

Abstract
Activation of the complement system occurs in a variety of neuroinflammatory diseases and neurodegenerative processes of the CNS. Studies in the last decade have demonstrated that essentially all of the activation components and receptors of the complement system are produced by astrocytes, microglia, and neurons. There is also rapidly growing evidence to indicate an active role of the complement system in cerebral ischemic injury. In addition to direct cell damage, regional cerebral ischemia and reperfusion (I/R) induces an inflammatory response involving complement activation and generation of active fragments, such as C3a and C5a anaphylatoxins, C3b, C4b, and iC3b. The use of specific inhibitors to block complement activation or their mediators such as C5a, can reduce local tissue injury after I/R. Consistent with therapeutic approaches that have been successful in models of autoimmune disorders, many of the same complement inhibition strategies are proving effective in animal models of cerebral I/R injury. One new form of therapy, which is less specific in its targeting of complement than monodrug administration, is the use of immunoglobulins. Intravenous immunoglobulin (IVIG) has the potential to inhibit multiple components of inflammation, including complement fragments, pro-inflammatory cytokine production and leukocyte cell adhesion. Thus, IVIG may directly protect neurons, reduce activation of intrinsic inflammatory cells (microglia) and inhibit transendothelial infiltration of leukocytes into the brain parenchyma following an ischemic stroke. The striking neuroprotective actions of IVIG in animal models of ischemic stroke suggest a potential therapeutic potential that merits consideration for clinical trials in stroke patients.
INTRODUCTION
In an attempt to further expand our understanding of neuronal injury in stroke and neurodegeneration, researchers have focused their efforts on one of the major elements of the inflammatory response, the complement cascade. The complement system is a component of the innate immune response comprised of multiple cascades that play an integrated role in the initiation and regulation of the inflammatory response. Furthermore, the complement cascade has been shown to play a critical role in ischemia/reperfusion (I/R) models of tissue damage (Arumugam et al., 2002; Arumugam et al., 2003; Arumugam et al., 2004b; Arumugam et al., 2004c; Woodruff et al., 2004; Arumugam et al., 2006), and is believed to have deleterious effects also in cerebral I/R injury (Mocco et al., 2006a; Arumugam et al., 2007). It has recently been suggested that the activation of the complement system is involved in the pathogenesis of several neurodegenerative diseases including Alzheimer's disease (AD) and Parkinson's disease (PD). A key finding regarding the mechanism of complement activation in AD was that Aβ, when aggregated, was a strong complement activator (Rogers et al., 1992) and this finding was supported by several other studies (Bradt and Kolb, 1998; Farkas et al., 2003). Recent immunochemical studies have shown that complement activation also occurs on Lewy bodies and melanized neurons in the PD substantia nigra (Loeffler et al., 2006). In addition, we recently showed that neuroinflammation in the form of complement activation and C5a generation plays a deleterious role in 3-Nitroproprionic Acid (3-NP)-induced striatal degeneration, an acute model of Huntington's disease (Woodruff et al., 2006). There is also rapidly growing evidence for an active role of the complement system in cerebral ischemic injury in animals. In fact, the 3-NP model of striatal degeneration is initiated by energy impairment of neuronal cells, in a similar manner to ischemia (Roberts, 2005; Garcia et al., 2002). In addition to direct cell damage, regional brain I/R induces an inflammatory response involving complement activation and generation of active fragments, such as C3a and C5a anaphylatoxins, C3b, C4b, and iC3b (D'Ambrosio et al., 2001). Expression of C3a and C5a receptors was found to be significantly increased after transient middle cerebral artery occlusion (MCAO) in the mouse (Nishino et al., 1994; Barnum et al., 2002). Direct deposits of different complement fragments have also been demonstrated in ischemic brain tissue (Mocco et al., 2006a) and complement depletion resulted in reduced post-ischemic brain injury in rats and mice (Atkinson et al., 2006; Costa et al., 2006; Mocco et al., 2006a; Arumugam et al., 2007). One study, in mice with traumatic brain cryoinjury resulted in complement-mediated inflammation and increased tissue damage, which was reduced by a C5a receptor antagonist (Sewel et al., 2004) developed in our laboratory (March et al., 2004). Further, in a different model of closed head traumatic brain injury, complement, at the level of C3, was shown to be a major mediator of brain damage (Leinhase et al., 2006) Taken together, these results provide compelling evidence for the activation and pathogenic role of complement in acute brain injury. Indeed, the relatively few studies using specific inhibitors of various complement components has enabled the dissection of the complement system to unravel which factors are pivotal in driving neural damage (Woodruff et al., 2008). It seems that the proinflammatory mediator, C5a, is likely a key initiator of events leading to neural damage and loss (Woodruff et al., 2008). However, there is much work still to be done to determine optimal targets for drug therapy.
One new form of therapy, which is less specific in its targeting of complement than monodrug administration, is the use of immunoglobulins. There is evidence that the use of hyperimmune serum, containing an enriched fraction of immunoglobulins (IVIG), may be of benefit in sepsis (Laupland et al., 2007), which is also thought to heavily involve the complement system in its pathology. Targeting complement activation in neuroinflammation with immunoglobulins has recently been shown to be beneficial in many neurological diseases (Hughes and Cornblath, 2005; Archelos and Fazekas, 2006; Ringel and Zettl, 2006; Stangel and Pul, 2006). IVIG preparations are fractionated from a plasma pool of healthy donors and contain both immune antibodies and physiologic autoantibodies. Initially used as replacement therapy for patients with primary and secondary immune deficiencies, IVIG is now widely used for the treatment of a large number of autoimmune and systemic inflammatory diseases (Bayry et al., 2007). IVIG has immunoregulatory effects on multiple components of the immune system. It was shown that interaction of IVIG with the complement system prevents the generation of the C5b-9 membrane attack complex and subsequent complement-mediated tissue damage, and by scavenging active complement components and diverting complement attack from cellular targets (Basta and Dalakas, 1994). IVIG is also shown to bind the activated components C3b and C4b, thus preventing the deposition of these fragments on target cell surfaces (Basta et al., 2003; Lutz et al., 2004; Arumugam et al., 2007). Because IVIG has the potential to inhibit multiple components of inflammation including complement fragments, it is not surprising that IVIG may provide neuroprotective effects in many neurological diseases. For example, we recently showed that administration of IVIG to mice subjected to experimental stroke almost entirely eliminated mortality and reduced the amount of brain damage (Arumugum et al., 2007) This review describes our current understanding of the neuroprotection by both complement inhibition and immunoglobulin therapy in ischemic stroke.
COMPLEMENT IN THE BRAIN
Complement can be activated by any one of four pathways: the antibody-dependent classical pathway, the alternative pathway, the mannose-binding lectin/ mannose-binding lectin-associated serine protease (MBL/MASP) pathway, and the recently discovered extrinsic protease pathway (Arumugam et al., 2004b; Thurman and Holers, 2006; Degn et al., 2007; Wills-Karp, 2007; Huber-Lang et al., 2006). The classical pathway is typically initiated when IgM or IgG antigen/antibody complexes bind to C1, the first component of complement. The alternative pathway is triggered by microbial cell surfaces as well as a variety of complex polysaccharides and is characterized by the slow generation of C3 (Fleming and Tsokos, 2006). The MBL/MASP pathway is initiated by the binding of the MBL protein to mannose and glucosamine residues on bacterial cell walls, which is associated with MASP- 1/MASP-2 (Degn et al., 2007). Activation of these proteins results in the cleavage of complement factors 4 and 2 (C4 and C2) and subsequent activation of C3. The principal biological functions of the complement system include cell lysis of invading foreign organisms by polymerization of C5b-9 and disruption of the integrity of the phospholipid bilayer, opsonization by C3b, as well as activation of the inflammation cascade in response to the generation of anaphylatoxins (Taylor and Fairlie, 2000; Haas and van Strijp, 2007). The complement system also promotes solubilization and phagocytic clearance of immune complexes and plays a significant role in promoting humoral immune responses by aiding in antigen presentation to lymphocytes (Kemper and Atkinson, 2007). C3a, C4a, and C5a are termed anaphylatoxins (ie a self-directed toxic principle promoting anaphylaxis) and induce the release of various mediators from mast cells and phagocytes, which in turn amplify inflammatory responses. C3a mediates the migration of eosinophils and mast cells but is not a chemoattractant for neutrophils and has lower potency and apparently fewer biological actions than C5a (Hugli, 1981; Taylor and Fairlie, 2000). Complement factor 5 (C5) is cleaved by C5 convertase to produce C5a and complement factor 5b (C5b). C5a is considered to be one of the most potent phlogistic peptides. It binds to the C5a receptor (C5aR) on PMNs, monocytes, and macrophages with high affinity (Taylor and Fairlie, 2000; Taylor et al., 2001) and increases neutrophil adhesiveness and evokes aggregation (Guo and Ward, 2002), stimulates oxidative metabolism and the production of reactive oxygen species (ROS) in neutrophils (Guo et al., 2003), also induces the secretion of lysosomal enzymes from macrophages and PMNs as well as the secretion of proinflammatory cytokines from monocytes and macrophages (Haynes et al., 2000). C5a may also bind to the recently described C5a-like receptor 2 (C5L2). C5L2 appears to be a non-signaling receptor for C5a, promoting its suggestion as a potential decoy receptor for C5a (Okinaga et al., 2003), However its precise physiological function still awaits comprehensive investigation (Scola et al., 2007; Lee et al., 2008), and its role in the CNS has not been investigated in any detail to date (Gavrilyuk et al., 2005). C5b sequentially binds to complement factor 6 (C6), complement factor 7 (C7), and complement factor 8 (C8) to form C5b-C8, which catalyzes the polymerization of complement factor 9 (C9) to form the membrane attack complex (MAC) (Morgan, 1999). The MAC inserts itself into foreign bacteria and viruses but, if host cells are inadequately protected, the MAC can damage these cells as well, in a process called ‘bystander lysis’. The MAC can also stimulate arachidonate metabolism resulting in the release of prostaglandin E2 from macrophages, leukotriene B4 from neutrophils, thromboxane B2 from human platelets, as well as the release of prostanoids, interleukin-1 (IL-1), and reactive oxygen species (ROS) from human monocytes (Hansch et al., 1984; Hansch et al., 1987; Morgan, 1999). With the current state of knowledge, the combined effects of C5a and the MAC seem to be largely responsible for the inflammation and tissue damage associated with over-exuberant complement activation.
The expression of the complement proteins was, until recently, thought to be restricted largely to cells of the immune system. However, in recent years, studies have demonstrated widespread localization of these complement proteins and complement receptors throughout many tissue and cell types outside the immune system. Recent findings demonstrating that brain cells such as astrocytes, microglia and, surprisingly, neurons possess complement proteins and complement receptors (Gasque et al., 1998; Rotshenker, 2003; Boos et al., 2005). C1q and C4 mRNAs are expressed in the brain at embryonic day 14 (E14) and are abundant in the adult brain (Johnson et al., 1994). Johnson and colleagues (1994) also found that C1qB and C4 mRNAs are present in high amounts in putative microglia in cortical marginal and intermediate zones, and hippocampal analge, but not in the neurogenic ventricular or sub-ventricular zones. This early study suggested that complement components may have novel roles during brain development that may be unrelated to normal cytotoxic actions of the activated complement cascade. Recent findings by Stevens and colleagues (2007) further support roles for complement in brain development (Stevens et al., 2007). They showed that mice deficient in complement protein C1q or the downstream complement protein C3 exhibit large sustained defects in CNS synapse elimination (Stevens et al., 2007). Other studies have also shown that C3a receptor (C3aR) and C5a receptor (C5aR) mRNA are present in most areas of the normal human CNS, including the cerebral cortex, cerebellum, spinal cord and a variety of CNS sub regions (Ames et al., 1996; Tornetta et al., 1997). There is now clear evidence that the both C3aR and C5aR are expressed in glial cells, notably in astrocytes and microglia (Lacy et al., 1995; Gasque et al., 1997; Gasque et al., 1998; Boos et al., 2005). Subsequently, neurons have been identified as being the predominant cell type that possesses C5aR and C3aR under physiological conditions (Davoust et al., 1999; O'Barr et al., 2001; Rahpeymai et al., 2006; Pedersen et al., 2007). More recently, it was demonstrated that neural progenitor cells and immature neurons express C5aR and C3aR (Rahpeymai et al., 2006). This study also showed that basal neurogenesis is decreased in C3 deficient mice and in mice lacking C3aR or mice treated with a C3aR antagonist. The C3 deficient mice also had impaired ischemia-induced neurogenesis both in the subventricular zone, the main source of neural progenitor cells in adult brain, and in the ischemic region (Rahpeymai et al., 2006).
Activation of the complement system occurs in a variety of neuroinflammatory diseases and neurodegenerative processes of the CNS. The hallmarks of AD are abnormal intracellular accumulations of paired helical filaments of the protein tau (neurofibrillary tangles), and extracellular deposits composed primarily of β-amyloid protein (Aβ) plaques within the brain. Complement proteins have been shown to be induced and associated with Aβ plaques in brains of AD patients and familial cerebral amyloid angiopathy patients, as well as in AD animal models, specifically those plaques containing the fibrillar form of the Aβ peptide (Rogers et al., 1992; Bradt and Kolb, 1998; Yasojima et al., 1999; Strohmeyer et al., 2000; Farkas et al., 2003; Fan et al., 2007). The overall effect of complement activation in AD depends on the balance of its detrimental and beneficial effects. On one side, complement activation could induce cell lysis and cause cell death (Shen et al., 1997; Fonseca et al., 2004); on the other side, complement components such as C1q and C3b can promote the clearance of cellular debris and apoptotic cells and enhance cell survival (Wyss-Coray et al., 2002; Rus et al., 2006). The immunohistochemical localization of the MAC was also demonstrated in Pick's Disease where Pick bodies were stained intensely for the MAC (Yasuhara et al., 1994). In contrast to AD, only a few studies have addressed complement involvement in PD. Yamada and colleagues have reported staining of Lewy bodies in the PD substantia nigra for C3b, C3d, C4d, C7 and C9 complement proteins (Yamada et al., 1992; Goldknopf et al., 2006). However, another study found no complement reactivity on Lewy bodies in the cingulate gyrus in PD (Rozemuller et al., 2000).
Complement activation products have also been observed in patients with motor neuron degenerative disease (amytrophic lateral schlerosis, ALS). Elevated levels of activated fragments of C3 and C4 are seen in the serum and CSF of patients with ALS (Annunziata et al., 1985; Tsuboi et al., 1994; Goldknopf et al., 2006). Protein for these activation fragments are also elevated and localized to various glia in the motor cortex and spinal cord of these patients (Kawamata et al., 1992; Tsuboi et al., 1994). Further, our group has demonstrated a specific pathogenic role for C5a in a rat SOD1G93A transgenic model of ALS (Woodruff et al., 2008). The role of complement in traumatic brain injury is also well reported. Clinical and experimental studies suggest a pathophysiologic role for intracerebral complement activation in contributing to inflammation, blood brain barrier (BBB) dysfunction, intracranial leukocyte recruitment, and neuronal cell death after traumatic brain injury (Kaczorowski et al., 1995; Nataf et al., 1999; Bellander et al., 2001; Stahel et al., 2001; You et al., 2007). One rather elegant study showed that the sequential infusion of individual proteins of the membrane attack pathway (C5b6, C7, C8, and C9) into the hippocampus of awake, freely moving rats induced both behavioral and electrographic seizures as well as cytotoxicity (Xiong et al., 2003). Several experimental and clinical studies point to the involvement of complement activation and the subsequent assembly of the C5b-9 complement complex in both neuroinflammation and neuroprotection in multiple sclerosis (MS) and in experimental autoimmune encephalomyelitis (EAE) (Barnum, 2002; Rus et al. 2006; Urich et al., 2006). The role of complement and the mechanistic overlap of the complement cascade with other biochemical events occurring in cerebral I/R injury is complex and is currently being investigated by several groups. These findings raise the possibility that complement activation following ischemic stroke might exert specific effects on glial cell activation and neuronal cell death.
THE ROLE OF COMPLEMENT IN STROKE
Soon after cerebral ischemia, energy-dependent pumps fail, resulting in the flow of ions down their concentration gradients. This results in cellular swelling and depolarization. Calcium ions enter the cell through voltage-dependent and ligand-gated ion channels, resulting in activation of a number of proteases, kinases, lipases and endonucleases, culminating in cell death (Pisani et al., 2004; Berliocchi et al., 2005). A significant portion of ischemia-induced neuronal damage is mediated by excessive accumulation of excitatory amino acids which activate ionotropic glutamate receptors resulting in toxic increases in intracellular sodium and calcium levels (Mehta et al., 2007; Ogawa et al., 2007). Numerous experimental and clinical studies have documented increased levels of oxidative stress during all forms of stroke injury. Free radicals involved in stroke-induced brain injury include superoxide anion radical, hydroxyl radical and nitric oxide (NO). Oxygen free radicals can also be generated by activated microglia and infiltrating peripheral leukocytes via the NADPH oxidase system following reperfusion of ischemic injury (Loh et al., 2006; Chang et al., 2007; Haberman et al., 2007). Both oxygen free radicals and reactive nitrogen species are involved in activating several pathways involved in cell death following stroke, including apoptosis and inflammation (McColl et al., 2008). There are several resident cell populations within brain tissue that are able to secrete proinflammatory mediators after an ischemic insult, including endothelial cells, astrocytes, microglia and neurons. Ischemic brain injury results in increased permeability of the BBB to vascular proteins and blood-borne leukocytes (lymphocytes, macrophages and monocytes) (Arumugam et al., 2005; Kerschensteiner et al., 2008; Klehmet et al., 2008). Ischemic stroke enhances the interactions of vascular endothelial cells with extravascular brain cells (astrocytes, microglia, neurons) and intravascular cells (platelets and leukocytes), interactions that may contribute to the injury process (Yenari et al., 2006; Urra et al., 2008). There is a large body of evidence that implicates leukocytes in the pathogenesis of stroke injury and we have recently reported the pathophysiological significance of lymphocyte recruitment into the brain after ischemic stroke (Arumugam et al., 2005; Ishikawa et al., 2005; Yilmaz et al., 2006).
The complement cascade is vital in initiating and modifying the inflammatory process through its influence on adhesion molecule upregulation, neutrophil chemotaxis, platelet activation, and generation of reactive oxygen species (D'Ambrosio et al., 2001; Arumugam et al., 2004b; Arumugam et al., 2006). The first evidence for involvement of complement in ischemic injuries was proposed by Hill and Ward over three decades ago (Hill and Ward, 1971). More recent experimental studies have shown strong evidence for involvement of complement in intestinal, cardiac, renal and hepatic I/R (Arumugam et al., 2004b). However, the role of complement activation in the pathogenesis of cerebral I/R has been less well understood, though a number of recent studies strongly implicate complement proteins in cerebral ischemia and hemorrhagic stroke.
Studies in the last decade have demonstrated that essentially all of the activation components and receptors of the complement system are produced by astrocytes, microglia, and neurons. The first study indicating a possible role of complement components in patients suffering from subarachnoid hemorrhage (SAH) showed that lumbar cerebrospinal fluid (CSF) levels of C3a and C4a were significantly elevated in the initial stage of SAH, but decreased rapidly (Kasuya and Shimizu, 1989). Within 48 hours after SAH, the mean C3a and C4a levels in the cisternal, lumbar, and ventricular CSF were significantly higher in patients with delayed ischemic neurological deficits (DIND) than in those without DIND (Kasuya and Shimizu, 1989). The serially measured plasma levels of C3a and C4a in patients with DIND were elevated more than in those without DIND. The higher CSF levels of C3a and C4a in patients with DIND indicated a relationship between these components and the pathogenesis of cerebral vasospasms. Another earlier study showed that complement activation by aged erythrocytes can result in MAC insertion into “innocent bystander” smooth-muscle cell membranes and that this mechanism may contribute to development of vasospasm after subarachnoid hemorrhage (Park et al., 1997). The earlier dogma that MAC could only attack non-nucleated cells (Whaley, 1987) now seems to be overturned, although it is still not clear what the final contribution of the MAC is to complement-mediated brain injury.
The effects of experimentally induced global ischemia in the CNS on the biosynthesis of C1q, the recognition subcomponent of the classical complement activation pathway, was demonstrated using semiquantitative in situ hybridization, immunohistochemistry, and confocal laser scanning microscopy. A dramatic and widespread increase of C1q biosynthesis in rat brain microglia (but not in astrocytes or neurons) within 24 h after the ischemic insult was observed (Schäfer et al., 2000). Additional evidence indicates a role for complement in ischemic stroke-induced brain injury. A study by Huang and colleagues (1999) showed that ischemic neurons strongly express C1q, which may target them for complement-mediated attack or alternatively, for the receptor for the complement component C1q (C1qRp)-mediated clearance (Huang et al., 1999). Another recent study also showed that C1q protein begins to accumulate in the ischemic hemisphere between 3 and 6 hr post-ischemia (Mack et al., 2006). Confocal microscopy demonstrated co-localization of C1q protein with neuronal cell bodies as well as necrotic cellular debris (Mack et al., 2006). The analysis of anaphylatoxin C3a and C5a receptors following cerebral ischemia demonstrated significant increases in the expression of C3aR and C5aR mRNAs in the ischemic cortex (Van Beek et al., 2000). The expression of C3aR and C5aR reached a peak at 2 days after MCAO (4.3- and 3.4-fold increases, respectively, compared with non-operated control cortical samples). C3aR and C5aR staining were found constitutively on neurons and astrocytes. In ischemic tissues, the authors also observed that C3aR and C5aR were expressed de novo on endothelial cells of blood vessels, at 6 h and 2 days after MCAO, respectively. C3aR and C5aR immunostaining were increased in macrophage-like cells and reactive astrocytes 7 days post occlusion (Van Beek et al., 2000). The above findings were further confirmed by Barnum and colleagues using a similar model system for both mice and rats. They showed C5aR expression was dramatically elevated within 3 h after MCAO, whereas C3aR expression was reduced to 25% of control animals. By 24 h post-occlusion, expression of both receptors was higher than at any other time point examined. This increased expression at the later time points after occlusion is most likely the result of a massive infiltration of leukocytes expressing the anaphylatoxin receptors (Barnum et al., 2002). Another group evaluated the contribution of complement activation to cerebral hypoxic-ischemic (HI) injury in the neonatal rat. They found that there was an eight-fold increase in the activation fragment, inactivated C3b at 16 hr. With immunofluorescence assays and confocal microscopy, both C3 and C9 were localized to injured neurons at 16 and 24 hr post-HI and the authors suggested that their results indicated that complement activation contributes to HI injury in neonatal rat brain (Cowell et al., 2003). In support of this, focal deposition of MAC has been detected in the brain infarcted brain areas of stroke patients (Lindsberg et al., 1996). Further evidence for systemic complement activation following human acute ischaemic stroke was demonstrated by Pedersen and colleagues as they found that the MAC increased significantly in plasma at 72 h, peaked at 7 days, was still significantly increased at 12 days and thereafter returned to normal levels (Pedersen et al., 2004). It was also shown recently that complement component 9 (C9) is deposited on neurons in the CNS of newborn infants who developed moderate to severe hypoxic-ischemic encephalopathy (Schultz et al., 2005). The evidence described above shows that complement components are actively expressed in the brain and their upregulation, in the setting of ischemic stroke-induced brain injury, now appears well-established. Given this, investigators have moved forward in attempts to inhibit complement activation to further elucidate the role of complement in the pathophysiology of cerebral ischemia. The next section focuses on complement inhibition strategies and their effectiveness in animal models of stroke.
NEUROPROTECTION BY COMPLEMENT INHIBITION
The use of specific inhibitors to block complement activation or their mediators such as C5a, has been shown to prevent local tissue injury after I/R (Arumugam et al., 2002; Arumugam et al., 2003; Arumugam et al., 2004b; Woodruff et al., 2004). Following much of what has proved successful in other organ models, many of the same complement inhibition strategies have been investigated in the brain. In an attempt to determine if abrogation of the complement system could provide protection for the brain during I/R injury, Vasthare and colleagues (1998) pretreated rats with cobra venom factor (CVF) one day prior to temporary cerebral ischemia. The magnitude of reactive hyperemia was significantly greater in the complement-depleted animals and there was also better preservation of somatosensory evoked potentials (SSEPs) in the complement-depleted animals (Vasthare et al., 1998). These differences were not associated with changes in leukocyte infiltration, as evidenced by myeloperoxidase and leukotriene B4 activity. These data demonstrated that depleting the complement system can actually improve blood flow and clinical outcome following cerebral ischemia with reperfusion (Vasthare et al., 1998). One weakness of this study was that complement depletion using CVF may disrupt other cellular events, due to its mechanism of initial complement overactivation (Proctor et al., 2006), and the authors failed to investigate this effect. As indicated by D'Ambrosio and colleagues (2001), the effects of CVF on the coagulation system might account for improved cerebral blood flow (CBF) and significant return of SSEPs in the reperfusion period (D'Ambrosio et al., 2001). Another group also used complement depletion via CVF to demonstrate a significantly reduced post-ischemic cerebral infarct volume and atrophy in adult and neonatal rats (Figueroa et al., 2005). This study found that CVF-induced decomplementation significantly reduced post-ischemic cerebral infarct volume in adult rats and post-hypoxic-ischemic cerebral atrophy in neonates (Figueroa et al., 2005). In contrast to the above two studies, a different study tested the effect of CVF-induced decomplementation in a rabbit model of thromboembolic stroke and found that infarct sizes in the complement-depleted rabbits were no different from the control group receiving no therapy (Lew et al., 1999). Similarly, no significant difference in regional cerebral blood flow or final intracranial pressure values were noted between any of the groups (Lew et al., 1999). The differences seen in these observations are most probably due to species selection and the differences in stroke models. In the study by Lew and colleagues (1999), only a modest return of CBF following thromboembolic stroke was seen. This may have minimized any potentially detrimental effect of leukocyte activation which known to play a negligible role in permanent ischemic stroke models, and as a result, of the complement cascade. Another study using intraperitoneal administration of the complement inhibitor soluble complement receptor type 1 (sCR1), or CVF-induced decomplementation in a model of HI in immature 21 day old rats, showed that inhibition of complement did not reduce the neuronal loss, edema, or atrophy (Lassiter et al., 2001). Lassiter and colleagues (2001) therefore suggested that complement activation did not contribute significantly to the cerebral injury observed in this immature rat model (Lassiter et al., 2001). These results are in contrast to results by Cowell and colleagues (2003), who have demonstrated a role for complement activation in the pathophysiology of unilateral forebrain HI injury neonatal (7 day old) rats (Cowell et al., 2003). This study found that CVF-administration depleted both systemic and brain C3 by the time of surgery and reduced infarct size. Analysis of lesioned, CVF-decomplemented animals demonstrated minimal neuronal C3 deposition but no reduction in C9 deposition localized to injured neurons. C3-immunoreactive microglia were particularly identified in injured areas. These results indicated that complement activation contributes to HI injury in neonatal rat brain, although systemic administration of CVF did not eliminate complement deposition within the injured brain (Cowell et al., 2003). The previous studies by Lassiter and colleagues (2001) failed to analyse complement proteins in 21 days old rats following CVF administration and the potential for complement factors to remain in the CNS following CVF-decomplementation may explain the differences seen between these two studies in neonatal rats. It is likely that CVF administration predominantly depletes systemic complement, without affecting cellular formation of complement factors within cells in the CNS. As such any conclusions on the role of complement factors in stroke obtained from these combined studies using CVF-induced decomplementation must be viewed with caution.
Subsequent studies using a more specific method to achieve complement inhibition, sCR1, demonstrated only modest reduction of cerebral infarct volume (Huang et al., 1999). This study demonstrated that sCR1 administration prior to I/R injury was moderately cerebroprotective in the setting of reperfusion stroke as demonstrated by a significant reduction in neurologic deficit, polymorphonuclear leukocytes (PMN) accumulation, and platelet accumulation. Trends towards smaller infarct volumes and improved CBF in sCR1-treated animals were observed (Huang et al., 1999). Additional benefit was conferred by sialyl Lewis x glycosylation of the unmodified parent sCR1 molecule (sCR1sLex) which was also able to inhibit selectin-mediated adhesion (Huang et al., 1999). However, conflicting data arose when Imm and colleagues (2002) found administration of a molecular hybrid of sCR1 the sCR1sLex did not affect the cerebral infarct volume compared to controls in 7 days old neonatal rats (Imm et al., 2002). Given the robust neuroprotection observed using this anti-complement strategy (Huang et al., 1999), the same group sought to translate sCR1 and sCR1sLex to the clinical arena for stroke. The first phase of this translation involved a double-blind, randomized, placebo-controlled trial of pre-ischemic sCR1 administration using well-characterized triple-vessel model of non-human primate reperfused stroke. The animals were subjected to 75 minutes of MCAO. Total serum complement activity was significantly depressed in the sCR1-treated animals compared with the controls. Immunostaining also demonstrated sCR1 deposition in the ischemic hemispheres of treated animals. This trial was halted following a planned interim analysis which suggested that administration of sCR1 could not achieve the expected efficacy using a rather limited number of animals (Mocco et al., 2006b). Given the more profound efficacy of the sCR1sLex protein in their rodent model, the same group evaluated this agent in a primate stroke model (Ducruet et al., 2007). In a preliminary cohort, infarct volume was greater in the drug-treated animals. As seen with sCR1 treatment, no difference in neurological score was found between groups. A hypotensive response was also observed in animals treated with sCR1sLex (Ducruet et al., 2007). Apart from the differences in age and animal species involved with the above studies using sCR1 and sCR1sLex, other differences such as models of ischemic stroke may explain the differences in results.
C1-inhibitor (C1-INH) is a major regulator of the complement classical pathway. Neuroprotection by C1-INH in brain ischemia has been reported (Heimann et al., 1999; Akita et al., 2003; De Simoni et al., 2003; De Simoni et al., 2004; Storini et al., 2005). The first study indicating the effect of C1-INH in a stroke model using a photochemical cortical vein occlusion model showed that administration of C1-INH had a significant protective effect against infarct development (Heimann et al., 1999). However, another study using same stroke model found that BSF468248, a novel potent C1-INH, was not effective in improving the CBF and the infarct volume following focal cerebral ischemia (Kaido et al., 2003). To investigate the mechanism of C1-INH action, De Simoni and colleagues (2003) evaluated neurodegeneration- and inflammation-related factors in mice subjected to cerebral I/R. C1-INH induced a dose-dependent reduction of ischemic volume and focal neurological deficit scores. Fluoro-Jade staining, a marker for neuronal degeneration showed that C1-INH-treated mice had a lower number of degenerating cells. Leukocyte infiltration, as assessed by CD45 immunostaining, was also markedly decreased. The authors then investigated the response to ischemia in C1q−/− mice. There was no decrease in infarct volume in C1q−/− mice compared to wild types (De Simoni et al., 2003). This observation in C1q−/− mice was recently confirmed by Mocco and colleagues who demonstrated that deletion of C1q reduces cerebral C5 immunopositivity; however, both C3 and C5 were detected in the infarcts of C1q-deficient mice (Mocco et al., 2006a). These results with adult knockout mice suggest that direct C1q-mediated mechanisms do not contribute especially to neurological injury following cerebral I/R. CR1, however, binds not only C1q but also C3b and C4b, facilitating factor I-mediated degradation of these proteins (Krych-Goldberg et al., 2001). Therefore, the modest cerebral protection afforded by sCR1 in adult mice (Mocco et al., 2006a; Mocco et al., 2006b) presumably occurs via non-C1q-mediated mechanisms. Furthermore, C1-INH significantly dampened the mRNA expression of the adhesion molecules P-selectin and ICAM-1 induced by the ischemic insult (De Simoni et al., 2003). C1-INH also significantly decreased certain pro-inflammatory cytokines (eg., TNF-α, IL-18) and increased certain protective cytokines (eg., IL-6, IL-10) gene expression. C1-INH treatment prevented the decrease of neurofilament (NFH) gene, a marker of cellular integrity, and also counteracted the increase of pro-caspase 3, an apoptosis index. In addition, C1-INH markedly inhibited the activation and/or recruitment of microglia/macrophages, as shown by immunohistochemistry (Storini et al., 2005). This study showed that C1-INH has a strong neuroprotective effect on brain I/R injury and that its action is may be independent from C1q-mediated activation of classical pathway.
Recent studies by Mocco and colleagues subjected mice, genetically deficient in selected complement components (C1q, C3, C5), to focal cerebral I/R injury. Of the strains investigated, only C3−/− mice were protected to any degree, as demonstrated by significant reductions in both infarct volume and neurological deficit score. C3−/− mice also manifested decreased granulocyte infiltration and reduced oxidative stress (Mocco et al., 2006a). C3-deficient mice also manifested decreased granulocyte infiltration and reduced oxidative stress with insult. The administration of a C3a-receptor antagonist SB290157 (Ames et al., 2001), which has been shown to specifically block C3a-mediated effects in rodent disease models of I/R injury (Proctor et al., 2004), resulted in commensurate neurological improvement and stroke volume reduction (Mocco et al., 2006a). It remains possible that the protection conferred by genetic C3 deletion was attributable to unknown effects of this manipulation; however, because reconstitution of C3−/− animals with C3 protein results in outcome similar to wild type mice, and C3a-receptor antagonist treated animals demonstrate significant neuroprotection, this is unlikely. An alternative interpretation may be that C3 deletion confers protection by preventing downstream C5 cleavage, forming the potent C5a anaphylatoxin the MAC (Mocco et al., 2006a). However, Ward and colleagues showed that C3 deletion promoting the formation of C5 convertase from the clotting system (Huber-Lang et al., 2006). To evaluate the role of C5 in stroke, we subjected C5 deficient mice (C5−/−) to I/R brain injury and found that they exhibited improved functional outcome and less brain damage compared with their wild-type littermates (Arumugam et al., 2007). We also found that C5−/− mice exhibited a reduced level of leukocytes recruitment in the infarcted area compared to wild-type controls (unpublished data). Although treatment of mice with a cyclic peptidic low molecular weight C5aR antagonist (Finch et al., 1999) to specifically block C5a-receptor interaction, did not result in a statistically significant improvement of the stroke outcome, there was seemingly a trend toward protection (Arumugam et al., 2007). Compared to our study, Mocco and colleagues found that neurons in C5−/− mice were not protected (Mocco et al., 2006a), and these differences in results were surprising given the same model and mice were used by us and this other group. Altogether, the available data suggest that complement activation is involved in the degeneration of neurons in models of cerebral I/R injury, but the degree is still of an uncertain nature, for a variety of reasons.
It should be noted that the recombinant complement inhibitors used in the vast majority of studies are very large molecules in terms of drug size, and distribution across the blood brain barrier (even under conditions of compromised function and intravenous administration with the attendant high blood levels of drug), may limit the therapeutic efficacy of these agents. Furthermore, if the critical complement components influencing CNS neuronal health are produced intracellularly in the CNS, and released in close proximity to target cells, then it may not be possible for such agents to be therapeutically effective, even if complement is a major pathogenic factor in the CNS. Pharmacokinetic considerations of a medicinal therapy in a privileged anatomical site, such as the CNS, may well trump pharmacodynamic aspects. These issues are rarely, if ever discussed, and the experimental literature does not take these factors into account in their specific reports; but in therapeutic medicine, these issues are both relevant and paramount.
INTRAVENOUS IMMUNOGLOBULIN
Immunoglobulin (Ig) has two major functional domains which are also structurally distinct. The Fab' domain is the amino-terminal region and contains the structural elements which recognize antigen. The Fab' domain contains elements of both the light and the heavy chains (Ibáñez and Montoro-Ronsano, 2003). The remainder of the Ig molecule is the hinge region and carboxy-terminal Fc domain which contains the constant domains. The Fc domain is responsible for the pharmacokinetics of antibody molecules and is critical for immunomodulatory function (Amzel and Poljak, 1979; Arumugam et al., 2008). IgG antibodies are the primary mediators of protective humoral immunity against pathogens, but they can also be pathogenic. Acting as immune complexes or as cytotoxic molecules, IgG autoantibodies are the principal mediators of autoimmune diseases such as immune thrombocytopenia, autoimmune hemolytic anemia, and systemic lupus erythematosus, and may contribute to other autoimmune diseases including rheumatoid arthritis, type I diabetes and multiple sclerosis (Bayary et al., 2006; Mix et al., 2006). Immunotherapy as a treatment method appeared soon after the discovery of adaptive immune responses and includes all methods that attempt to overcome abnormal immune responses by inducing clonal deletion, anergy, immune tolerance or immune deviation (Peakman and Dayan, 2001; Verhagen et al., 2006).
Immunoglobulin antibodies have been used therapeutically for over a century. They were first used as antitoxins in the pre-antibiotic era for the treatment of infectious diseases (Eibl and Wedgwood, 1989). IVIG is a purified polyclonal preparation of human immunoglobulin obtained from human plasma and first licensed for the treatment of immunocompromised individuals such as those with primary immune deficiency (Knapp and Colburn, 1990). The donor pool size is typically around several thousand individuals, which provides a sufficiently large sample number to ensure homogenic consistency but functional heterogeneity (Nimmerjahn and Ravetch, 2008a). IVIG is used to treat various inflammatory and autoimmune diseases, and has been approved by the FDA for the treatment of immune thrombocytopenia, Kawasaki's disease, humoral immunodeficiency and bone marrow transplantation (Gold et al., 2007; Nimmerjahn and Ravetch, 2008a). Off-label uses of IVIG include the treatment of rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis and scleroderma (Darabi et al., 2006; Kumar et al., 2006). Commercial formulations of IVIG contain mostly polyspecific IgG, low amounts of IgA, and traces of IgM (Lemieux et al., 2005). There has been an increase in the use of IVIG for immunomodulation in the last two decades. IVIG has also become established therapy for several neurological conditions including Guillain-Barre syndrome, chronic inflammatory demyelinating polyneuropathy and multifocal motor neuropathy (Misra et al., 2005).
MECHANISMS OF ACTIONS OF INTRAVENOUS IMMUNOGLOBULIN
The mechanisms by which IVIG exerts a therapeutic effect in numerous diseases with diverse causes are not well understood, but likely involve multiple immunomodulatory processes. Many groups have proposed immunoregulatory mechanisms of action for IVIG. Whilst some of these immunoregulatory mechanisms have been demonstrated, many others are still an enigma, or at least not completely explored. Some of these mechanisms include Fc-receptor blockade, neutralization of pathogenic autoantibodies via idiotypic and anti-idiotypic antibodies, effects on the Fas apoptotic pathway via agonistic and antagonistic anti-Fas autoantibodies, regulation of complement components, modulation of cytokine secretion, hindrance of natural-killer cell activity, inhibition of matrix metalloproteinase-9, suppression of nuclear factor kappa B (NFκB) activation and IκB degradation, G1 cell cycle arrest, prevention of tumor growth, decrease in leukocyte recruitment, attenuation of T-cell stimulation, effects on antibody kinetics and also effects on dendritic cells (Sapir and Shoenfeld, 2005). Here we analyse some of the demonstrated mechanisms of action of IVIG in inflammatory conditions.
Efficacy with IVIG therapy is thought to depend on two general types of mechanisms. The first is antigen binding and modification of various effector functions which is mediated by the antigen binding fragment (Fab) of IVIG (Latremouille et al., 1997; Basta et al., 2003; Ibáñez and Montoro-Ronsano, 2003). The second involves IVIG binding to the various crystallizable fragment (Fc) receptors resulting in modulation of expression and function of Fc receptors, complement activation, and anti-inflammatory effects resulting from interference with proinflammatory cytokines, provision of anti-idiotypic antibodies and modulation of T- and B-cell activation (Anthony et al., 2008; Nimmerjahn and Ravetch, 2008a; Nimmerjahn and Ravetch, 2008b). IVIG also contains natural anti-Fas receptor autoantibodies that block Fas ligand/Fas receptor interactions and prevent apoptosis (Sapir and Shoenfeld, 2005; Janke et al., 2006). In contrast, IVIG has been demonstrated to induce apoptosis in leukemic lymphocytes and monocytes as well as normal tonsillar B cells, an effect mediated at least in part by anti-CD95 antibodies present within the IVIG preparations (Prasad et al., 1998). This latter effect may support anti-inflammatory effects of IVIG, at least in part, by promoting apoptosis of activated leukocytes.
Modulation of the production of proinflammatory cytokines is another major mechanism by which IVIG exerts its anti-inflammatory effects in vivo in various neuromuscular disorders. The anti-inflammatory effects of IVIG involving modulation of cytokine production are not restricted to monocytic cytokines, but are also reliant on the ability of IVIG to increase T helper (Th)1 and Th2 cytokine gene expression and production (Sewell et al., 1999; Zhu et al., 2007). IVIG contains high-affinity neutralizing antibodies against IL-1, IL-6 and tumor necrosis factor-α (TNF-α) in quantities sufficient to suppress circulating proinflammatory pathogenic cytokines or downregulate the synthesis of cytokines by the T cells (Toungouz et al., 1995). Modulation of cytokines and cytokine antagonists by IVIG is another major mechanism by which immunoglobulin exerts its anti-inflammatory effects. IVIG was shown to selectively trigger the production of IL-1 receptor antagonist (IL-1ra), the natural antagonist of IL-1 (Craciun et al., 2007; Crow et al., 2007). There is also evidence to indicate that IVIG causes down-regulation of endothelial cell intercellular adhesion molecules ICAM-I, and lymphocyte function-associated antigen-1 (LFA-1) on activated T-cells (Rigal et al., 1994). Using a simple system of observing leukocytes under flow conditions on immobilized protein (P-selectin or E-selectin), Gill and colleagues (2005) found that IVIG inhibited leukocyte rolling (Gill et al., 2005). From their study it also appears that IVIG targets the P-selectin/P-selectin ligand interaction more than the β2- integrin/ICAM-1 interaction. Using an in vitro flow chamber assay, the same group also demonstrated that IVIG disrupts α4-integrin-dependent leukocyte–VCAM-1 interactions (Lapointe et al., 2004).
The binding of i.v. infused IgG molecules to Fc gamma receptors (FcγR) on the surface of phagocytic cells that invade the target tissues of patients with various autoimmune neurologic diseases can prevent FcγR-mediated phagocytosis of antigen-bearing target cells and/or inhibit antibody-dependent cell-mediated cytotoxicity by saturating or altering the affinity of the FcγR (Boros et al., 2005). IgG molecules bind via their Fc region to FcγR on macrophages, neutrophils, eosinophils, platelets, mast cells, natural killer cells, and B cells. FcγR are immunoglobulin superfamily members and are of 3 main types - FcγRI (CD64), FcγRII (CD32) and FcγRIII (CD16). FcγR are of the activating type, and receptor engagement leads to a variety of functions depending on the type of effector cell, and these functions include phagocytosis, degranulation, antibody-dependent cell-mediated cytotoxicity, cytokine release, and regulation of antibody production. It is also important to note that the two-step model proposed by Ravetch (Nimmerjahn and Ravetch, 2007) established a framework for the cellular basis of a sustained anti-inflammatory IVIG effect. IVIG indirectly upregulated the expression of inhibitory FcγRIIB on proinflammatory macrophages, which would be expected to oppose activating FcγR signaling, thereby suppressing the antibody-triggered inflammatory response (Nimmerjahn and Ravetch, 2007). The therapeutic efficacy of IVIG in some disorders, such as idiopathic thrombocytopenic purpura (ITP) and other autoantibody-induced cytopenias, is believed to be mediated by the blockade of the Fc receptor on macrophages, which prevents the removal of sensitized platelets by the reticulo-endothelial system (Park-Min et al., 2007; Siragam et al., 2006). Finally, it has been shown that interaction of IVIG with the complement system prevents the generation of C5b-9 (MAC) and subsequent complement-mediated tissue damage, and by scavenging active complement components and diverting complement attack from cellular targets (Basta and Dalakas et al., 1994). IVIG binds the activated components C3b and C4b in both a C1q-independent and a C1q-dependent manner, thus preventing the deposition of these fragments on target cell surfaces (Vivanco et al., 1999; Basta et al., 2003; Lutz et al., 2004). In the next section of this review article we will discuss the neuroprotective effect of IVIG in stroke.
NEUROPROTECTION BY IMMUNOGLOBULIN IN STROKE
IVIG has been used for several neurological disorders such as chronic inflammatory demyelinating polyneuropathy and Guillain-Barre syndrome (Hughes and Cornblath, 2005; Antel and Bar-Or, 2006; Sorensen, 2008). The effect of IVIG has also been tested in AD patients (Dodel et al., 2004; Relkin et al., 2008). Because healthy individuals have circulating auto-antibodies against Aβ-peptide, IVIG contains antibodies against Aβ-peptide as well. For this reason, Dodel and colleagues (2004) evaluated the effect of this therapeutic approach in patients with AD. Total levels of Aβ-peptide decreased in CSF in the five patients evaluated, whereas levels of the peptide in serum increased. In addition, stabilisation or even a mild improvement in cognitive function was observed in the patients (Dodel et al., 2004). The latter findings were confirmed by another human AD clinical study (Relkin et al., 2008). Relkin and colleagues (2008) carried out an open label dose-ranging study in 8 mild AD patients in which IVIg was added to approved AD therapies for 6 months, discontinued, and then resumed for another 9 months. Plasma Aβ levels increased transiently after each infusion. Cerebrospinal fluid Aβ decreased significantly at 6 months, returned to baseline after washout and decreased again after IVIG was re-administered for an additional 9 months. Mini-mental state scores increased an average of 2.5 points after 6 months, returned to baseline during washout and remained stable during subsequent IVIG treatment (Relkin et al., 2008). However, use of IVIG in other neurodegenerative conditions such as in PD has not yet been reported. Clearly, there seems potential for the exploratory use of IVIG in a variety of neurodegenerative diseases.
Because IVIG has the potential to inhibit multiple components of inflammation, including complement fragments, proinflammatory cytokine production and leukocyte cell adhesion, we tested the effect of IVIG in experimental models of ischemic stroke. Administration of IVIG to mice subjected to experimental stroke almost entirely eliminated mortality and reduced the size of brain infarction by 50-60 % (Arumugam et al., 2007). Moreover, not only was the infarcted area reduced, but also within this ischemic region, neurons were spared and only occasional cell loss was observed. This had a therapeutic impact on functional outcome, which was significantly improved over control animals. Because stroke survivors often suffer long-term disabilities, our findings imply that the neuron-sparing effect of IVIG could reduce the neurological consequences of stroke in humans, particularly when given as soon as possible after the stroke insult. The efficacy of IVIG against stroke-induced brain injury in our study was due, in part, to its ability to selectively bind complement components. Our findings, and those of other groups, showed that the levels of the C3b fragment, which is known to be crucial for continuation of the complement cascade and generation of cell-destroying terminal membrane complex, were increased at the site of injury following ischemic stroke (Stahel et al., 2001; De Simoni et al., 2003; De Simoni et al., 2004; Mocco et al., 2006a and Arumugam et al., 2007). We also evaluated C3 immunoreactivity in brain sections that included infarcted regions, and found that C3 levels associated with neuronal plasma membranes are increased in the ischemic tissue. IVIG treatment significantly reduced ischemic stroke-induced increases in the complement fragment C3b (Arumugam et al., 2007). In addition, immunoprecipitation analysis of brain samples from the ischemic hemisphere of IVIG-treated mice showed that human IgG binds mouse C3b, suggesting that IVIG targeting ischemic stroke induced complement activation.
IVIG may also provide therapeutic effects in stroke by reducing cell adhesion molecule production and subsequent infiltration of inflammatory cells, thus reducing inflammation in the infarcted region. There is considerable evidence that supports a role for inflammation in the pathogenesis of ischemic stroke. First, an increased expression of endothelial cell adhesion molecules such as ICAM-1, E-selectin and P-selectin leads to the recruitment of a large number of adherent leukocytes and platelets in cerebral venules followed by an accumulation of cytokines in postischemic brain tissue (Arumugam et al., 2005). It is interesting to note that each of these molecules are upregulated by the complement activation product, C5a (Ward refs). The possibility that inflammation contributes to ischemic brain injury, rather that being an epiphenomenon, is supported by a large number of reports that describe a reduction in stroke-induced infarct size and brain edema in animals treated with blocking antibodies against specific cell adhesion molecules that mediate leukocyte recruitment (Arumugam et al., 2004a; Yilmaz et al., 2006). Similar protection against ischemic brain injury has been demonstrated in mice that are genetically deficient in adhesion molecules such as ICAM-1, CD11b/CD18, or P-selectin (Arumugam et al., 2005). It is also well established that active complement fragments can induce expression of proinflammatory molecules that play a role in the inflammatory reaction following I/R injuries (Arumugam et al., 2004b; Ishikawa et al., 2004). Recent studies by Lapointe and colleagues demonstrated a direct positive impact of IVIG on α4-integrin-dependent leukocyte-endothelial cell interactions in EAE but failed to demonstrate any benefit in a mouse model of stroke. In fact, in their stroke model a far more severe thrombosis was observed with IVIG, providing some mechanistic insight for the few negative reports with respect to IVIG. However, the study by Lapointe and colleagues was limited to 2 hours of reperfusion where the effects of IVIG against stroke-induced leukocyte adhesion were limited since I/R-induced leukocyte adhesion becomes prominent in infarct development after 24 hours of reperfusion (Lapointe et al., 2004). In our studies, we found that IVIG pre- and post-treatment suppressed cerebral ischemia-induced increases in the levels of ICAM-1, CD11a and CD11b, markers of endothelial cell adhesion, lymphocyte infiltration and microglial activation, respectively, in the ischemic cortex adjacent to the brain infarction. This reduction in adhesion molecules following cerebral-ischemia induction is a further mechanism by which IVIG treatment is able to reduce neurological damage following stroke. We have also found that in the ipsilateral (injured) ischemic cortex that human IgG (from the IVIG treatment) is present not only in cerebral vessels, but also in the brain parenchyma as the result of disruption of the blood-brain barrier (Arumugam et al., 2007). The parenchymal human IgG was also associated with activated microglia. This observation, along with reduced level of CD11b resulting from IVIG treatment suggests that suppression of microglial activation is one mechanism by which IVIG protects neurons against ischemic injury.
In additional studies we have provided evidence that IVIG can also directly protect neurons against ischemia-like conditions. We found that oxygen and glucose deprivation (OGD) in cultured neurons caused an increase of C3 levels as compared to control neurons exposed to normal culture conditions. Increased expression of C3 was associated with a parallel increase in levels of cleaved (ie., enzymatically active) caspase-3 (a marker of apoptosis) and a progressive decrease in neuronal viability (Arumugam et al., 2007), suggesting the involvement of C3 in apoptotic cell death. Treatment with IVIG suppressed the OGD-induced increases in C3 as well as activated caspase-3 levels. Taken together, these findings indicate that complement fragments generated under ischemic conditions most probably lead to either: direct neuronal necrosis via generation of terminal membrane attack complex (mediated by C3b/C5b), and/or apoptotic cell death as indicated by the rise in levels of activated caspase-3 that were linked to increased levels of neuronal C3. IVIG seems to have the ability to protect against injury or cell death by targeting both injury mechanisms. However, it has been reported that, when infused at high rate and optimal doses (that can reach up to 2 g/Kg of body weight), IVIG can substantially increase blood viscosity. In certain at-risk patients with high blood pressure and advanced atherosclerosis, occurrence of thrombotic events was associated with changes in viscosity after high-dose IVIG (Orbach et al., 2005). In our previous experiments, we used IVIG at a dose that is four times lower than the maximal dose used in humans, and found it was equally protective against the effects of I/R injury. This finding eliminates the concern of increased blood viscosity and possible adverse effects due to therapy itself.
SUMMARY
The complement system is a component of the immune response comprised of multiple cascades that play an integrated role in the initiation and regulation of the inflammatory response. The involvement of the complement system in promoting acute brain injury and/or neurodegeneration seems assured. With the current state of knowledge and understanding, the application and limitations of modern scientific techniques, there is little doubt that complement plays a deleterious role in brain health, especially following insult, injury, infection, stroke or chronic immune malfunction.
At present, given our parlous insight and therapies of dubious efficacy, but with our rapidly increasing understanding of brain injury and the consequent inflammatory processes that exacerbate the original insult, there seems a clear path forward. Inhibition of complement: this may be achieved with precise pharmacological tools which target specific various factors or processes in the complement cascade, and/or the use of multifactorial agents such as IVIG, which have a wide variety of activities against the complement system, and presumably against other parts of the inflammatory cascade. Given the now apparently clear involvement of the complement system in immunoinflammatory pathogenic processes throughout the body, including the CNS, and coupled with the current lack of anti-complement therapeutic agents, there is a pressing clinical requirement for this new class of therapies.
With regards to the CNS, it is not too radical to propose that a cocktail of agents, comprising either C5 inhibitors and/or C5aR antagonists, together with the administration of IVIG - administered optimally within the “golden hour” or shortly thereafter following ischemic brain insult - may offer the best therapeutic prospect for limiting the degree of brain injury and the magnified motor and cognitive decrements that can result. Pharmacokinetic considerations of delivering novel anti-complement agents to cellular or even intracellular sites in the CNS are unavoidable. There are few greater imperatives in modern neurological clinical medicine than restoring brain-injured patients to their former selves. The tools are here, or almost here, and the combined understanding of the multifaceted treatment issues are relatively straightforward; what is required is a formidable will of purpose amongst health professionals involved in this arena, to drive forward, to give real new hope with real new therapies, where previously there was little-to-none.
Figure 1.
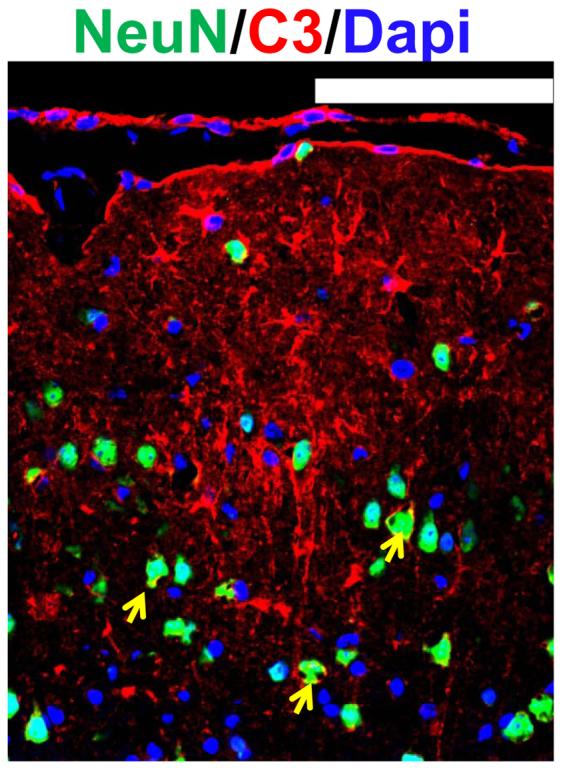
Up-regulation of C3 in neurons as well as other brain cells in infarcted brain regions following ischemic stroke. Staining and counterstaining with nuclear marker DAPI indicate colocalization of the neuronal nuclear marker NeuN (green fluorescence) and membranous type of C3 immunoreactivity (yellow arrows), whereas nonneuronal brain cell exhibit cytoplasmic type of C3 up-regulation.
Figure 2.

Schematic representation of ischemic stroke injury pathways that could be targeted by IVIG. LAM: Leukocyte adhesion molecules.
Table 1.
Effect of complement inhibition in stroke
CVF
|
C1 Inhibition
|
C3 Inhibition
|
C5 Inhibition
|
MAC Inhibition
|
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute on Aging, NIH, USA (Mark P Mattson) and a NH&MRC grant to Stephen M Taylor, The School of Biomedical Sciences, The University of Queensland, Australia.
Abbreviations
- 3-NP
3-nitroproprionic Acid
- Aβ
β-amyloid protein
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- BBB
blood brain barrier
- C1
complement 1
- C1-INH
C1-inhibitor
- C3
complement 3
- C3aR
C3a receptor
- C5
complement 5, C5aR, C5a receptor
- C5L2
C5a-like receptor 2
- CBF
cerebral blood flow
- CNS
central nervous system
- CSF
cerebrospinal fluid
- CVF
cobra venom factor
- DIND
delayed ischemic neurological deficits
- EAE
experimental autoimmune encephalomyelitis
- FcγR
Fc gamma receptors
- ICAM
intercellular adhesion molecules
- IL
interleukin
- I/R
ischemia-reperfusion
- ITP
idiopathic thrombocytopenic purpura
- IVIG
intravenous immunoglobulin
- HI
hypoxic-ischemic
- LFA-1
lymphocyte function-associated antigen-1
- MAC
membrane attack complex
- MCAO
middle cerebral artery occlusion
- MBL/MASP
mannose-binding lectin/ mannose-binding lectin-associated serine protease
- MS
multiple sclerosis
- NFH
neurofilament, NFκB, nuclear factor kappa B
- NO
nitric oxide
- OGD
oxygen and glucose deprivation
- PD
Parkinson's disease
- PMN
polymorphonuclear leukocytes
- ROS
reactive oxygen species
- SAH
subarachnoid hemorrhage
- sCR1
soluble complement receptor type 1
- sCR1sLex
Lewis x glycosylation of the sCR1 molecule
- SSEPs
somatosensory evoked potentials
- TNF-α
tumor necrosis factor-α
References
- Ames RS, Li Y, Sarau HM, Nuthulaganti P, Foley JJ, Ellis C, Zeng Z, Su K, Jurewicz AJ, Hertzberg RP, Bergsma DJ, Kumar C. Molecular cloning and characterization of the human anaphylatoxin C3a receptor. J Biol Chem. 1996;271:20231–20234. doi: 10.1074/jbc.271.34.20231. [DOI] [PubMed] [Google Scholar]
- Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science. 2008;320:373–376. doi: 10.1126/science.1154315. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita N, Nakase H, Kaido T, Kanemoto Y, Sakaki T. Protective effect of C1 esterase inhibitor on reperfusion injury in the rat middle cerebral artery occlusion model. Neurosurgery. 2003;52:395–400. doi: 10.1227/01.neu.0000043710.61233.b4. [DOI] [PubMed] [Google Scholar]
- Ames RS, Lee D, Foley JJ, Jurewicz AJ, Tornetta MA, Bautsch W, Settmacher B, Klos A, Erhard KF, Cousins RD, Sulpizio AC, Hieble JP, McCafferty G, Ward KW, Adams JL, Bondinell WE, Underwood DC, Osborn RR, Badger AM, Sarau HM. Identification of a selective nonpeptide antagonist of the anaphylatoxin C3a receptor that demonstrates antiinflammatory activity in animal models. J Immunol. 2001;166:6341–6348. doi: 10.4049/jimmunol.166.10.6341. [DOI] [PubMed] [Google Scholar]
- Amzel LM, Poljak RJ. Three-dimensional structure of immunoglobulins. Annu Rev Biochem. 1979;48:961–997. doi: 10.1146/annurev.bi.48.070179.004525. [DOI] [PubMed] [Google Scholar]
- Antel J, Bar-Or A. Roles of immunoglobulins and B cells in multiple sclerosis: from pathogenesis to treatment. J Neuroimmunol. 2006;180:3–8. doi: 10.1016/j.jneuroim.2006.06.032. [DOI] [PubMed] [Google Scholar]
- Annunziata P, Volpi N. High-levels of C3c in the cerebrospinal-fluid from amyotrophic lateral sclerosis patients. Acta Neurol Scand. 1985;72:61–64. doi: 10.1111/j.1600-0404.1985.tb01548.x. [DOI] [PubMed] [Google Scholar]
- Archelos JJ, Fazekas F. IVIG therapy in neurological disorders of childhood. J Neurol. 2006;253(Suppl 5):V80–86. doi: 10.1007/s00415-006-5014-y. [DOI] [PubMed] [Google Scholar]
- Arumugam TV, Granger DN, Mattson MP. Stroke and T-cells. Neuromolecular Med. 2005;7:229–242. doi: 10.1385/NMM:7:3:229. [DOI] [PubMed] [Google Scholar]
- Arumugam TV, Magnus T, Woodruff TM, Proctor LM, Shiels IA, Taylor SM. Complement mediators in ischemia-reperfusion injury. Clin Chim Acta. 2006;374:33–45. doi: 10.1016/j.cca.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Arumugam TV, Salter JW, Chidlow JH, Ballantyne CM, Kevil CG, Granger DN. Contributions of LFA-1 and Mac-1 to brain injury and microvascular dysfunction induced by transient middle cerebral artery occlusion. Am J Physiol Heart Circ Physiol. 2004a;287:H2555–2560. doi: 10.1152/ajpheart.00588.2004. [DOI] [PubMed] [Google Scholar]
- Arumugam TV, Selvaraj PK, Woodruff TM, Mattson MP. Targeting ischemic brain injury with intravenous immunoglobulin. Expert Opin Ther Targets. 2008;12:19–29. doi: 10.1517/14728222.12.1.19. [DOI] [PubMed] [Google Scholar]
- Arumugam TV, Shiels IA, Strachan AJ, Abbenante G, Fairlie DP, Taylor SM. A small molecule C5a receptor antagonist protects kidneys from ischemia/reperfusion injury in rats. Kidney Int. 2003;63:134–142. doi: 10.1046/j.1523-1755.2003.00737.x. [DOI] [PubMed] [Google Scholar]
- Arumugam TV, Shiels IA, Woodruff TM, Granger DN, Taylor SM. The role of the complement system in ischemia-reperfusion injury. Shock. 2004b;21:401–409. doi: 10.1097/00024382-200405000-00002. [DOI] [PubMed] [Google Scholar]
- Arumugam TV, Shiels IA, Woodruff TM, Reid RC, Fairlie DP, Taylor SM. Protective effect of a new C5a receptor antagonist against ischemia-reperfusion injury in the rat small intestine. J Surg Res. 2002;103:260–267. doi: 10.1006/jsre.2002.6369. [DOI] [PubMed] [Google Scholar]
- Arumugam TV, Tang SC, Lathia JD, Cheng A, Mughal MR, Chigurupati S, Magnus T, Chan SL, Jo DG, Ouyang X, Fairlie DP, Granger DN, Vortmeyer A, Basta M, Mattson MP. Intravenous immunoglobulin (IVIG) protects the brain against experimental stroke by preventing complement-mediated neuronal cell death. Proc Natl Acad Sci U S A. 2007;104:14104–14109. doi: 10.1073/pnas.0700506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam TV, Woodruff TM, Stocks SZ, Proctor LM, Pollitt S, Shiels IA, Reid RC, Fairlie DP, Taylor SM. Protective effect of a human C5a receptor antagonist against hepatic ischaemia-reperfusion injury in rats. J Hepatol. 2004c;40:934–941. doi: 10.1016/j.jhep.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Atkinson C, Zhu H, Qiao F, Varela JC, Yu J, Song H, Kindy MS, Tomlinson S. Complement-dependent P-selectin expression and injury following ischemic stroke. J Immunol. 2006;177:7266–7274. doi: 10.4049/jimmunol.177.10.7266. [DOI] [PubMed] [Google Scholar]
- Barnum SR, Ames RS, Maycox PR, Hadingham SJ, Meakin J, Harrison D, Parsons AA. Expression of the complement C3a and C5a receptors after permanent focal ischemia: An alternative interpretation. Glia. 2002;38:169–173. doi: 10.1002/glia.10069. [DOI] [PubMed] [Google Scholar]
- Barnum SR. Complement in central nervous system inflammation. Immunol Res. 2002;26:7–13. doi: 10.1385/IR:26:1-3:007. [DOI] [PubMed] [Google Scholar]
- Basta M, Dalakas MC. High-dose intravenous immunoglobulin exerts its beneficial effect in patients with dermatomyositis by blocking endomysial deposition of activated complement fragments. J Clin Invest. 1994;94:1729–1735. doi: 10.1172/JCI117520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basta M, Van Goor F, Luccioli S, Billings EM, Vortmeyer AO, Baranyi L, Szebeni J, Alving CR, Carroll MC, Berkower I, Stojilkovic SS, Metcalfe DD. F(ab)′2-mediated neutralization of C3a and C5a anaphylatoxins: a novel effector function of immunoglobulins. Nat Med. 2003;9:431–438. doi: 10.1038/nm836. [DOI] [PubMed] [Google Scholar]
- Bayry J, Lacroix-Desmazes S, Kazatchkine MD, Kaveri SV. Monoclonal antibody and intravenous immunoglobulin therapy for rheumatic diseases: rationale and mechanisms of action. Nat Clin Pract Rheumatol. 2007;3:262–272. doi: 10.1038/ncprheum0481. [DOI] [PubMed] [Google Scholar]
- Bayary J, Dasgupta S, Misra N, Ephrem A, Van Huyen JP, Delignat S, Hassan G, Caligiuri G, Nicoletti A, Lacroix-Desmazes S, Kazatchkine MD, Kaveri S. Intravenous immunoglobulin in autoimmune disorders: an insight into the immunoregulatory mechanisms. Int Immunopharmacol. 2006;6:528–534. doi: 10.1016/j.intimp.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Bellander BM, Singhrao SK, Ohlsson M, Mattsson P, Svensson M. Complement activation in the human brain after traumatic head injury. J Neurotrauma. 2001;18:1295–1311. doi: 10.1089/08977150152725605. [DOI] [PubMed] [Google Scholar]
- Berliocchi L, Bano D, Nicotera P. Ca2+ signals and death programmes in neurons. Philos Trans R Soc Lond B Biol Sci. 2005;360:2255–2258. doi: 10.1098/rstb.2005.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos L, Szalai AJ, Barnum SR. C3a expressed in the central nervous system protects against LPS-induced shock. Neurosci Lett. 2005;387:68–71. doi: 10.1016/j.neulet.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Boros P, Gondolesi G, Bromberg JS. High dose intravenous immunoglobulin treatment: mechanisms of action. Liver Transpl. 2005;11:1469–1480. doi: 10.1002/lt.20594. [DOI] [PubMed] [Google Scholar]
- Bradt BM, Kolb WP, Cooper NR. Complement-dependent proinflammatory properties of the Alzheimer's disease beta-peptide. J Exp Med. 1998;188:431–438. doi: 10.1084/jem.188.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia M, Vanhoutte P, Pages C, Besson MJ, Brouillet E, Caboche J. The mitochondrial toxin 3-nitropropionic acid induces striatal neurodegeneration via a c-Jun N-terminal kinase/c-Jun module. J Neurosci. 2002;22:2174–2184. doi: 10.1523/JNEUROSCI.22-06-02174.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CK, Chang CP, Liu SY, Lin MT. Oxidative stress and ischemic injuries in heat stroke. Prog Brain Res. 2007;162:525–546. doi: 10.1016/S0079-6123(06)62025-6. [DOI] [PubMed] [Google Scholar]
- Costa C, Zhao L, Shen Y, Su X, Hao L, Colgan SP, Stahl GL, Zhou T, Wang Y. Role of complement component C5 in cerebral ischemia/reperfusion injury. Brain Res. 2006;1100:142–151. doi: 10.1016/j.brainres.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Cowell RM, Plane JM, Silverstein FS. Complement activation contributes to hypoxic-ischemic brain injury in neonatal rats. J Neurosci. 2003;23:9459–9468. doi: 10.1523/JNEUROSCI.23-28-09459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craciun LI, DiGiambattista M, Laub R, Goldman M, Dupont E. Apoptosis: a target for potentiation of UV-induced IL-1Ra synthesis by IVIg. Immunol Lett. 2007;110:36–41. doi: 10.1016/j.imlet.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Crow AR, Song S, Semple JW, Freedman J, Lazarus AH. A role for IL-1 receptor antagonist or other cytokines in the acute therapeutic effects of IVIg? Blood. 2007;109:155–158. doi: 10.1182/blood-2006-05-023796. [DOI] [PubMed] [Google Scholar]
- D'Ambrosio AL, Pinsky DJ, Connolly ES. The role of the complement cascade in ischemia/reperfusion injury: implications for neuroprotection. Mol Med. 2001;7:367–382. [PMC free article] [PubMed] [Google Scholar]
- Darabi K, Abdel-Wahab O, Dzik WH. Current usage of intravenous immune globulin and the rationale behind it: the Massachusetts General Hospital data and a review of the literature. Transfusion. 2006;46:741–753. doi: 10.1111/j.1537-2995.2006.00792.x. [DOI] [PubMed] [Google Scholar]
- Davoust N, Jones J, Stahel PF, Ames RS, Barnum SR. Receptor for the C3a anaphylatoxin is expressed by neurons and glial cells. Glia. 1999;26:201–211. doi: 10.1002/(sici)1098-1136(199905)26:3<201::aid-glia2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Degn SE, Thiel S, Jensenius JC. New perspectives on mannan-binding lectin-mediated complement activation. Immunobiology. 2007;212:301–311. doi: 10.1016/j.imbio.2006.12.004. [DOI] [PubMed] [Google Scholar]
- De Simoni MG, Storini C, Barba M, Catapano L, Arabia AM, Rossi E, Bergamaschini L. Neuroprotection by complement (C1) inhibitor in mouse transient brain ischemia. J Cereb Blood Flow Metab. 2003;23:232–239. doi: 10.1097/01.WCB.0000046146.31247.A1. [DOI] [PubMed] [Google Scholar]
- De Simoni MG, Rossi E, Storini C, Pizzimenti S, Echart C, Bergamaschini L. The powerful neuroprotective action of C1-inhibitor on brain ischemia-reperfusion injury does not require C1q. Am J Pathol. 2004;164:1857–1863. doi: 10.1016/S0002-9440(10)63744-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodel RC, Du Y, Depboylu C, Hampel H, Frölich L, Haag A, Hemmeter U, Paulsen S, Teipel SJ, Brettschneider S, Spottke A, Nölker C, Möller HJ, Wei X, Farlow M, Sommer N, Oertel WH. Intravenous immunoglobulins containing antibodies against beta-amyloid for the treatment of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2004;75:1472–1474. doi: 10.1136/jnnp.2003.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducruet AF, Mocco J, Mack WJ, Coon AL, Marsh HC, Pinsky DJ, Hickman ZL, Kim GH, Connolly ES., Jr Pre-clinical evaluation of an sLe x-glycosylated complement inhibitory protein in a non-human primate model of reperfused stroke. J Med Primatol. 2007;36:375–380. doi: 10.1111/j.1600-0684.2007.00213.x. [DOI] [PubMed] [Google Scholar]
- Eibl MM, Wedgwood RJ. Intravenous immunoglobulin: a review. Immunodefic Rev. 1989;1(Suppl):1–42. [PubMed] [Google Scholar]
- Fan R, DeFilippis K, Van Nostrand WE. Induction of complement proteins in a mouse model for cerebral microvascular A beta deposition. J Neuroinflammation. 2007;4:22. doi: 10.1186/1742-2094-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas I, Takahashi M, Fukuda A, Yamamoto N, Akatsu H, Baranyi L, Tateyama H, Yamamoto T, Okada N, Okada H. Complement C5a receptor-mediated signaling may be involved in neurodegeneration in Alzheimer's disease. J Immunol. 2003;170:5764–5771. doi: 10.4049/jimmunol.170.11.5764. [DOI] [PubMed] [Google Scholar]
- Figueroa E, Gordon LE, Feldhoff PW, Lassiter HA. The administration of cobra venom factor reduces post-ischemic cerebral injury in adult and neonatal rats. Neurosci Lett. 2005;380:48–53. doi: 10.1016/j.neulet.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Finch AM, Wong AK, Paczkowski NJ, Wadi SK, Craik DJ, Fairlie DP, Taylor SM. Low-molecular-weight peptidic and cyclic antagonists of the receptor for the complement factor C5a. J Med Chem. 1999;42:1965–1974. doi: 10.1021/jm9806594. [DOI] [PubMed] [Google Scholar]
- Fleming SD, Tsokos GC. Complement, natural antibodies, autoantibodies and tissue injury. Autoimmun Rev. 2006;5:89–92. doi: 10.1016/j.autrev.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer's disease. J Neurosci. 2004;24:6457–6465. doi: 10.1523/JNEUROSCI.0901-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasque P, Singhrao SK, Neal JW, Götze O, Morgan BP. Expression of the receptor for complement C5a (CD88) is up-regulated on reactive astrocytes, microglia, and endothelial cells in the inflamed human central nervous system. Am J Pathol. 1997;150:31–41. [PMC free article] [PubMed] [Google Scholar]
- Gasque P, Singhrao SK, Neal JW, Wang P, Sayah S, Fontaine M, Morgan BP. The receptor for complement anaphylatoxin C3a is expressed by myeloid cells and nonmyeloid cells in inflamed human central nervous system: analysis in multiple sclerosis and bacterial meningitis. J Immunol. 1998;160:3543–3554. [PubMed] [Google Scholar]
- Gavrilyuk V, Kalinin S, Hilbush BS, Middlecamp A, McGuire S, Pelligrino D, Weinberg G, Feinstein DL. Identification of complement 5a-like receptor (C5L2) from astrocytes: characterization of anti-inflammatory properties. J Neurochem. 2005;92:1140–1149. doi: 10.1111/j.1471-4159.2004.02942.x. [DOI] [PubMed] [Google Scholar]
- Gill V, Doig C, Knight D, Love E, Kubes P. Targeting adhesion molecules as a potential mechanism of action for intravenous immunoglobulin. Circulation. 2005;112:2031–2039. doi: 10.1161/CIRCULATIONAHA.105.546150. [DOI] [PubMed] [Google Scholar]
- Gold R, Stangel M, Dalakas MC. Drug Insight: the use of intravenous immunoglobulin in neurology--therapeutic considerations and practical issues. Nat Clin Pract Neurol. 2007;3:36–44. doi: 10.1038/ncpneuro0376. [DOI] [PubMed] [Google Scholar]
- Goldknopf IL, Sheta EA, Bryson J, Folsom B, Wilson C, Duty J, Yen AA, Appel SH. Complement C3c and related protein biomarkers in amyotrophic lateral sclerosis and Parkinson's disease. Biochem Biophys Res Commun. 2006;342:1034–1039. doi: 10.1016/j.bbrc.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Guo RF, Riedemann NC, Bernacki KD, Sarma VJ, Laudes IJ, Reuben JS, Younkin EM, Neff TA, Paulauskis JD, Zetoune FS, Ward PA. Neutrophil C5a receptor and the outcome in a rat model of sepsis. FASEB J. 2003;17:1889–1891. doi: 10.1096/fj.03-0009fje. [DOI] [PubMed] [Google Scholar]
- Guo RF, Ward PA. Mediators and regulation of neutrophil accumulation in inflammatory responses in lung: insights from the IgG immune complex model. Free Radic Biol Med. 2002;33:303–310. doi: 10.1016/s0891-5849(02)00823-7. [DOI] [PubMed] [Google Scholar]
- Haas PJ, van Strijp J. Anaphylatoxins: their role in bacterial infection and inflammation. Immunol Res. 2007;37:161–175. doi: 10.1007/BF02697367. [DOI] [PubMed] [Google Scholar]
- Haberman F, Tang SC, Arumugam TV, Hyun DH, Yu QS, Cutler RG, Guo Z, Holloway HW, Greig NH, Mattson MP. Soluble neuroprotective antioxidant uric acid analogs ameliorate ischemic brain injury in mice. Neuromolecular Med. 2007;9:315–323. doi: 10.1007/s12017-007-8010-1. [DOI] [PubMed] [Google Scholar]
- Hansch GM, Seitz M, Betz M. Effect of the late complement components C5b-9 on human monocytes: release of prostanoids, oxygen radicals and of a factor inducing cell proliferation. Int Arch Allergy Appl Immunol. 1987;82:317–320. doi: 10.1159/000234216. [DOI] [PubMed] [Google Scholar]
- Hansch GM, Seitz M, Martinotti G, Betz M, Rauterberg EW, Gemsa D. Macrophages release arachidonic acid, prostaglandin E2, and thromboxane in response to late complement components. J Immunol. 1984;133:2145–2150. [PubMed] [Google Scholar]
- Haynes DR, Harkin DG, Bignold LP, Hutchens MJ, Taylor SM, Fairlie DP. Inhibition of C5a-induced neutrophil chemotaxis and macrophage cytokine production in vitro by a new C5a receptor antagonist. Biochem Pharmacol. 2000;60:729–733. doi: 10.1016/s0006-2952(00)00361-0. [DOI] [PubMed] [Google Scholar]
- Heimann A, Takeshima T, Horstick G, Kempski O. C1-esterase inhibitor reduces infarct volume after cortical vein occlusion. Brain Res. 1999;838:210–213. doi: 10.1016/s0006-8993(99)01740-0. [DOI] [PubMed] [Google Scholar]
- Hetland G, del Zoppo GJ, Mori E, Thomas WS, Hugli TE. Uptake of C5a by polymorphonuclear leukocytes (PMNs) after focal cerebral ischemia. I. Effect of tirilazad mesylate intervention on C5a uptake by PMNs. Immunopharmacology. 1994;27:191–198. doi: 10.1016/0162-3109(94)90015-9. [DOI] [PubMed] [Google Scholar]
- Hill JH, Ward PA. The phlogistic role of C3 leukotactic fragments in myocardial infarcts of rats. J Exp Med. 1971;133:885–900. doi: 10.1084/jem.133.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Kim LJ, Mealey R, Marsh HC, Jr, Zhang Y, Tenner AJ, Connolly ES, Jr, Pinsky DJ. Neuronal protection in stroke by an sLex-glycosylated complement inhibitory protein. Science. 1999;285:595–599. doi: 10.1126/science.285.5427.595. [DOI] [PubMed] [Google Scholar]
- Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- Hughes RA, Cornblath DR. Guillain-Barre syndrome. Lancet. 2005;366:1653–1666. doi: 10.1016/S0140-6736(05)67665-9. [DOI] [PubMed] [Google Scholar]
- Hugli TE. The structural basis for anaphylatoxin and chemotactic functions of C3a, C4a, and C5a. Crit Rev Immunol. 1981;1:321–366. [PubMed] [Google Scholar]
- Ibáñez C, Montoro-Ronsano JB. Intravenous immunoglobulin preparations and autoimmune disorders: mechanisms of action. Curr Pharm Biotechnol. 2003;4:239–247. doi: 10.2174/1389201033489775. [DOI] [PubMed] [Google Scholar]
- Imm MD, Feldhoff PW, Feldhoff RC, Lassiter HA. The administration of complement component C9 augments post-ischemic cerebral infarction volume in neonatal rats. Neurosci Lett. 2002;325:175–178. doi: 10.1016/s0304-3940(02)00271-9. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Vowinkel T, Stokes KY, Arumugam TV, Yilmaz G, Nanda A, Granger DN. CD40/CD40 ligand signaling in mouse cerebral microvasculature after focal ischemia/reperfusion. Circulation. 2005;111:1690–1696. doi: 10.1161/01.CIR.0000160349.42665.0C. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Zhang JH, Nanda A, Granger DN. Inflammatory responses to ischemia and reperfusion in the cerebral microcirculation. Front Biosci. 2004;9:1339–1347. doi: 10.2741/1330. [DOI] [PubMed] [Google Scholar]
- Janke AD, Giuliani F, Yong VW. IVIg attenuates T cell-mediated killing of human neurons. J. Neuroimmunol. 2006;177:181–188. doi: 10.1016/j.jneuroim.2006.04.023. [DOI] [PubMed] [Google Scholar]
- Johnson SA, Pasinetti GM, Finch CE. Expression of complement C1qB and C4 mRNAs during rat brain development. Brain Res Dev Brain Res. 1994;80:163–174. doi: 10.1016/0165-3806(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Kaido T, Heimann A, Kempski O. Novel complement C1 inhibitor BSF468248 does not improve brain damage after cortical vein occlusion. Methods Find Exp Clin Pharmacol. 2003;25:611–616. doi: 10.1358/mf.2003.25.8.778081. [DOI] [PubMed] [Google Scholar]
- Kaczorowski SL, Schiding JK, Toth CA, Kochanek PM. Effect of soluble complement receptor-1 on neutrophil accumulation after traumatic brain injury in rats. J Cereb Blood Flow Metab. 1995;15:860–864. doi: 10.1038/jcbfm.1995.107. [DOI] [PubMed] [Google Scholar]
- Kasuya H, Shimizu T. Activated complement components C3a and C4a in cerebrospinal fluid and plasma following subarachnoid hemorrhage. J Neurosurg. 1989;71:741–746. doi: 10.3171/jns.1989.71.5.0741. [DOI] [PubMed] [Google Scholar]
- Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol. 1992;140:691–707. [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner, et al. Neuro-immune crosstalk in CNS diseases. Neuroscience. 2008 doi: 10.1016/j.neuroscience.2008.09.009. [DOI] [PubMed] [Google Scholar]
- Klehmet, et al. Stroke-induced immunodepression and post-stroke infections: Lessons from the PANTHERIS trial. Neuroscience. 2008 doi: 10.1016/j.neuroscience.2008.07.044. [DOI] [PubMed] [Google Scholar]
- Krych-Goldberg M, Atkinson JP. Structure-function relationships of complement receptor type 1. Immunol Rev. 2001;180:112–122. doi: 10.1034/j.1600-065x.2001.1800110.x. [DOI] [PubMed] [Google Scholar]
- Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7:9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]
- Knapp MJ, Colburn PA. Clinical uses of intravenous immune globulin. Clin Pharm. 1990;9:509–529. [PubMed] [Google Scholar]
- Kumar A, Teuber SS, Gershwin ME. Intravenous immunoglobulin: striving for appropriate use. Int Arch Allergy Immunol. 2006;140:185–198. doi: 10.1159/000093204. [DOI] [PubMed] [Google Scholar]
- Lacy M, Jones J, Whittemore SR, Haviland DL, Wetsel RA, Barnum SR. Expression of the receptors for the C5a anaphylatoxin, interleukin-8 and FMLP by human astrocytes and microglia. J Neuroimmunol. 1995;61:71–78. doi: 10.1016/0165-5728(95)00075-d. [DOI] [PubMed] [Google Scholar]
- Lassiter HA, Feldhoff RC, Dabhia N, Parker JC, Jr, Feldhoff PW. Complement inhibition does not reduce post-hypoxic-ischemic cerebral injury in 21-day-old rats. Neurosci Lett. 2001;302:37–40. doi: 10.1016/s0304-3940(01)01653-6. [DOI] [PubMed] [Google Scholar]
- Latremouille C, Genevaz D, Hu MC, Schussler O, Goussef N, Mandet C, Bruneval P, Haeffner-Cavaillon N, Carpentier A, Glotz D. Normal human immunoglobulins for intravenous use (IVIg) delay hyperacute xenograft rejection through F(ab′)2-mediated anti-complement activity. Clin Exp Immunol. 1997;110:122–126. doi: 10.1046/j.1365-2249.1997.4591358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laupland KB, Kirkpatrick AW, Delaney A. Polyclonal intravenous immunoglobulin for the treatment of severe sepsis and septic shock in critically ill adults: a systematic review and meta-analysis. Crit Care Med. 2007;35:2686–2692. [PubMed] [Google Scholar]
- Lee H, Whitfeld PL, Mackay CR. Receptors for complement C5a. The importance of C5aR and the enigmatic role of C5L2. Immunol Cell Biol. 2008;86:153–160. doi: 10.1038/sj.icb.7100166. [DOI] [PubMed] [Google Scholar]
- Leinhase I, Schmidt OI, Thurman JM, Hossini AM, Rozanski M, Taha ME, Scheffler A, John T, Smith WR, Holers VM, Stahel PF. Pharmacological complement inhibition at the C3 convertase level promotes neuronal survival, neuroprotective intracerebral gene expression, and neurological outcome after traumatic brain injury. Exp Neurol. 2006;199:454–464. doi: 10.1016/j.expneurol.2006.01.033. [DOI] [PubMed] [Google Scholar]
- Lemieux R, Bazin R, Neron S. Therapeutic intravenous immunoglobulins. Mol Immunol. 2005;42:839–848. doi: 10.1016/j.molimm.2004.07.046. [DOI] [PubMed] [Google Scholar]
- Lew SM, Gross CE, Bednar MM, Russell SJ, Fuller SP, Ellenberger CL, Howard D. Complement depletion does not reduce brain injury in a rabbit model of thromboembolic stroke. Brain Res Bull. 1999;48:325–331. doi: 10.1016/s0361-9230(99)00004-0. [DOI] [PubMed] [Google Scholar]
- Lindsberg PJ, Ohman J, Lehto T, Karjalainen-Lindsberg ML, Paetau A, Wuorimaa T, Carpén O, Kaste M, Meri S. Complement activation in the central nervous system following blood-brain barrier damage in man. Ann Neurol. 1996;40:587–596. doi: 10.1002/ana.410400408. [DOI] [PubMed] [Google Scholar]
- Loeffler DA, Camp DM, Conant SB. Complement activation in the Parkinson's disease substantia nigra: an immunocytochemical study. J Neuroinflammation. 2006;3:29. doi: 10.1186/1742-2094-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KP, Huang SH, De Silva R, Tan BK, Zhu YZ. Oxidative stress: apoptosis in neuronal injury. Curr Alzheimer Res. 2006;3:327–337. doi: 10.2174/156720506778249515. [DOI] [PubMed] [Google Scholar]
- Lapointe BM, Herx LM, Gill V, Metz LM, Kubes P. IVIg therapy in brain inflammation: etiology-dependent differential effects on leucocyte recruitment. Brain. 2004;127:2649–2656. doi: 10.1093/brain/awh297. [DOI] [PubMed] [Google Scholar]
- Lutz HU, Stammler P, Bianchi V, Trüeb RM, Hunziker T, Burger R, Jelezarova E, Späth PJ. Intravenously applied IgG stimulates complement attenuation in a complement-dependent autoimmune disease at the amplifying C3 convertase level. Blood. 2004;103:465–472. doi: 10.1182/blood-2003-05-1530. [DOI] [PubMed] [Google Scholar]
- Mack WJ, Sughrue ME, Ducruet AF, Mocco J, Sosunov SA, Hassid BG, Silverberg JZ, Ten VS, Pinsky DJ, Connolly ES., Jr Temporal pattern of C1q deposition after transient focal cerebral ischemia. J Neurosci Res. 2006;83:883–889. doi: 10.1002/jnr.20775. [DOI] [PubMed] [Google Scholar]
- March DR, Proctor LM, Stoermer MJ, Sbaglia R, Abbenante G, Reid RC, Woodruff TM, Wadi K, Paczkowski N, Tyndall JD, Taylor SM, Fairlie DP. Potent cyclic antagonists of the complement C5a receptor on human polymorphonuclear leukocytes. Relationships between structures and activity. Mol Pharmacol. 2004;65:868–879. doi: 10.1124/mol.65.4.868. [DOI] [PubMed] [Google Scholar]
- McColl, et al. Systemic infection, inflammation and acute ischaemic stroke. Neuroscience. 2008 doi: 10.1016/j.neuroscience.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Misra N, Bayry J, Ephrem A, Dasgupta S, Delignat S, Van Huyen JP, Prost F, Lacroix-Desmazes S, Nicoletti A, Kazatchkine MD, Kaveri SV. Intravenous immunoglobulin in neurological disorders: a mechanistic perspective. J Neurol. 2005;252:I1–6. doi: 10.1007/s00415-005-1102-7. [DOI] [PubMed] [Google Scholar]
- Mix E, Goertsches R, Zett UK. Immunoglobulins--basic considerations. J Neurol. 2006;253:V9–17. doi: 10.1007/s00415-006-5002-2. [DOI] [PubMed] [Google Scholar]
- Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007;54:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Mocco J, Mack WJ, Ducruet AF, Sosunov SA, Sughrue ME, Hassid BG, Nair MN, Laufer I, Komotar RJ, Claire M, Holland H, Pinsky DJ, Connolly ES., Jr Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circ Res. 2006a;99:209–217. doi: 10.1161/01.RES.0000232544.90675.42. [DOI] [PubMed] [Google Scholar]
- Mocco J, Mack WJ, Ducruet AF, King RG, Sughrue ME, Coon AL, Sosunov SA, Sciacca RR, Zhang Y, Marsh HC, Jr, Pinsky DJ, Connolly ES., Jr Preclinical evaluation of the neuroprotective effect of soluble complement receptor type 1 in a nonhuman primate model of reperfused stroke. J Neurosurg. 2006b;105:595–601. doi: 10.3171/jns.2006.105.4.595. [DOI] [PubMed] [Google Scholar]
- Morgan BP. Regulation of the complement membrane attack pathway. Crit Rev Immunol. 1999;19:173–198. [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. The antiinflammatory activity of IgG: the intravenous IgG paradox. J Exp Med. 2007;204:11–15. doi: 10.1084/jem.20061788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Anti-Inflammatory Actions of Intravenous Immunoglobulin. Annu Rev Immunol. 2008a;26:513–533. doi: 10.1146/annurev.immunol.26.021607.090232. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008b;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- Nishino H, Czurkó A, Fukuda A, Hashitani T, Hida H, Karadi Z, Lénárd L. Pathophysiological process after transient ischemia of the middle cerebral artery in the rat. Brain Res Bull. 1994;35:51–56. doi: 10.1016/0361-9230(94)90215-1. [DOI] [PubMed] [Google Scholar]
- O'Barr SA, Caguioa J, Gruol D, Perkins G, Ember JA, Hugli T, Cooper NR. Neuronal expression of a functional receptor for the C5a complement activation fragment. J Immunol. 2001;166:4154–4162. doi: 10.4049/jimmunol.166.6.4154. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Kitao Y, Hori O. Ischemia-induced neuronal cell death and stress response. Antioxid Redox Signal. 2007;9:573–587. doi: 10.1089/ars.2006.1516. [DOI] [PubMed] [Google Scholar]
- Okinaga S, Slattery D, Humbles A, Zsengeller Z, Morteau O, Kinrade MB, Brodbeck RM, Krause JE, Choe HR, Gerard NP, Gerard C. C5L2, a nonsignaling C5A binding protein. Biochemistry. 2003;42:9406–9415. doi: 10.1021/bi034489v. [DOI] [PubMed] [Google Scholar]
- Orbach H, Katz U, Sherer Y, Shoenfeld Y. Intravenous immunoglobulin: adverse effects and safe administration. Clin Rev Allergy Immunol. 2005;29:173–184. doi: 10.1385/CRIAI:29:3:173. [DOI] [PubMed] [Google Scholar]
- Park CC, Shin ML, Simard JM. The complement membrane attack complex and the bystander effect in cerebral vasospasm. J Neurosurg. 1997;87:294–300. doi: 10.3171/jns.1997.87.2.0294. [DOI] [PubMed] [Google Scholar]
- Park-Min KH, Serbina NV, Yang W, Ma X, Krystal G, Neel BG, Nutt SL, Hu X, Ivashkiv LB. FcgammaRIII-dependent inhibition of interferon-gamma responses mediates suppressive effects of intravenous immune globulin. Immunity. 2007;26:67–78. doi: 10.1016/j.immuni.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Peakman M, Dayan CM. Antigen-specific immunotherapy for autoimmune disease: fighting fire with fire? Immunology. 2001;104:361–366. doi: 10.1046/j.1365-2567.2001.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen ED, Frøyland E, Kvissel AK, Pharo AM, Skålhegg BS, Rootwelt T, Mollnes TE. Expression of complement regulators and receptors on human NT2-N neurons--effect of hypoxia and reoxygenation. Mol Immunol. 2007;44:2459–2468. doi: 10.1016/j.molimm.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Pedersen ED, Waje-Andreassen U, Vedeler CA, Aamodt G, Mollnes TE. Systemic complement activation following human acute ischaemic stroke. Clin Exp Immunol. 2004;137:117–122. doi: 10.1111/j.1365-2249.2004.02489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Bonsi P, Calabresi P. Calcium signaling and neuronal vulnerability to ischemia in the striatum. Cell Calcium. 2004;36:277–284. doi: 10.1016/j.ceca.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Prasad NK, Papoff G, Zeuner A, Bonnin E, Kazatchkine MD, Ruberti G, Kaveri SV. Therapeutic preparations of normal polyspecific IgG (IVIg) induce apoptosis in human lymphocytes and monocytes: a novel mechanism of action of IVIg involving the Fas apoptotic pathway. J Immunol. 1998;161:3781–3790. [PubMed] [Google Scholar]
- Proctor LM, Arumugam TV, Shiels I, Reid RC, Fairlie DP, Taylor SM. Comparative anti-inflammatory activities of antagonists to C3a and C5a receptors in a rat model of intestinal ischaemia/reperfusion injury. Br J Pharmacol. 2004;142:756–764. doi: 10.1038/sj.bjp.0705819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor LM, Strachan AJ, Woodruff TM, Mahadevan IB, Williams HM, Shiels IA, Taylor SM. Complement inhibitors selectively attenuate injury following administration of cobra venom factor to rats. Int Immunopharmacol. 2006;6:1224–1232. doi: 10.1016/j.intimp.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Rahpeymai Y, Hietala MA, Wilhelmsson U, Fotheringham A, Davies I, Nilsson AK, Zwirner J, Wetsel RA, Gerard C, Pekny M, Pekna M. Complement: a novel factor in basal and ischemia-induced neurogenesis. EMBO J. 2006;25:1364–1374. doi: 10.1038/sj.emboj.7601004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relkin NR, Szabo P, Adamiak B, Burgut T, Monthe C, Lent RW, Younkin S, Younkin L, Schiff R, Weksler ME. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2007.12.021. In Press. [DOI] [PubMed] [Google Scholar]
- Rigal D, Vermot-Desroches C, Heitz S, Bernaud J, Alfonsi F, Monier JC. Effects of intravenous immunoglobulins (IVIG) on peripheral blood B, NK, and T cell subpopulations in women with recurrent spontaneous abortions: specific effects on LFA-1 and CD56 molecules. Clin Immunol Immunopathol. 1994;71:309–314. doi: 10.1006/clin.1994.1091. [DOI] [PubMed] [Google Scholar]
- Ringel I, Zettl UK. Intravenous immunoglobulin therapy in neurological diseases during pregnancy. J Neurol. 2006;253:V70–74. doi: 10.1007/s00415-006-5012-0. [DOI] [PubMed] [Google Scholar]
- Roberts TJ. 3-nitropropionic acid model of metabolic stress: assessment by magnetic resonance imaging. Methods Mol Med. 2005;104:203–220. doi: 10.1385/1-59259-836-6:203. [DOI] [PubMed] [Google Scholar]
- Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P, Ivan Lieberburg. Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1992;89:10016–10020. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotshenker S. Microglia and macrophage activation and the regulation of complement-receptor-3 (CR3/MAC-1)-mediated myelin phagocytosis in injury and disease. J Mol Neurosci. 2003;21:65–72. doi: 10.1385/JMN:21:1:65. [DOI] [PubMed] [Google Scholar]
- Rozemuller AJ, Eikelenboom P, Theeuwes JW, Jansen Steur EN, de Vos RA. Activated microglial cells and complement factors are unrelated to cortical Lewy bodies. Acta Neuropathol (Berl) 2000;100:701–708. doi: 10.1007/s004010000225. [DOI] [PubMed] [Google Scholar]
- Rus H, Cudrici C, Niculescu F, Shin ML. Complement activation in autoimmune demyelination: dual role in neuroinflammation and neuroprotection. J Neuroimmunol. 2006;180:9–16. doi: 10.1016/j.jneuroim.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Sapir T, Shoenfeld Y. Facing the enigma of immunomodulatory effects of intravenous immunoglobulin. Clin Rev Allergy Immunol. 2005;29:185–199. doi: 10.1385/CRIAI:29:3:185. [DOI] [PubMed] [Google Scholar]
- Sayah S, Patte C, Gasque P, Chan P, Ischenko A, Vaudry H, Fontaine M. Characterization of rat C5a anaphylatoxin receptor (C5aR): cloning of rat C5aR cDNA and study of C5aR expression by rat astrocytes. Brain Res Mol Brain Res. 1997;48:215–222. doi: 10.1016/s0169-328x(97)00094-6. [DOI] [PubMed] [Google Scholar]
- Schäfer MK, Schwaeble WJ, Post C, Salvati P, Calabresi M, Sim RB, Petry F, Loos M, Weihe E. Complement C1q is dramatically up-regulated in brain microglia in response to transient global cerebral ischemia. J Immunol. 2000;164:5446–5452. doi: 10.4049/jimmunol.164.10.5446. [DOI] [PubMed] [Google Scholar]
- Schultz SJ, Aly H, Hasanen BM, Khashaba MT, Lear SC, Bendon RW, Gordon LE, Feldhoff PW, Lassiter HA. Complement component 9 activation, consumption, and neuronal deposition in the post-hypoxic-ischemic central nervous system of human newborn infants. Neurosci Lett. 2005;378:1–6. doi: 10.1016/j.neulet.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Scola AM, Higginbottom A, Partridge LJ, Reid RC, Woodruff T, Taylor SM, Fairlie DP, Monk PN. The role of the N-terminal domain of the complement fragment receptor C5L2 in ligand binding. J Biol Chem. 2007;282:3664–3671. doi: 10.1074/jbc.M609178200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell WA, North ME, Cambronero R, Webster AD, Farrant J. In vivo modulation of cytokine synthesis by intravenous immunoglobulin. Clin Exp Immunol. 1999;116:509–515. doi: 10.1046/j.1365-2249.1999.00924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell DL, Nacewicz B, Liu F, Macvilay S, Erdei A, Lambris JD, Sandor M, Fabry Z. Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J Neuroimmunol. 2004;155:55–63. doi: 10.1016/j.jneuroim.2004.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Halperin JA, Benzaquen L, Lee CM. Characterization of neuronal cell death induced by complement activation. Brain research. 1997;1:186–194. doi: 10.1016/s1385-299x(96)00026-8. [DOI] [PubMed] [Google Scholar]
- Siragam V, Crow AR, Brinc D, Song S, Freedman J, Lazarus AH. Intravenous immunoglobulin ameliorates ITP via activating Fc gamma receptors on dendritic cells. Nat Med. 2006;12:688–692. doi: 10.1038/nm1416. [DOI] [PubMed] [Google Scholar]
- Sorensen PS. Intravenous polyclonal human immunoglobulins in multiple sclerosis. Neurodegener Dis. 2008;5:8–15. doi: 10.1159/000109932. [DOI] [PubMed] [Google Scholar]
- Stahel PF, Morganti-Kossmann MC, Perez D, Redaelli C, Gloor B, Trentz O, Kossmann T. Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood-brain barrier dysfunction in patients with traumatic brain injury. J Neurotrauma. 2001;18:773–781. doi: 10.1089/089771501316919139. [DOI] [PubMed] [Google Scholar]
- Stangel M, Pul R. Basic principles of intravenous immunoglobulin (IVIg) treatment. J Neurol. 2006;253:V18–24. doi: 10.1007/s00415-006-5003-1. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Stoltzner SE, Grenfell TJ, Mori C, Wisniewski KE, Wisniewski TM, Selkoe DJ, Lemere CA. Temporal accrual of complement proteins in amyloid plaques in Down's syndrome with Alzheimer's disease. Am J Pathol. 2000;156:489–499. doi: 10.1016/S0002-9440(10)64753-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storini C, Rossi E, Marrella V, Distaso M, Veerhuis R, Vergani C, Bergamaschini L, De Simoni MG. C1-inhibitor protects against brain ischemia-reperfusion injury via inhibition of cell recruitment and inflammation. Neurobiol Dis. 2005;19:10–17. doi: 10.1016/j.nbd.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Strohmeyer R, Shen Y, Rogers J. Detection of complement alternative pathway mRNA and proteins in the Alzheimer's disease brain. Brain research. 2000;81:7–18. doi: 10.1016/s0169-328x(00)00149-2. [DOI] [PubMed] [Google Scholar]
- Taylor SM, Fairlie DA. Regulators of the anaphylatoxin C5a. Exp Opin Ther Patents. 2000;10:449–458. [Google Scholar]
- Taylor SM, Sherman SA, Kirnarsky L, Sanderson SD. Development of response-selective agonists of human C5a anaphylatoxin: conformational, biological, and therapeutic considerations. Curr Med Chem. 2001;8:675–684. doi: 10.2174/0929867013373156. [DOI] [PubMed] [Google Scholar]
- Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol. 2006;176:1305–1310. doi: 10.4049/jimmunol.176.3.1305. [DOI] [PubMed] [Google Scholar]
- Tornetta MA, Foley JJ, Sarau HM, Ames RS. The mouse anaphylatoxin C3a receptor: molecular cloning, genomic organization, and functional expression. J Immunol. 1997;158:5277–5282. [PubMed] [Google Scholar]
- Toungouz M, Denys CH, De Groote D, Dupont E. In vitro inhibition of tumour necrosis factor-alpha and interleukin-6 production by intravenous immunoglobulins. Br J Haematol. 1995;89:698–703. doi: 10.1111/j.1365-2141.1995.tb08404.x. [DOI] [PubMed] [Google Scholar]
- Tsuboi Y, Yamada T. Increased concentration of C4d complement protein in CSF in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1994;57:859–861. doi: 10.1136/jnnp.57.7.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urich E, Gutcher I, Prinz M, Becher B. Autoantibody-mediated demyelination depends on complement activation but not activatory Fc-receptors. Proc Natl Acad Sci U S A. 2006;103:18697–18702. doi: 10.1073/pnas.0607283103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urra, et al. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience. 2008 doi: 10.1016/j.neuroscience.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Van Beek J, Bernaudin M, Petit E, Gasque P, Nouvelot A, MacKenzie ET, Fontaine M. Expression of receptors for complement anaphylatoxins C3a and C5a following permanent focal cerebral ischemia in the mouse. Exp Neurol. 2000;161:373–382. doi: 10.1006/exnr.1999.7273. [DOI] [PubMed] [Google Scholar]
- Vasthare US, Barone FC, Sarau HM, Rosenwasser RH, DiMartino M, Young WF, Tuma RF. Complement depletion improves neurological function in cerebral ischemia. Brain Res Bull. 1998;45:413–419. doi: 10.1016/s0361-9230(97)00408-5. [DOI] [PubMed] [Google Scholar]
- Verhagen J, Blaser K, Akdis CA, Akdis M. Mechanisms of allergen-specific immunotherapy: T-regulatory cells and more. Immunol Allergy Clin North Am. 2006;26:207–231. doi: 10.1016/j.iac.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Vivanco F, Muñoz E, Vidarte L, Pastor C. The covalent interaction of C3 with IgG immune complexes. Mol Immunol. 1999;36:843–852. doi: 10.1016/s0161-5890(99)00105-4. [DOI] [PubMed] [Google Scholar]
- Wills-Karp M. Complement activation pathways: a bridge between innate and adaptive immune responses in asthma. Proc Am Thorac Soc. 2007;4:247–251. doi: 10.1513/pats.200704-046AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff TM, Arumugam TV, Shiels IA, Reid RC, Fairlie DP, Taylor SM. Protective effects of a potent C5a receptor antagonist on experimental acute limb ischemia-reperfusion in rats. J Surg Res. 2004;116:81–90. doi: 10.1016/j.jss.2003.04.001. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Crane JW, Proctor LM, Buller KM, Shek AB, de Vos K, Pollitt S, Williams HM, Shiels IA, Monk PN, Taylor SM. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J. 2006;20:1407–1417. doi: 10.1096/fj.05-5814com. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Denny KJ, Taylor SM, Noakes PN. Role of complement in motor neuron disease: animal models and therapeutic potential of complement inhibitors. Current Topics in Complement II: Advances in Experimental Medicine and Biology. 2008 Submitted. [PubMed] [Google Scholar]
- Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci U S A. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZQ, Qian W, Suzuki K, McNamara JO. Formation of complement membrane attack complex in mammalian cerebral cortex evokes seizures and neurodegeneration. J Neurosci. 2003;23:955–960. doi: 10.1523/JNEUROSCI.23-03-00955.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, McGeer PL, McGeer EG. Lewy bodies in Parkinson's disease are recognized by antibodies to complement proteins. Acta Neuropathol (Berl) 1992;84:100–104. doi: 10.1007/BF00427222. [DOI] [PubMed] [Google Scholar]
- Yasojima K, Schwab C, McGeer EG, McGeer PL. Up-regulated production and activation of the complement system in Alzheimer's disease brain. The American journal of pathology. 1999;154:927–936. doi: 10.1016/S0002-9440(10)65340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara O, Aimi Y, McGeer EG, McGeer PL. Expression of the complement membrane attack complex and its inhibitors in Pick disease brain. Brain Res. 1994;652:346–349. doi: 10.1016/0006-8993(94)90246-1. [DOI] [PubMed] [Google Scholar]
- Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. 2006;37:1087–1093. doi: 10.1161/01.STR.0000206281.77178.ac. [DOI] [PubMed] [Google Scholar]
- Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- You Z, Yang J, Takahashi K, Yager PH, Kim HH, Qin T, Stahl GL, Ezekowitz RA, Carroll MC, Whalen MJ. Reduced tissue damage and improved recovery of motor function after traumatic brain injury in mice deficient in complement component C4. J Cereb Blood Flow Metab. 2007;27:1954–1964. doi: 10.1038/sj.jcbfm.9600497. [DOI] [PubMed] [Google Scholar]
- Zhu KY, Feferman T, Maiti PK, Souroujon MC, Fuchs S. Intravenous immunoglobulin suppresses experimental myasthenia gravis: immunological mechanisms. J Neuroimmunol. 2006;176:187–197. doi: 10.1016/j.jneuroim.2006.04.011. [DOI] [PubMed] [Google Scholar]