breast carcinoma cell-line and MCA 12A, the immortalized breast epithelial cell-line, as well as the 143B osteosarcoma cell-line without mtDNA (rho0 cells) obtained from wild-type cells by chemical treatment as described under material and methods. Figure 1A demonstrates that, as expected, the mitochondrially encoded COX II was absent from the three tested types of rho0 cells and present in parental cells.
Figure 1.
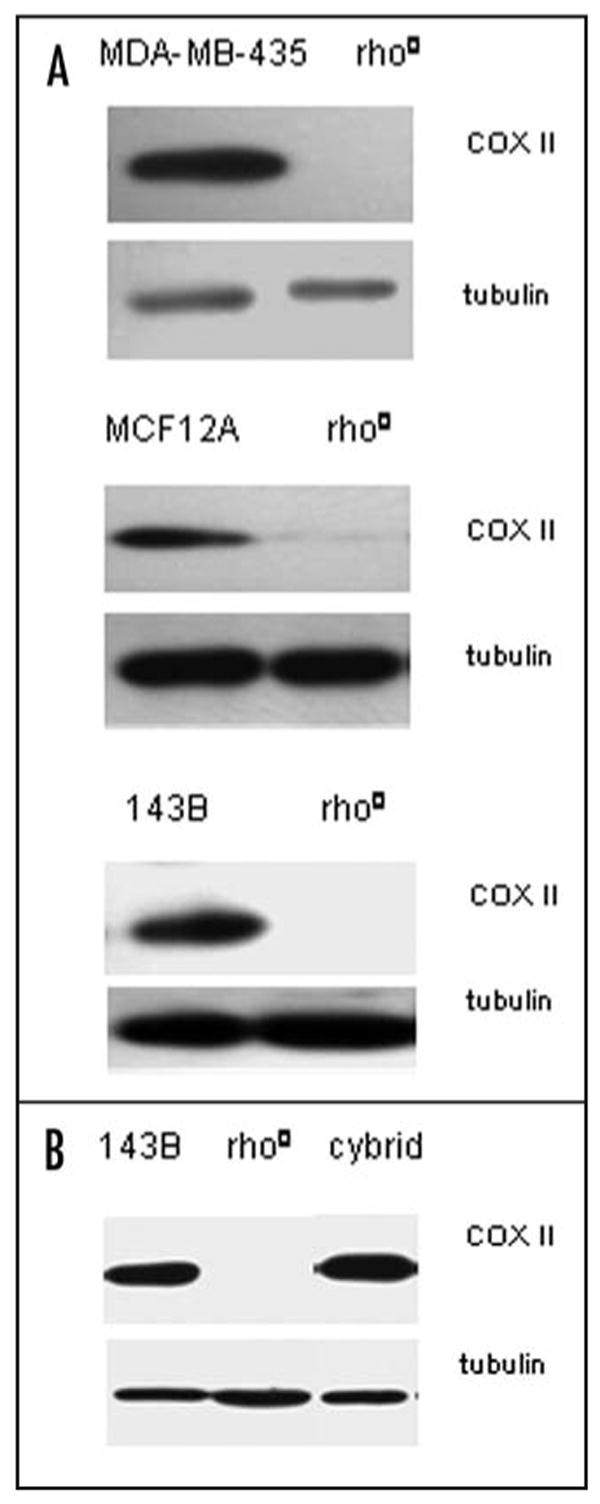
Mitochondrial COXII expression in rho0 and cybrid cells. Western blot analysis of expression of subunit II of respiratory chain enzyme complex IV (cytochrome c oxidase: COX II) in cultured cells lacking mtDNA (rho0 cells) derived either chemically (A) or genetically after transfection of parental cells with HSV UL12.5 gene (B) characterized by absence of mtDNA encoded COXII expression. Repopulation of 143B rho0 cells with human platelet mitochondria resulted in reexpression of COX II expression level in cybrids cells. The tubulin antibody was used as a loading control.
We took a complementary approach to deplete mtDNA. This approach utilized a proven genetic method recently, described by Saffran et al. (2007). Saffran et al. demonstrated that expression of herpes-simplex virus (HSV) 12.5 gene in human cells leads to depletion of mtDNA.46 We obtained genetically engineered mtDNA-depleted 143B cells that were generated after the transfection of cells with the HSV UL12.5 gene.46 This genetically engineered mtDNA-depleted 143B cell-line was used to generate cybrid cell lines after reintroducing wild-type mtDNA from human blood platelets. Figure 1B demonstrates that mitochondrially-encoded COX II protein is present in cybrid cells but absent in rho0 cells, thus confirming successful reintroduction of wild type mtDNA in rho0 cells.
We performed a restriction landmark genomic-scanning (RLGS) analysis to assess whether CpG island methylation changes were associated with the loss of mtDNA. In each case, the pattern of RLGS spots in the parental cell-line was compared to the rho0 cell-line. The technical aspects of RLGS are fairly straightforward conceptually52,56-58 and have been described in detail.59 High-molecular-weight DNA is digested with the “landmark” enzyme. The landmark enzyme determines the sites of the genome that will be labeled by filling in the enzyme half-site with radioactive nucleotides and thus is responsible for the pattern visualized on autoradiography. Using a methylation-sensitive landmark enzyme such as NotI is what gives RLGS its methylation scanning abilities. Only unmethylated sites can be cut and labeled. Therefore, the methylated sites do not contribute to the two-dimensional pattern seen on autoradiography. Absence of a spot in an RLGS profile is indicative of methylation of the NotI restriction site, while the appearance of a new spot is indicative of hypomethylation of the restriction site.60-63
Figure 2 shows representative regions of the RLGS profiles, comparing the three parental lines to the chemically generated rho0 lines. We found that depletion of mtDNA in these cell-lines led to a number of methylation changes, both hypomethylation events and hypermethylation events. Examples of three RLGS spot changes between parental MCF12A and the rho0 line are shown in Figure 2A. Spots 1 and 2 are present in the parental line, but absent from the rho0 line, indicative of hypermethylation in the rho0 line. Spot 3 is absent from the parental line, but becomes visible in the rho0 line, indicative of hypomethylation. Examples of two RLGS spots hypermethylated in the MDA-MB-435 rho0 line are also shown (Fig. 2B).
Figure 2.
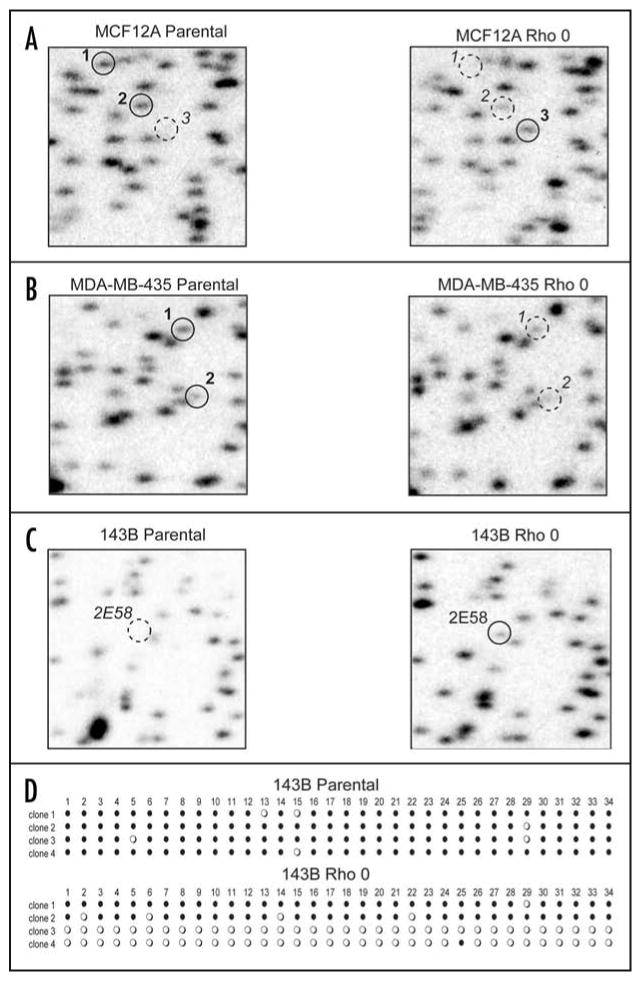
Aberrant CpG island methylation in rho0 cells. Representative examples of RLGS analysis are shown comparing three parental cell lines to their corresponding rho0 cell lines. RLGS spots of interest are shown by solid-line circles to indicate presence of the spot (lack of methylation), while dashed-line circles indicate absence of a spot (methylation). (A) MCF12A cell-line showing spots 1 and 2 exhibiting hypermethylation in the rho0 cell line and spot 3 exhibiting hypomethylation in the rho0 cell line. (B) MDA-MB-435 cell line showing spot 1 and 2 exhibiting hypermethylation in the rho0 cell line. (C) 143B cell line showing RLGS spot 2E58 exhibiting hypomethylation in the rho0 cell line. (D) Bisulfite-sequencing of 2E58 from both the 143B parental and rho0 cell lines. Four clones each were sequenced across a 285 bp region containing 34 CpG dinucleotides. Each horizontal line of circles represents the sequencing data from an individual clone. Each circle represents one CpG: closed circle = methylation; open circle = no methylation.
In the 143B line, we studied the methylation change of RLGS spot 2E58 more closely. This spot was completely absent from the parental line, but appeared in the rho0 line at approximately half of its normal intensity (Fig. 2C). This is indicative of partial hypomethylation of the NotI site in the rho0 cells. We have previously identified the genomic region represented by this spot to be on chromosome 2q31.1 and to be found in a CpG island at the 5′ end of mRNA BC030713.53 We performed standard bisulfite-sequencing of this CpG island in a 285 bp region spanning the transcriptional start site and encompassing 34 CpG dinucleotides. Four clones were sequenced from each cell-line. We found that the parental cell-line was nearly completely methylated across this span in all four clones, but in the rho0 cell line, two of the four clones were nearly completely hypomethylated (Fig 2D). These data confirm the RLGS results demonstrating partial hypomethylation of this CpG island resulting from depletion of the mtDNA.
Table 1 shows the number of hyper- and hypomethylation events detected in each rho0 cell-line relative to the parental lines. The most changes were seen in the MCF12A rho0 line. More than 100 RLGS spots differed between the parental and rho0 MCF12A lines. This large number of changes is high compared to most human cancer RLGS profiles60 and is more typical of the number of differences seen between cell-lines and normal tissues.64 Approximately half the number of changes was seen in the 143B rho0 lines, and half again in the MDA-MB-435 rho0 cells. Interestingly, the 143B rho0 cell line that was created genetically showed only 40% of the hypo- and hypermethylation events than when the rho0 cell line was created chemically.
Table 1. Number of RLGS methylation changes in rho0 cells compared to parental line.
| 1Hypomethylation | 2Hypermethylation | |
|---|---|---|
| MDA-MB-435 Rho0-Chem | 13/1426 (0.9%) | 19/1426 (1.3%) |
| MCF12A Rho0-Chem | 64/1138 (5.6%) | 50/1138 (4.4%) |
| 143B Rho0-Chem | 44/1564 (2.8%) | 14/1564 (0.9%) |
| 143B Rho0-Gen | 16/1437 (1.1%) | 7/1437 (0.5%) |
RLGS spot absent in parental line, but present in rho0 line;
RLGS spot present in parental line, but absent from rho0 line.
Repletion of mtDNA reverses some, but not all DNA methylation changes
Creation of the 143B rho0 line by the genetic approach is superior to the chemical approach for a number of reasons. The chemical approach involves long-term (several weeks) treatment of cells that could contribute directly to methylation changes. In contrast, the genetic approach to develop rho0 cells is quick and requires only 24–48 hours. We complemented the above study by using the genetically generated rho0 cell line and the isogenic cybrid cell-line to determine if the DNA methylation changes we observed could be reversed if wild type mtDNA was re-introduced into the rho0 cells.
A cybrid version of the genetically engineered 143B rho0 cell-line was generated and an RLGS profile was analyzed. Table 2 shows the total set of RLGS spots that differed between the 143B parental and the genetically generated 143B rho0 and/or the cybrid cell lines. We found that 17 of the changes seen between the parental and the rho0 lines were preserved in the cybrid cell lines. Figure 3 shows an example of such a spot. RLGS spot 4B15 is completely methylated in the parental line, but partially hypomethylated in the rho0 line. This partial hypmethylation is maintained, or even slightly enhanced, in the cybrid line. We confirmed this observation by quantitative bisulfite-sequencing using Mass Array Quantitative Methylation analysis (MAQMA) on the Sequenome platform.55 We confirmed heavy methylation of spot 4B15 in the parental line and partial hypomethylation in both the rho0 and cybrid lines (Fig. 2B). Interestingly, we also found five RLGS spots such as 5C32 (Fig. 3C and E) that were hypomethylated in the rho0 lines but remethylated in the cybrid line. MAQMA analysis confirmed the RLGS results demonstrating partial methylation of the 5C32 CpG island in the 143B parental line, hypomethylation in the rho0 line and, in the cybrid line, reversal back to the same level of methylation observed for the parental (Fig. 3D and E). In addition, we identified one hypo- and one hypermethylation event novel to the cybrid lines, in spots 3C15 and 2F77, respectively.
Table 2. RLGS methylation in genetically engineered 143B rho0 and cybrid compared to the parental line.
| Spot | 143B parental | 143B Rho0-genetic | 143B Cybrid-genetic | 3NotI +/- 200 bp | Chr | CpG island | Gene | Context |
|---|---|---|---|---|---|---|---|---|
| 3C15 | 1M | M | U | 4- | - | - | - | - |
| 5C32 | M | 2U | M | chr1:206150742-206151142 | 1q32.2 | Y | CD34 | Body |
| 2B47 | M | U | M | chr2:106048354-106048754 | 2q12.2 | Y | ECRG4 | 5′ End |
| 2E23 | M | U | M | chr4:48037991-48038391 | 4p12 | Y | SLAIN2 | 5′ End |
| 2D46 | M | U | M | chr5:170669561-170669961 | 5q35.1 | Y | TLX3 | Body |
| 3DX1 | M | U | M | - | - | - | - | - |
| 5F13 | M | U | U | chr18:71296405-71296805 | 18q22.3 | Y | BI830246 | 5′ End |
| 4D26 | M | U | U | chr16:48446206-48446606 | 16q12.1 | Y | BX403293 | 5′ End |
| 4B15 | M | U | U | chr16:23954123-23954523 | 16p12.2 | N | PRKCB1 | Body |
| 3CX2 | M | U | U | - | - | - | - | - |
| 2FX1 | M | U | U | - | - | - | - | - |
| 4CX1 | M | U | U | - | - | - | - | - |
| 3FX1 | M | U | U | - | - | - | - | - |
| 3F58 | M | U | U | - | - | - | - | - |
| 3D72 | M | U | U | - | - | - | - | - |
| 3D39 | M | U | U | - | - | - | - | - |
| 2FX1 | M | U | U | - | - | - | - | - |
| 2C61 | M | U | U | - | - | - | - | - |
| 2D53 | U | M | M | chr16:4105987-4106387 | 16p13.3 | Y | ADCY9 | 5′ End |
| 5B04 | U | M | M | chr6:91063383-91063783 | 6q15 | Y | BACH2 | 5′ End |
| 2F68 | U | M | M | chr19:368638-369038 | 19p13.3 | N | SHC2 | Body |
| 3CX1 | U | M | M | - | - | - | - | - |
| 5E12 | U | M | M | - | - | - | - | - |
| 2F77 | U | U | M | chr13:101850696-101851096 | 13q33.1 | Y | FGF14 | Body |
RLGS spot is methylated;
RLGS spot is unmethylated;
Position of the NotI site +/- 200 bp based on the March 2006 freeze of the human genome;
RLGS spot is unidentified.
Figure 3.
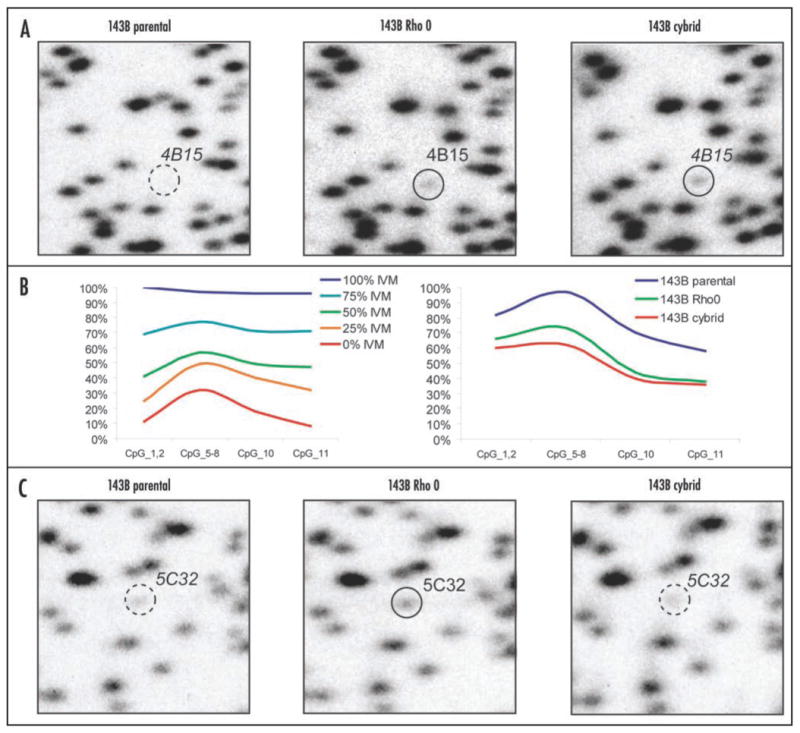
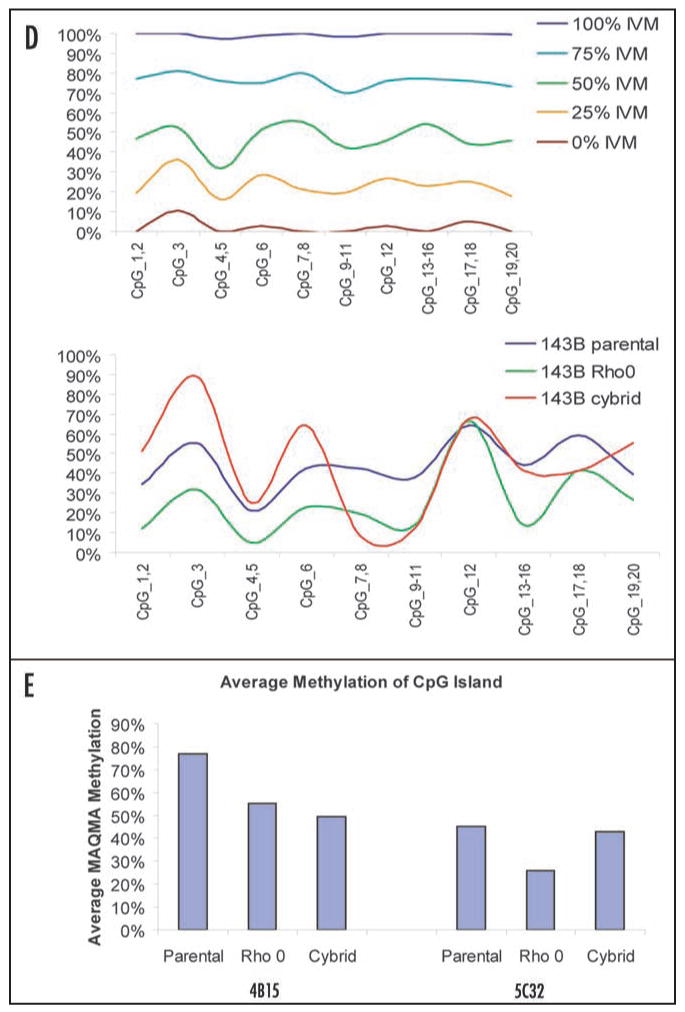
Figure 3. (A–C) Reversal of aberrant CpG island methylation in cybrid cell-line. (A) Representative examples of RLGS analysis are shown comparing spot 4B15 in the 143B parental, genetically engineered rho0 cell line, and cybrid cell lines. The dashed-line circle indicates methylation of 4B15 in the parental line, while the solid-line circles indicate partial hypomethylation of 4B15 in both the rho0 and cybrid lines. (B) Smoothed-line graph of Mass Array Quantitative Methylation Analysis (MAQMA) data for 4B15. The level of methylation at each informative CpG dinucleotide is shown. CpGs 3, 4 and 9 were non-informative because of inappropriate fragment size for MALDI-TOF detection. The left panel shows the results on control DNAs with the indicated ratio of in vitro methylated (IVM) DNA. The right panel shows the results of the three cell-lines, with both the rho0 and the cybrid lines showing hypomethylation relative to the parental line. (C) Representative examples of RLGS analysis comparing spot 5C32 in the 143B parental, genetically rho0 and cybrid cell-lines. The dashed-line circle indicates partial methylation of 5C32 in both the parental and cybrid lines, while the solid-line circle indicates hypomethylation of 4B15 in the rho0 cell line.
Figure 3. (D and E) Reversal of aberrant CpG island methylation in cybrid cell-line. (D) Smoothed-line graph of MAQMA data for 5C32. The level of methylation at each of 20 CpG dinucleotides is shown. The top panel shows the results on control DNAs with the indicated ratio of in vitro methylated (IVM) DNA. The bottom panel shows the results of the three cell-lines, with the rho0 cell line showing hypomethylation relative to the parental line and cybrid lines, except at CpGs 9–11. (E) Histograph analysis of MAQMA data. To obtain a single value for each locus in each cell-line, the average level of methylation at which all informative CpG dinucleotides for 4B15 and 5C32 were calculated is shown.
Discussion
We have previously reported that depletion of mtDNA is a frequent event in breast and ovarian tumors.22 Consistent with our finding in primary tumors, we also reported that depletion of mtDNA from human cells results in tumorigenesis,35 changes in gene-expression,37 nucleotide pools36 and increased chromosomal instability in the form of translocations.35 Here we have demonstrated that the rho0 cell-lines exhibit a significant degree of aberrant CpG island methylation compared to their parental cell-lines. We observed both hypo- and hypermethylation. We analyzed between 1138 and 1564 RLGS spots in each cell line and the number of changes we observed ranged from 114 (∼10%) in the chemically generated MCF12A rho0 cell line to 23 (∼2%) in the genetically engineered 143B rho0 cell line (Table 1). These data suggest that mitochondrial impairment may play a significant role in the aberrant CpG island methylation that is found in nearly all cancers.
Many of the methylation changes that we observed were partial changes. Furthermore, the changes seen in the genetically generated 143B rho0 line were less complete than those seen in any of the three chemically generated rho0 lines. We interpret the partial methylation changes to represent heterogeneity in the population of rho0 cells. The chemical generation of rho0 cells requires a minimum of 12 weeks of treatment, while the genetic generation of rho0 cells is completed within only 1–2 days. We argue that the longer time of treatment using the chemical method leads to significant selection of specific populations of cells and, therefore, less overall heterogeneity in the population of rho0 cells. This explains why the methylation changes in the chemically generated rho0cells are more pronounced than those observed in the genetically generated rho0 cells.
The observation that only a subpopulation of the rho0 cells exhibits a change in the methylation of a particular CpG island suggests that the island was not specifically targeted for aberrant methylation consequent to mitochondrial impairment. This suggests that either the biological consequence of the aberrant methylation, allowing for cell survival and growth under the rho0 conditions, can be accomplished through means other than changes in DNA methylation in some cells, or that other affected loci can substitute for aberrant methylation at the locus in question. This interpretation implies that changes in methylation of the genomic DNA actively contribute to the cells' ability to grow with severe mitochondrial dysfunction. The fact that five of the RLGS spots that were hypomethylated in the genetically induced 143B rho0 line could be reversed by re-introducing wild-type mitochondria to create a cybrid line supports the notion that those loci became hypomethylated in the rho0 line as a direct and specific response to the mitochondrial dysfunction. Once mitochondrial dysfunction was reversed in the cybrid line, the methylation of those loci returned. However, 18 other RLGS loci that exhibited a change in methylation between the parental line and the 143B rho0 line were not reversed by the re-introduction of mitochondria. This may suggest either that some of the methylation changes confer neither an advantage nor a disadvantage once the cells reacquire mitochondrial function, or that those methylation changes had little significance in allowing the cells to survive and grow. These data also suggest that some of the methylation changes induced by the rho0 status of the cells may be random events not specifically selected for.
We do not yet know the mitochondrial signal(s) that triggers epigenetic changes in the nucleus. MtDNA depletion leads to changes in redox status, membrane potential and level of ATP. It is unclear at this time whether a single intracellular change associated with mitochondrial dysfunction or a combination signals methylation changes. Further studies involving specific inhibition of various steps in oxidative phosphorylation may reveal the exact nature of the mitochondrial signal that triggers the epigenetic response. These studies may also aid in understanding the mechanism involving changes in the mtDNA copy number, mitochondrial OXPHOS and epigenetic changes associated with tumorigenesis and treatment of cancer
Studies reported in this paper are also relevant to cytoplasmic/ooplasmic transfer techniques frequently used in assisted reproduction in humans.65 In this technique, cytoplasm is transferred from a healthy fertile donor into the oocytes of a patient who is unable to conceive children. Recent studies demonstrate that the cytoplasmic transfer from fertile oocytes leads to the birth of children.65,66 Our studies suggest a potential epigenetic modification when mitochondria are transferred into the recipient oocytes. Our study also paves the way in identifying the role of epigenetic changes induced by mtDNA mutation(s) involved in pathogenesis of mitochondrial diseases.
Materials and Methods
Cell-lines
MCF12A human epithelial cells were obtained from the American Type Culture Collection and maintained in a 1:1 Dulbecco's modified Eagle's medium (DMEM)/HAMS F12 medium (Mediatech, Manassas, VA) with hydrocortisone (Sigma-Aldrich, St. Louis, MO) (10 mg/ml), cholera toxin (1 mg/ml), (Sigma-Aldrich, St. Louis, MO), insulin (20 mg/ml) (Sigma-Aldrich, St. Louis, MO), epidermal growth factor (100 mg/ml) (Peprotech, Rocky Hill, NJ) and 10% (v/v) horse serum (Mediatech, Manassas, VA). MDA-MB-435 cells (estrogen receptor-negative human breast cancer cells) and 143B human osteosarcoma cells were maintained in the DMEM medium, supplemented with 10% (v/v) FBS (Mediatech, Manassas, VA). All media were supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin. The cells were grown at 37°C in a humidified atmosphere containing 10% CO2 and passaged weekly using 0.25% trypsin.
143B rho0 MCF12A rho0, MDA-MB-435 rho0 (mtDNA-less) cells were derived from 143B, MCF12A and MDA-MB-435 cells, respectively, by the chemical method, as described.37,45 A genetic method involving expression of the HSV 12.5 gene was also used to generate 143B rho0 cells.46 This cell line was kindly provided by Dr. JR Smiley.46 Complete depletion of mtDNA in this cell line was achieved by transient expression of HSV UL12.5 followed by clone isolation. Cells without mtDNA were maintained in culture media supplemented with 100 μg/mL sodium pyruvate and 50 μg/ml uridine. Cybrids of the rho0 cells were constructed as described.47 Platelets were isolated from normal human blood and fused with rho0 cells using polyethylene glycol (PEG) 1500. The resulting cybrid cells were selected in a uridine-free culture medium. Single cybrid clones were isolated by ring cloning. The cybrid status was confirmed by COX II immunoblotting.
Western blot analysis
Western blot was used to confirm the expression of the mtDNA-encoded second subunit of cytochrome C oxidase (COX II). Protein samples (30 μg) were prepared using a RIPA lysis buffer [50 mM tris Ph 7.4, 150 mM NaCl, 1 mM PMSF, 1 mM EDTA, 1% triton x-100, 1% sodium deoxycholate and 0.1% SDS]. This preparation was resolved by electrophoresis on 12% SDS (w/v) polyacrylamide gel by electrophoresis. The separated proteins were transferred to an Immuno-Blot PVDF Membrane (Bio-Rad, Hercules, CA) followed by incubation for one hour at room temperature with PBS containing 1% casein to block nonspecific binding. Blots were incubated overnight at 4°C with 0.2 μg/ml anti-COX II, (Molecular Probes, Eugene, OR) antibody, followed by washing and incubation with a 1:5000 anti-mouse IgG (H + L) antibody (Vector Laboratories, Burlingame, CA).
The bound secondary antibody was detected using an enhanced chemiluminescence solution {1:1 mixture of solution A and B [A: 9.9 ml of 0.1 M Tris (pH 8.5) + 100 μl luminol + 44 μl P-coumaric; B: 10 ml of 0.1 M Tris (pH 8.5) + 6 μl hydrogen peroxide]. The membranes were exposed to CL-XPosure film (ThermoScientific, Rockford, IL). Anti-bovine α-tubulin, mouse monoclonal antibody (Molecular Probes, Eugene, OR) was used to determine equal protein-loading.
Restriction landmark genomic scanning
The protocol for extraction of genomic DNA from tissues was described previously.48 The published protocol of Dai et al. (2003)49 was followed for RLGS gels. RLGS spots of interest were cloned, as previously described.50-53
Sodium-bisulfite treatment
Sodium-bisulfite treatment to convert unmethylated cytosine to thymidine was completed following the manufacturer's protocol EZ-96 DNA Methylation Kit (Zymo Research, Orange, CA), using 750 ng in 50 μl of distilled water and M-Dilution Buffer. The treated samples were re-suspended in 75 μl of M-Elution Buffer and stored at -20°C. A set of five known methylated and unmethylated control DNA samples were included in each round of bisulfite treatment (100%, 75%, 50%, 25% and 0% methylated). The modified DNA was then subjected to bisulfite PCR and subsequent MassARRAY Quantitative Methylation Analysis (MAQMA). All bisulfite PCR reactions were set up using the buffers and conditions originally defined by Herman et al.54 Bisulfite PCR products were cloned for bisulfite sequencing using the Invitrogen (Carlsbad, CA) TOPO cloning kit. The Qiagen miniprep kit was used to isolate individual clone DNAs, and sequencing was performed by the Core facility at Roswell Park Cancer Institute.
MassARRAY quantitative methylation analysis (MAQMA)
MassArray Quantitative Methylation Analysis (MAQMA) was performed using the MassARRAY Compact system developed by the Sequenome Company, as previously described,55 using the primers shown in Supplemental Table 2. This system utilizes mass spectrometry (MS) for the detection and quantitative analysis of DNA methylation using Homogeneous MassCLEAVE (hMC) base-specific cleavage and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS. Methylation calls were performed using the Quantitative Methylation software (Sequenome).
Acknowledgments
We thank Ms. Paula Jones for editing this manuscript. The research in our laboratory was supported by NIH grant RO1 121904 and RO1CA116430 (to KKS) and NY State Department of Health-Breast Cancer Program Contract # CO21336 (MK and KKS). It was also supported, in part, by the National Cancer Institute Support Grant to Roswell Park Cancer Institute, CA 16056.
References
- 1.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–70. [PubMed] [Google Scholar]
- 2.Damiani S, Dina R, Eusebi V. Eosinophilic and granular cell tumors of the breast. Semin Diagn Pathol. 1999;16:117–25. [PubMed] [Google Scholar]
- 3.Damiani S, Eusebi V, Losi L, D'Adda T, Rosai J. Oncocytic carcinoma (malignant oncocytoma) of the breast. Am J Surg Pathol. 1998;22:221–30. doi: 10.1097/00000478-199802000-00011. [DOI] [PubMed] [Google Scholar]
- 4.Singh KK. Mitochondrial DNA mutations in aging, disease and cancer. New York, USA: Springer; 1998. [Google Scholar]
- 5.Singh KK, Russell J, Sigala B, Zhang Y, Williams J, Keshav KF. Mitochondrial DNA determines the cellular response to cancer therapeutic agents. Oncogene. 1999;18:6641–6. doi: 10.1038/sj.onc.1203056. [DOI] [PubMed] [Google Scholar]
- 6.Modica-Napolitano JS, Kulawiec M, Singh KK. Mitochondria and human cancer. Curr Mol Med. 2007;7:121–31. doi: 10.2174/156652407779940495. [DOI] [PubMed] [Google Scholar]
- 7.Modica-Napolitano JS, Singh KK. Mitochondria as targets for detection and treatment of cancer. Expert Rev Mol Med. 2002;4:1–19. doi: 10.1017/S1462399402004453. [DOI] [PubMed] [Google Scholar]
- 8.Modica-Napolitano JS, Singh KK. Mitochondrial dysfunction in cancer. Mitochondrion. 2004;4:755–62. doi: 10.1016/j.mito.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 9.Penta JS, Johnson FM, Wachsman JT, Copeland WC. Mitochondrial DNA in human malignancy. Mutation Research/Reviews in Mutation Research. 2001;488:119–33. doi: 10.1016/s1383-5742(01)00053-9. [DOI] [PubMed] [Google Scholar]
- 10.Rempel A, Mathupala SP, Griffin CA, Hawkins AL, Pedersen PL. Glucose catabolism in cancer cells: amplification of the gene encoding type II hexokinase. Cancer Res. 1996;56:2468–71. [PubMed] [Google Scholar]
- 11.Tallini G. Oncocytic tumours. Virchows Arch. 1998;433:5–12. doi: 10.1007/s004280050209. [DOI] [PubMed] [Google Scholar]
- 12.Bianchi MS, Bianchi NO, Bailliet G. Mitochondrial DNA mutations in normal and tumor tissues from breast cancer patients. Cytogenet Cell Genet. 1995;71:99–103. doi: 10.1159/000134072. [DOI] [PubMed] [Google Scholar]
- 13.Bianchi NO, Bianchi MS, Richard SM. Mitochondrial genome instability in human cancers. Mutat Res. 2001;488:9–23. doi: 10.1016/s1383-5742(00)00063-6. [DOI] [PubMed] [Google Scholar]
- 14.Pedersen PL. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers' most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr. 2007;39:211–22. doi: 10.1007/s10863-007-9094-x. [DOI] [PubMed] [Google Scholar]
- 15.Richard SM, Bailliet G, Paez GL, Bianchi MS, Peltomaki P, Bianchi NO. Nuclear and mitochondrial genome instability in human breast cancer. Cancer Res. 2000;60:4231–7. [PubMed] [Google Scholar]
- 16.Park SY, Chang I, Kim JY, Kang SW, Park SH, Singh K, Lee MS. Resistance of mitochondrial DNA-depleted cells against cell death: Role of mitochondrial superoxide dismutase. J Biol Chem. 2004;279:7512–20. doi: 10.1074/jbc.M307677200. [DOI] [PubMed] [Google Scholar]
- 17.Schatz G. The protein import system of mitochondria. J Biol Chem. 1996;271:31763–6. doi: 10.1074/jbc.271.50.31763. [DOI] [PubMed] [Google Scholar]
- 18.Grossman LI, Shoubridge EA. Mitochondrial genetics and human disease. Bioessays. 1996;18:983–91. doi: 10.1002/bies.950181208. [DOI] [PubMed] [Google Scholar]
- 19.Johns DR. Seminars in medicine of the Beth Israel Hospital, Boston. Mitochondrial DNA and disease. N Engl J Med. 1995;333:638–44. doi: 10.1056/NEJM199509073331007. [DOI] [PubMed] [Google Scholar]
- 20.Clayton DA. Transcription and replication of mitochondrial DNA. Hum Reprod. 2000;15:11–7. doi: 10.1093/humrep/15.suppl_2.11. [DOI] [PubMed] [Google Scholar]
- 21.Clayton DA. Replication and transcription of vertebrate mitochondrial DNA. Annu Rev Cell Biol. 1991;7:453–78. doi: 10.1146/annurev.cb.07.110191.002321. [DOI] [PubMed] [Google Scholar]
- 22.Desouki MM, Kulawiec M, Bansal S, Das GM, Singh KK. Cross talk between mitochondria and superoxide generating NADPH oxidase in breast and ovarian tumors. Cancer Biol Ther. 2005;4:1367–73. doi: 10.4161/cbt.4.12.2233. [DOI] [PubMed] [Google Scholar]
- 23.Tseng LM, Yin PH, Chi CW, Hsu CY, Wu CW, Lee LM, Wei YH, Lee HC. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer. 2006;45:629–38. doi: 10.1002/gcc.20326. [DOI] [PubMed] [Google Scholar]
- 24.Lee HC, Yin PH, Lin JC, Wu CC, Chen CY, Wu CW, Chi CW, Tam TN, Wei YH. Mitochondrial genome instability and mtDNA depletion in human cancers. Ann N Y Acad Sci. 2005;1042:109–22. doi: 10.1196/annals.1338.011. [DOI] [PubMed] [Google Scholar]
- 25.Selvanayagam P, Rajaraman S. Detection of mitochondrial genome depletion by a novel cDNA in renal cell carcinoma. Lab Invest. 1996;74:592–9. [PubMed] [Google Scholar]
- 26.Yin PH, Lee HC, Chau GY, Wu YT, Li SH, Lui WY, Wei YH, Liu TY, Chi CW. Alteration of the copy number and deletion of mitochondrial DNA in human hepatocellular carcinoma. Br J Cancer. 2004;90:2390–6. doi: 10.1038/sj.bjc.6601838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu CW, Yin PH, Hung WY, Li AF, Li SH, Chi CW, Wei YH, Lee HC. Mitochondrial DNA mutations and mitochondrial DNA depletion in gastric cancer. Genes Chromosomes Cancer. 2005;44:19–28. doi: 10.1002/gcc.20213. [DOI] [PubMed] [Google Scholar]
- 28.Simonnet H, Alazard N, Pfeiffer K, Gallou C, Beroud C, Demont J, Bouvier R, Schagger H, Godinot C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis. 2002;23:759–68. doi: 10.1093/carcin/23.5.759. [DOI] [PubMed] [Google Scholar]
- 29.Kakuda TN. Pharmacology of nucleoside and nucleotide reverse transcriptase inhibitor-induced mitochondrial toxicity. Clin Ther. 2000;22:685–708. doi: 10.1016/S0149-2918(00)90004-3. [DOI] [PubMed] [Google Scholar]
- 30.Larosche I, Letteron P, Fromenty B, Vadrot N, Abbey-Toby A, Feldmann G, Pessayre D, Mansouri A. Tamoxifen inhibits topoisomerases, depletes mitochondrial DNA, and triggers steatosis in mouse liver. J Pharmacol Exp Ther. 2007;321:526–35. doi: 10.1124/jpet.106.114546. [DOI] [PubMed] [Google Scholar]
- 31.Yu M, Zhou Y, Shi Y, Ning L, Yang Y, Wei X, Zhang N, Hao X, Niu R. Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life. 2007;59:450–7. doi: 10.1080/15216540701509955. [DOI] [PubMed] [Google Scholar]
- 32.Butow RA, Avadhani NG. Mitochondrial signaling: The retrograde response. Mol Cell. 2004;14:1–15. doi: 10.1016/s1097-2765(04)00179-0. [DOI] [PubMed] [Google Scholar]
- 33.Kulawiec M, Arnouk H, Desouki MM, Kazim L, Still I, Singh KK. Proteomic analysis of mitochondria-to-nucleus retrograde response in human cancer. Cancer Biol Ther. 2006;5:967–75. doi: 10.4161/cbt.5.8.2880. [DOI] [PubMed] [Google Scholar]
- 34.Rasmussen AK, Chatterjee A, Rasmussen LJ, Singh KK. Mitochondria-mediated nuclear mutator phenotype in Saccharomyces cerevisiae. Nucleic Acids Res. 2003;31:3909–17. doi: 10.1093/nar/gkg446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh KK, Kulawiec M, Still I, Desouki MM, Geradts J, Matsui S. Inter-genomic cross talk between mitochondria and the nucleus plays an important role in tumorigenesis. Gene. 2005;354:140–6. doi: 10.1016/j.gene.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 36.Desler C, Munch-Petersen B, Stevnsner T, Matsui S, Kulawiec M, Singh KK, Rasmussen LJ. Mitochondria as determinant of nucleotide pools and chromosomal stability. Mutat Res. 2007;625:112–24. doi: 10.1016/j.mrfmmm.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 37.Delsite R, Kachhap S, Anbazhagan R, Gabrielson E, Singh KK. Nuclear genes involved in mitochondria-to-nucleus communication in breast cancer cells. Mol Cancer. 2002;1:6. doi: 10.1186/1476-4598-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karpf AR, Matsui S. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res. 2005;65:8635–9. doi: 10.1158/0008-5472.CAN-05-1961. [DOI] [PubMed] [Google Scholar]
- 39.Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, Ehrlich M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983;11:6883–94. doi: 10.1093/nar/11.19.6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gama-Sosa MA, Wang RY, Kuo KC, Gehrke CW, Ehrlich M. The 5-methylcytosine content of highly repeated sequences in human DNA. Nucleic Acids Res. 1983;11:3087–95. doi: 10.1093/nar/11.10.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–96. [PubMed] [Google Scholar]
- 42.Costello JF, Plass C. Methylation matters. J Med Genet. 2001;38:285–303. doi: 10.1136/jmg.38.5.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones PA. Cancer. Death and methylation. Nature. 2001;409:141–4. doi: 10.1038/35051677. [DOI] [PubMed] [Google Scholar]
- 44.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–66. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 45.Inoue K, Takai D, Hosaka H, Ito S, Shitara H, Isobe K, LePecq JB, Segal-Bendirdjian E, Hayashi J. Isolation and characterization of mitochondrial DNA-less lines from various mammalian cell lines by application of an anticancer drug, ditercalinium. Biochem Biophys Res Commun. 1997;239:257–60. doi: 10.1006/bbrc.1997.7446. [DOI] [PubMed] [Google Scholar]
- 46.Saffran HA, Pare JM, Corcoran JA, Weller SK, Smiley JR. Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep. 2007;8:188–93. doi: 10.1038/sj.embor.7400878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chomyn A. Platelet-mediated transformation of human mitochondrial DNA-less cells. Methods Enzymol. 1996;264:334–9. doi: 10.1016/s0076-6879(96)64031-2. [DOI] [PubMed] [Google Scholar]
- 48.Smiraglia DJ, Fruhwald MC, Costello JF, McCormick SP, Dai Z, Peltomaki P, O'Dorisio MS, Cavenee WK, Plass C. A new tool for the rapid cloning of amplified and hypermethylated human DNA sequences from restriction landmark genome scanning gels. Genomics. 1999;58:254–62. doi: 10.1006/geno.1999.5840. [DOI] [PubMed] [Google Scholar]
- 49.Dai Z, Zhu WG, Morrison CD, Brena RM, Smiraglia DJ, Raval A, Wu YZ, Rush LJ, Ross P, Molina JR, Otterson GA, Plass C. A comprehensive search for DNA amplification in lung cancer identifies inhibitors of apoptosis cIAP1 and cIAP2 as candidate oncogenes. Hum Mol Genet. 2003;12:791–801. doi: 10.1093/hmg/ddg083. [DOI] [PubMed] [Google Scholar]
- 50.Yu L, Liu C, Bennett K, Wu YZ, Dai Z, Vandeusen J, Opavsky R, Raval A, Trikha P, Rodriguez B, Becknell B, Mao C, Lee S, Davuluri RV, Leone G, Van den Veyver IB, Caligiuri MA, Plass C. A NotI-EcoRV promoter library for studies of genetic and epigenetic alterations in mouse models of human malignancies. Genomics. 2004;84:647–60. doi: 10.1016/j.ygeno.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 51.Zardo G, Tiirikainen MI, Hong C, Misra A, Feuerstein BG, Volik S, Collins CC, Lamborn KR, Bollen A, Pinkel D, Albertson DG, Costello JF. Integrated genomic and epigenomic analyses pinpoint biallelic gene inactivation in tumors. Nat Genet. 2002;32:453–8. doi: 10.1038/ng1007. [DOI] [PubMed] [Google Scholar]
- 52.Smiraglia DJ, Frühwald MC, Costello JF, McCormick SP, Dai Z, Peltomäki P, OD MS, Cavenee WK, Plass C. A new tool for the rapid cloning of amplified and hypermethylated human DNA sequences from restriction landmark genome scanning gels. Genomics. 1999;58:254–62. doi: 10.1006/geno.1999.5840. [DOI] [PubMed] [Google Scholar]
- 53.Smiraglia DJ, Kazhiyur-Mannar R, Oakes CC, Wu YZ, Liang P, Ansari T, Su J, Rush LJ, Smith LT, Yu L, Liu C, Dai Z, Chen SS, Wang SH, Costello J, Ioshikhes I, Dawson DW, Hong JS, Teitell MA, Szafranek A, Camoriano M, Song F, Elliott R, Held W, Trasler JM, Plass C, Wenger R. Restriction landmark genomic scanning (RLGS) spot identification by second generation virtual RLGS in multiple genomes with multiple enzyme combinations. BMC Genomics. 2007;8:446. doi: 10.1186/1471-2164-8-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, Cantor CR, Field JK, van den Boom D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci USA. 2005;102:15785–90. doi: 10.1073/pnas.0507816102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hatada I, Hayashizaki Y, Hirotsune S, Komatsubara H, Mukai T. A genomic scanning method for higher organisms using restriction sites as landmarks. Proc Natl Acad Sci USA. 1991;88:9523–7. doi: 10.1073/pnas.88.21.9523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okazaki Y, Okuizumi H, Sasaki N, Ohsumi T, Kuromitsu J, Hirota N, Muramatsu M, Hayashizaki Y. An expanded system of restriction landmark genomic scanning (RLGS Ver. 1.8) Electrophoresis. 1995;16:197–202. doi: 10.1002/elps.1150160134. [DOI] [PubMed] [Google Scholar]
- 58.Hirotsune S, Hatada I, Komatsubara H, Nagai H, Kuma K, Kobayakawa K, Kawara T, Nakagawara A, Fujii K, Mukai T, et al. New approach for detection of amplification in cancer DNA using restriction landmark genomic scanning. Cancer Res. 1992;52:3642–7. [PubMed] [Google Scholar]
- 59.Costello JF, Smiraglia DJ, Plass C. Restriction landmark genome scanning. Methods. 2002;27:144–9. doi: 10.1016/s1046-2023(02)00067-1. [DOI] [PubMed] [Google Scholar]
- 60.Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomaki P, Lang JC, Schuller DE, Yu L, Bloomfield CD, Caligiuri MA, Yates A, Nishikawa R, Su Huang H, Petrelli NJ, Zhang X, O'Dorisio MS, Held WA, Cavenee WK, Plass C. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000;24:132–8. doi: 10.1038/72785. [DOI] [PubMed] [Google Scholar]
- 61.Smiraglia DJ, Plass C. The study of aberrant methylation in cancer via restriction landmark genomic scanning. Oncogene. 2002;21:5414–26. doi: 10.1038/sj.onc.1205608. [DOI] [PubMed] [Google Scholar]
- 62.Smiraglia DJ, Szymanska J, Kraggerud SM, Lothe RA, Peltomaki P, Plass C. Distinct epigenetic phenotypes in seminomatous and nonseminomatous testicular germ cell tumors. Oncogene. 2002;21:3909–16. doi: 10.1038/sj.onc.1205488. [DOI] [PubMed] [Google Scholar]
- 63.Smiraglia DJ, Smith LT, Lang JC, Rush LJ, Dai Z, Schuller DE, Plass C. Differential targets of CpG island hypermethylation in primary and metastatic head and neck squamous cell carcinoma (HNSCC) J Med Genet. 2003;40:25–33. doi: 10.1136/jmg.40.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smiraglia DJ, Rush LJ, Fruhwald MC, Dai Z, Held WA, Costello JF, Lang JC, Eng C, Li B, Wright FA, Caligiuri MA, Plass C. Excessive CpG island hypermethylation in cancer cell lines versus primary human malignancies. Hum Mol Genet. 2001;10:1413–9. doi: 10.1093/hmg/10.13.1413. [DOI] [PubMed] [Google Scholar]
- 65.Barritt JA, Brenner CA, Malter HE, Cohen J. Mitochondria in human offspring derived from ooplasmic transplantation. Hum Reprod. 2001;16:513–6. doi: 10.1093/humrep/16.3.513. [DOI] [PubMed] [Google Scholar]
- 66.Hawes SM, Sapienza C, Latham KE. Ooplasmic donation in humans: The potential for epigenic modifications. Hum Reprod. 2002;17:850–2. doi: 10.1093/humrep/17.4.850. [DOI] [PubMed] [Google Scholar]