

Abstract
Post-transcriptional regulation of gene expression by microRNAs has been implicated in the regulation of chronic physiological and pathological responses. In this report, we demonstrate that changes in the expression of microRNAs (miRNAs) can also regulate acute inflammatory responses in human lung alveolar epithelial cells. Thus, stimulation with interleukin (IL)-1β results in a rapid time- and concentration-dependent increase in miRNA-146a and, to a lesser extent, miRNA-146b expression although these increases were only observed at high IL-1β concentration. Examination of miRNA function by over-expression and inhibition showed that increased miRNA-146a expression negatively regulated the release of the pro-inflammatory chemokines, IL-8 and RANTES. Subsequent examination of the mechanism demonstrated that the action of miRNA-146a was mediated at the translational level and not through down-regulation of proteins involved in the IL-1β signalling pathway or chemokine transcription or secretion. Overall, these studies indicate that rapid increase in miRNA-146a expression provides a novel mechanism for negative regulation of severe inflammation during the innate immune response.
Keywords: inflammation, chemokines, cytokines, gene regulation, lung
Introduction
The innate immune response, mediated by epithelial and immune cells such as macrophages and dendritic cells, is the first line of defence against infection. This response is commonly mediated through activation of members of the Toll/interleukin-1 receptor (TIR) superfamily, which can be divided into two groups: the interleukin-1 receptors (IL-1R) and the Toll-like receptors (TLR). The IL-1R family is known to consist of ten receptors that mediate the responses to IL-1α, IL-1β and IL-18, whilst the TLR family contains at least eleven members that are involved in the recognition of conserved molecular patterns upon invading micro-organisms, entitled pathogen-associated molecular patterns (PAMPs) (1,2). Significantly, all members of this receptor superfamily contain the TIR cytoplasmic domain and are thought to signal through a similar intracellular pathway. Thus, signalling through the TIR domain involves an association with the adaptor protein MyD88, which recruits IL-1R associated kinase 1 (IRAK1) and TNF receptor-associated factor 6 (TRAF6) following ligand binding. Dissociation of IRAK1 from MyD88 following phosphorylation causes the activation of TRAF6, which subsequently stimulates the formation and activation of the TAK1/TAB1/TAB2/TAB3 complex. This in turn activates a variety of pro-inflammatory transcription factors such as nuclear factor (NF)-κB and activator protein (AP)-1 via the IκB kinase (IKK) complex and c-jun N-terminal kinase (JNK) respectively. To prevent an inappropriate inflammatory response following activation of the TIR receptors a variety of extracellular and intracellular negative feedback pathways have evolved to regulate this process. These include the production of soluble TLRs, which compete with membrane receptors for ligand binding, regulation of TLR/IL-1 receptor expression, production of dominant negative splice variants of MyD88 and IRAK, as well as post-translational modifications such as phosphorylation, ubiquitination and degradation (3).
Recent investigations have identified microRNA (miRNA)-mediated RNA interference (RNAi) as a novel, evolutionary conserved mechanism for the regulation of gene expression at the post-transcriptional level (4,5) and have identified a host of endogenous mammalian genes that are processed to produce ~ 700 miRNAs (miRNA registry at www.sanger.ac.uk/Software/Rfam/mirna/). miRNA biogenesis involves the initial transcription of primary miRNAs by RNA polymerase II, which are subsequently cleaved by the RNase III enzyme Drosha, in combination with DGCR8, to produce a hairpin RNA of ~ 65-nucleotides known as pre-miRNAs (6). These are then exported into the cytoplasm by exportin 5 and further cleaved by the RNase III enzyme Dicer, to produce the 21- to 23-nucleotide double stranded RNA duplexes. The actions of miRNAs are mediated by the miRNA-induced silencing complex (miRISC) which uses the mature miRNA guide strand as a template to identify target mRNA. At present, miRNAs are believed to either block mRNA translation or reduce mRNA stability, following imperfect binding of the guide strand to miRNA-recognition elements (MRE) within the 3′ untranslated region (UTR) of target genes. Specificity of the guide strand is thought to be primarily mediated by the ‘seed’ region localised at residues 2-8 at the 5′ end; although this also appears to be influenced by additional factors such as the presence and cooperation between multiple MREs (7,8), the spacing between MREs (8,9), proximity to the stop codon (8), position within the 3′ UTR (8), AU composition (8) and target mRNA secondary structure (10). The mechanism underlying the repression of protein synthesis by miRNAs is currently an area of intense investigation, although it appears that individual miRNAs might employ divergent pathways and that these are integrated with the mRNA degradation process associated with P-bodies (11). Thus, recent reports have suggested that the actions of miRNAs might function through mRNA destabilisation and degradation (12,13) or alternatively, following the repression of both translational initiation (14-17) and elongation steps (18).
The physiological role of the majority of identified miRNAs is unknown, although their constitutive expression in tissues and changes during development indicate their importance in the maintenance of cellular phenotype. Thus, islet selective miRNA-375 expression has been shown to regulate insulin secretion (19), liver specific miRNA-122 is involved in cholesterol metabolism in vivo (20,21) and muscle specific miRNA-1 and miRNA-133 are known to control heart development and physiological responses (22-24). At present, little is known of the function of miRNAs during the inflammatory response. A potential role of miRNA-155 in the adaptive immune response was provided from studies using knockout mice. These showed reduced CD3/CD28-induced IFN-γ release from CD4+ T-cells (25) and BCR-mediated TNF-α production from B-cells (26), which indicates that miRNA-155 facilitates or positively regulates cytokine release in lymphoid cells. Similarly, increased miRNA-181a expression was also shown to augment IL-2 release following activation of T-cells through the down-regulation of phosphatases (27). Interestingly, recent reports have shown increased miRNA-155, miRNA-146a and miRNA-146b expression following activation of the innate immune response in monocytes/macrophages (28,29). The functional role of miRNA-146a and miRNA-146b are presently unknown, although it has been speculated that they might ‘fine-tune’ negative feedback regulation of inflammation through down-regulation of IRAK1 and TRAF6, two proteins involved in TIR signalling (29). However, there is little experimental evidence to support this hypothesis except from studies of CMV-driven over-expression of miRNA-146a and miRNA-146b in HEK293 cells, which was shown to inhibit the activity of luciferase reporter plasmids that contained either the 3′ UTR of IRAK1 or TRAF6 (29). For this reason, we have investigated the functional link between IL-1β induced miRNA-146a and miRNA-146b expression and the release of the pro-inflammatory chemokines, IL-8 and RANTES. Significantly, rather than ‘fine-tune’ the inflammatory response, we have shown that miRNA-146a and to a lesser extent miRNA-146b, are central to the negative feedback regulation of IL-1β-induced inflammation. Furthermore, we report that the expression and action of miRNA-146a is observed at high IL-1β concentration which indicates that this negative feedback mechanism is only activated during severe inflammation. Examination of the mechanism of action of miRNA-146a and miRNA-146b showed this was unlikely to be mediated through down-regulation of the IL-1β signalling proteins, IRAK-1 and TRAF-6. Instead, the fact that miRNA-146a and miRNA-146b also did not appear to act upon either IL-8 and RANTES transcription or secretion implies that their action is upon chemokine translation. Overall, these investigations show that in addition to their role in the maintenance of cellular phenotype, rapid changes in miRNA levels are involved in the regulation of acute biological responses such as inflammation. Furthermore, given the observation that miRNA-146a and miRNA-146b expression is also increased following exposure of monocytes and macrophages to a range of microbial stimuli that act through the TIR family (29), this implies that these miRNAs are important regulators of the innate immune response.
Material and Methods
Cell Studies
A549 cells were grown in DMEM containing 10% FCS and 2 mM L-glutamine and then plated at 75% confluence in 96-, 24- or 6-well plates, cultured for an additional 4 h and then stimulated with the indicated concentration of IL-1β for 6 h and/or 24 h. Beas2B (ATCC CRL-9609) cells that were at passage 41-50 were grown in Keratinocyte media with 5% L-glutamine, EGF and bovine pituitary whilst THP-1 cells were cultured in RPMI medium 1640 supplemented with 10% FCS, 100 units/ml penicillin, 100 units/ml streptomycin and 2 mM L-glutamine. Normal human bronchial epithelial cells were obtained from Lonza Ltd and were cultured to a maximum of 5 passages in bronchial epithelial medium (BEGM®) containing a cocktail of growth factors, cytokines, and supplements (BulletKit®). For the determination of cytokine/chemokine release, supernatant were removed and IL-8, RANTES and IFN-α levels determined by DuoSet ELISA (R&D Systems). The viability of the remaining cells was determined by MTT assay.
Transfection with miRNA mimics and inhibitors
miRNA-146a/b mimics and controls (Pre™-miRNA-146a/b) were obtained from Ambion/Applied Biosystems Ltd whilst miRNA-146a/b inhibitors and controls were obtained from Exiqon Ltd (Denmark). For studies in 96-well plates, miRNA mimics and inhibitors were resuspended in 50 μl Lipofectamine 2000/Opti-MEM (Invitrogen), added to the relevant plates prior to the addition of A549 cells and incubation at 37°C for 4 h.
Measurement of miRNA and mRNA expression
Total RNA was extracted using the mirVana™ miRNA isolation kit (Ambion Europe) according to the manufacturer's instructions. RNA was eluted in 50 μl RNase-free water (Promega UK, Southampton, UK) and stored at −70°C. RNA content and purity was measured using a BioTek PowerWave XS (SSi Robotics, Tustin, CA, U.S.A.) spectrophotometer. miRNA expression profiling was carried out on total RNA extracts by two-step TaqMan® reverse transcription polymerase chain reaction protocol (RT-PCR), normalised to 18S, as previously described (30). The separate well, 2−(ΔΔCt) method (31) was used to determine relative-quantitative levels of individual miRNAs, and these were expressed as the fold-difference to the relevant controls. mRNA expression levels of IRAK1, TRAF6, RANTES and IL-8 was determined using semi-quantitative two-step RT-PCR as previously described (32) using Assay on Demand primer/probe sets obtained from Applied Biosystems, UK.
Western Blotting
Proteins were extracted from A549 cells as previously described (33), separated upon 10% SDS-PAGE or 4-12% PAGE gels (Invitrogen) and transferred to nitrocellulose (Amersham Ltd). Protein (5 -10 μg) were detected by Western blotting using a rabbit anti-TRAF6 antibody (H-274) (34), rabbit anti-IRAK1 antibody (H-273)(35) and goat anti-synaptotagmin-1 (N-19) were obtained from Santa Cruz Biotechnology, rabbit anti-syntaxin-3 was obtained from Calbiochem (Nottingham, UK) and rabbit anti-Sec23 interacting protein was obtained from Novus Biologicals (Littleton, USA) All primary antibodies were used a concentration of 1:200 or 1:400 and were incubated overnight. Labelling of the first antibody was detected using relevant secondary antibodies conjugated to HRP (Dako Ltd) and detected using ECL reagents (Amersham Ltd., UK).
Statistical Analysis
Statistical changes in IL-8, RANTES, IFN-α, miRNA-146a and miRNA-146b expression were determined using either a two-tailed Student's t-test or ANOVA with α set to 0.05 using Prism 4 for Windows (version 4.03). miRNA expression data was analysed and displayed using the Genesis (version 1.7.0) designed by Alexander Sturn and obtained from the Institute of Genomics and Bioinformatics at the Graz Institute of Technology. Using this software package, significant differences was determined using analysis of variance (ANOVA) followed by Dunn's post-test with α set to 0.01.
Results
IL-1β-induced IL-8 and RANTES response
Initial studies were undertaken to characterise the mechanism of IL-1β-induced production of IL-8 and RANTES. Exposure to IL-1β (1 ng/ml) induced a differential time- and concentration-dependent release of the inflammatory chemokines, IL-8 and RANTES. Although we observed rapid release of both chemokines, the IL-8 response reached a plateau at approximately 6-8 h whilst RANTES release continued to increase throughout the 24 h period (Fig. 1a). Similarly, examination of the effect of increasing IL-1β concentration at 6 h showed that IL-8 release occurred at low IL-1β levels (EC50 ~ 0.01 ng/ml), whilst the RANTES response was elicited at higher IL-1β concentrations (EC50 ~ 0.3 ng/ml) (Fig. 1b).
Figure 1.
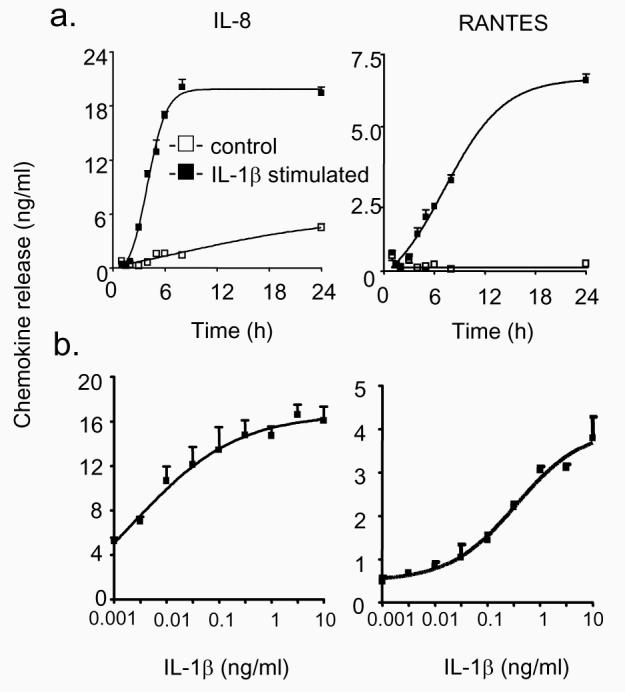
Time- and concentration-dependency of IL-1β-induced IL-8 and RANTES release. A549 cells were exposed to either buffer or 1 ng/ml IL-1β for the indicated time (a) or to the indicated IL-1β concentration for 24 h (b) prior to measurement of IL-8 and RANTES release. The results are expressed as the mean ± SEM of 3 individual samples and are representative of 3 independent experiments.
Effect of IL-1β on the profile of miRNA expression
To determine if miRNA levels were affected by IL-1β challenge, we measured the expression of 156 miRNAs using an RT-PCR based approach (30). This showed that 137 out of 156 miRNAs were expressed in untreated A549 cells (data not shown). In general, treatment with IL-1β produced an overall reduction in the miRNA expression profile at 3 h, although we only observed significant reduction (p < 0.01) in the levels of let-7g, miRNA-26b, miR-104, miRNA-195, miR-296 and miRNA-299 and a large, 24-fold increase in the expression of miRNA-146a (Fig. 2). Following this initial observation, an additional miRNA-146-encoding gene named miRNA-146b was characterised, which differs from miR-146a by 2 residues at the 3′ end of the guide strand. Semi-quantitative determination of miRNA-146a and miRNA-146b expression in untreated cells by RT-PCR showed that these were almost identical, giving ΔCt (−18S as control) of 14.5 ± 1.5 and 13.1 ± 1.1 (n = 5 independent experiments, p = not significant), respectively.
Figure 2.
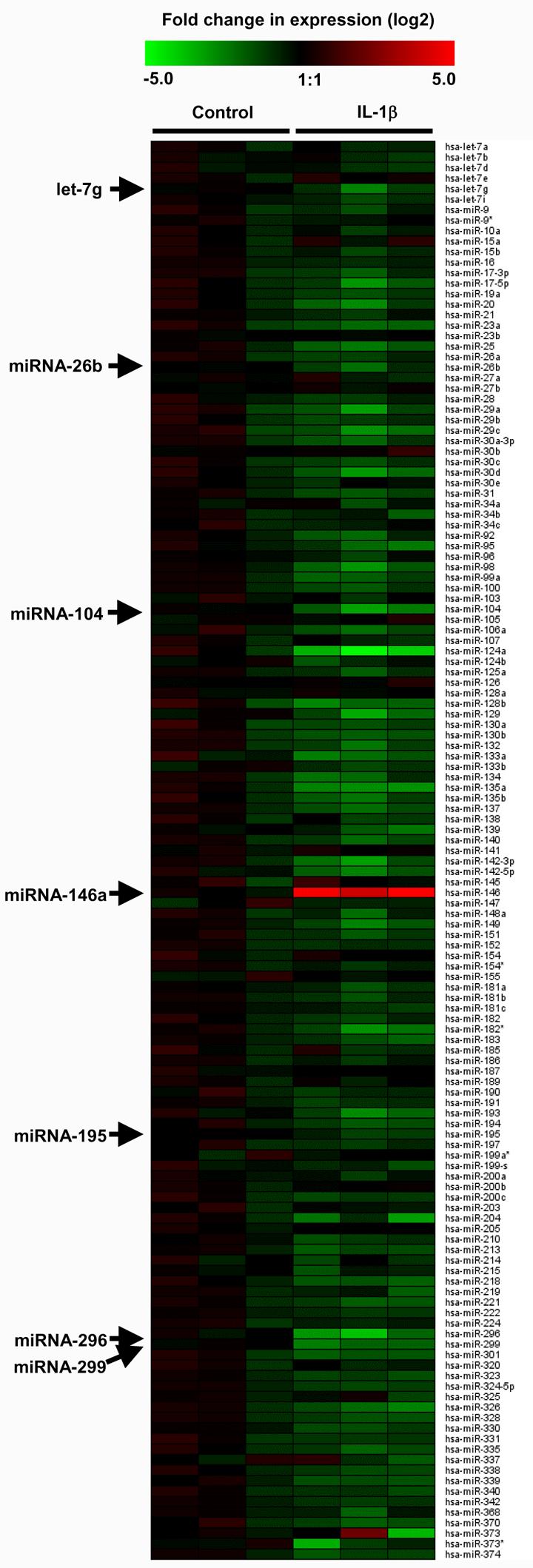
IL-1β-induced changes in miRNA expression. A549 cells were exposed to IL-1β (1 ng/ml) for 3 h and the profile of expression of 156 miRNAs was measured using TaqMan RT-PCR. The log2 transformed values of the fold-change in expression compared to time-matched saline controls are represented as a heat map where red and green indicate an increase and decrease in miRNA expression, respectively.
To determine the potential role of miRNA-146a and miRNA-146b in the inflammatory response, we examined the time course of their expression. IL-1β stimulated a time-dependent 70- and 20-fold increase in the expression of miRNA-146a and miRNA-146b, which reached a plateau at approximately 3h and 6h, respectively (Fig. 3a). Examination of the effect of increasing IL-1β concentration showed that miRNA-146a and miRNA-146b expression occurred at high IL-1β levels, giving similar EC50 values of ~ 0.3 ng/ml (Fig. 3b). Since miRNA-146a and miRNA-146b differ by only 2 nucleotides, we determined the extent of cross-reactivity between the respective RT-PCR probes. Using miRNA mimics, we showed that there was limited cross-reactivity, with the probes for miRNA-146a (< 10% versus miRNA-146b) being less selective than miRNA-146b (< 0.4% versus miRNA-146a).
Figure 3.

Characterisation of the mechanism of miRNA-146a and miRNA-146b expression. The time- and concentration-dependent induction of miRNA-146a and miRNA-146b in A549 cells was determined following exposure to 1 ng/ml IL-1β for the indicated time (a) or to the indicated IL-1β concentration for 6 h (b) and the increases in miR-146a and miR-146b expression were determined by RT-PCR. To confirm the observations in A549 cells, the levels of miRNA-146a and miR-146b were measured at 6 h in IL-1β-stimulated primary human bronchial epithelial cells and transformed human bronchial Beas2B epithelial cells (c) or in LPS-stimulated monocytic THP-1 cells (d). Alternatively, control- and IL-1β-(1ng/ml) stimulated A549 cells were pre-treated with dexamethasone (1 μM) for 60 min, and the expression of miRNA-146a and miRNA-146b was determined at the indicated time points (e). These results are expressed as the mean ± SEM of 3 independent experiments. ND = not detected.
To ensure that these changes in miRNA expression were not restricted to the lung A549 epithelial cell line, we also examined the response in primary bronchial epithelial cells and in the transformed bronchial Beas2B epithelial cell line. These studies showed a comparable but selective increase in miRNA-146a but not miRNA-146b expression in both cell types following IL-1β stimulation (Fig. 3c). Indeed, we were unable to detect the expression of miRNA-146b in the primary human bronchial epithelial cells. In addition, studies showed that this IL-1β-induced response was also comparable to that observed following LPS stimulation of the human monocytic THP-1 cell line, which resulted in an approximate 30- and 3-fold increase in the expression of miRNA-146a and miRNA-146b, respectively (Fig. 3d).
The previous study by Taganov et al. demonstrated that miRNA-146a transcription was regulated by NF-κB (29). To investigate the role of this pathway in the regulation of the IL-1β induced response, we have also examined the effect of pre-incubation with dexamethasone (1 μM). This corticosteroid, which is known to attenuate the action of multiple pro-inflammatory transcription factors including NF-κB, was found to inhibit both IL-1β-induced IL-8/RANTES release (data not shown) and miRNA-146a/miRNA-146b expression by 70-80% (Fig. 3e). This therefore suggests that expression of both miRNA-146a and miRNA-146b is regulated by pro-inflammatory transcription factors, possibly NF-κB, that are corticosteroid sensitive.
Overall, on the basis of the rapid time- and concentration-dependent increases in miRNA-146a and miRNA-146b, we hypothesised that these miRNAs might be involved in the regulation of inflammation at high IL-1β concentration.
Inhibition of miRNA-146a and miRNA-146b increases IL-1β-induced IL-8 and RANTES release
The functional relevance of changes in miRNA-146a and miRNA-146b expression during the IL-1β–induced IL-8 and RANTES release was assessed using miRNA inhibitors (20,21,36,37). Locked nucleic acid/DNA based antisense inhibitors and controls were transfected in A549 cells for 4 h. We had previously shown that this was sufficient time to obtain oligonucleotide delivery in A549 cells when examining the inhibition of the IL-1β-induced increases in IL-8 production using an siRNA smartpool (Dharmacon Inc) (data not shown). The miRNA-146a inhibitor increased IL-1β-induced (1 ng/ml) IL-8 and RANTES release in a concentration-dependent manner with an EC50 ~ 3-10 nM (Fig. 4a). In contrast, the inhibitor of miRNA-146b was less effective at increasing IL-8 release and had no significant effect upon RANTES release. This implied that the IL-1β-induced increase in miRNA-146a but not miRNA-146b has the greater capacity to modulate this mechanism. This contention was supported by investigations on the effect of miRNA-146 inhibitors (10 nM) upon chemokine release at increasing IL-1β concentration (Fig. 4b). Thus, the IL-1β-induced IL-8 and RANTES release was not significantly affected by the control or miRNA-146b inhibitor. However, as might be expected given that IL-1β–induced miRNA-146a expression has an EC50 ~ 0.3ng/ml (Fig. 2b), we observed a significant increase in IL-8 and RANTES release at high IL-1β concentrations (> 1 ng/ml). To eliminate the possibility that the actions of miRNA inhibitors were secondary to the induction of an anti-viral response and/or toxicity, we measured the extracellular release of interferon-α (IFN-α) and cellular viability at 24 h and found no significant changes (Fig. 5). Overall, these inhibitor studies implied that IL-1β-induced increases in miRNA-146a expression at high IL-1β concentrations were involved in the negative feedback regulation of the inflammatory response.
Figure 4.

Effect of inhibitors and mimics of miRNA-146a and miRNA-146b upon IL-1β–induced IL-8 and RANTES release. A549 cells were exposed to transfection reagent alone (blank), control inhibitor or mimic, miR-146a inhibitor or mimic and miR-146b inhibitor or mimic for 4 h and then exposed to either 1 ng/ml IL-1β (a,c) or the indicated IL-1β concentration (b,d) and the concentration of IL-8 and RANTES in the supernatant was determined at 6 h and 24 h, respectively. The results are expressed as the mean ± SEM of 3 individual samples and are representative of at least 3 independent experiments. * p < 0.05 versus time-matched (a,c) or concentration-matched (b,d) transfected controls.
Figure 5.
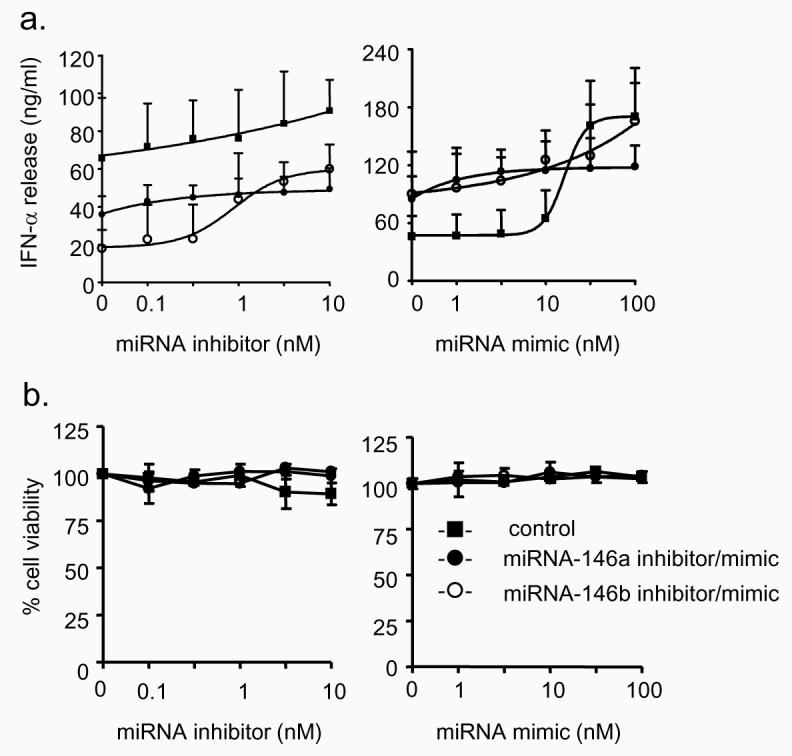
Effect of miRNA-146a and miRNA-146b inhibitors and mimics upon IFN-α release and cell viability. A549 cells exposed to control miRNA inhibitor/mimic, miR-146a inhibitor/mimic or miR-146b inhibitor/mimic for 4 h and then exposed to 1 ng/ml IL-1β for 24h. Supernatant samples were then removed for measurement of IFN-α (a) and the cell viability of the remaining cells was determined (b). The results are expressed as the mean ± SEM of 3 individual samples and the graphs are representative of 3 independent experiments.
miRNA-146a and miRNA-146b mimics decreases IL-1β-induced IL-8 and RANTES release
To provide additional evidence on the role of miRNA-146a in negative feedback regulation, we examined the effect of miRNA over-expression by use of miRNA mimics. Cells were once again transfected with the miRNA mimics at 4 h prior to IL-1β stimulation and IL-8 and RANTES release was measured at 6 h and 24 h, respectively. Measurement of the concentration dependency showed that when the cells were stimulated with 1ng/ml of IL-1β, mimics of both miRNA-146a and miRNA-146b significantly attenuated IL-8 and RANTES release by approximately 60-80% at 100 nM (Fig. 4c). Measurement of the effect of 100 nM mimic upon absolute miRNA-146a and miRNA-146b levels showed variability in the transfection efficacy but resulted in ~ 500-6000-fold increases above the baseline levels for these miRNAs (data not shown). We next examined the action of the miRNA mimics at 100 nM concentrations upon chemokine release at increasing IL-1β concentrations. Interestingly, this suggested that miRNA over-expression resulted in non-competitive inhibition since both miRNA-146a and miRNA-146b mimics (100nM) inhibited IL-8 and RANTES release by 60 – 90% across the IL-1β concentration range (0.001 – 10 ng/ml) (Fig. 4d). Once again, measurement of IFN-α release and cell viability showed no significant action of miRNA mimics at all the concentrations tested (Fig. 5).
miRNA-146a and miRNA-146b do not target chemokine transcription
To determine their potential mechanism, we used the most recently released database for the prediction of miRNAs targets (TargetScan database (www.targetscan.org) (38)). In addition to multiple transcription factors, this identified five additional proteins, two involved in IL-1β signalling (IRAK1 and TRAF6) and three implicated in secretion (syntaxin-3, synaptotagmin-1 and sec23 interacting protein (sec23IP)).
Initially we determined whether the actions of miRNA-146a and miRNA-146b were mediated through down-regulation of proteins involved in the IL-1β signalling pathway. Measurement of protein levels during the 24 h following IL-1β stimulation showed no change in the expression of TRAF6 but an upward shift in the IRAK1 band at 1-3 h, followed by a reduction in protein expression at 6 h and 24 h (Fig. 6a). Measurement of IRAK1 and TRAF6 mRNA expression showed no significant changes under these conditions (Fig. 6b). Previous studies have shown that this increase in the IRAK1 molecular weight is caused by protein phosphorylation and ubiquitination, that subsequently results in proteolytic degradation (39,40). To determine whether increased miRNA-146a and miRNA-146b might contribute towards IRAK1 protein degradation, we examined the effect of the miRNA inhibitors and mimics. Following 4 h transfection (i.e. time = 0 h), we observed no effect upon IRAK1 protein expression whilst miRNA inhibitors and mimics also had no effect upon IL-1β–induced IRAK1 degradation at 6 h (Fig. 6c). This protein expression data therefore indicated that the action of miRNA-146a and miRNA-146b were not mediated through an action upon proteins involved in the IL-1β signalling pathway. To support this conclusion and to ascertain whether their actions might instead involve down-regulation of transcription factors, we determined the action of miRNA inhibitors on IL-1β-induced IL-8 and RANTES mRNA expression. Initial measurement of the time course of IL-1β-induced changes in IL-8 and RANTES mRNA levels showed a rapid increase in both transcripts that peaked at 6-8 h and remained elevated at 24 h (Fig. 7a). In subsequent studies, we found that inhibitors (10 nM) of miRNA-146a and miRNA-146b had no effect upon IL-1β–induced IL-8 and RANTES mRNA production at either 6 h or 24 h (Fig. 7b). In contrast, transfection with miRNA-146a and miRNA-146b mimics at 100 nM significantly attenuated IL-8 and RANTES mRNA expression at 6 h and 24 h (Fig. 7b). Interestingly, despite the fact that IL-1β stimulation alone did not significantly increase IRAK1 and TRAF6 mRNA expression (Fig. 6b), we observed a significant reduction in IRAK1 but not TRAF6 mRNA in the presence of miRNA mimics (Fig. 7b). Since we had previously shown that transfection with miRNA mimics increased intracellular miRNA-146a and miRNA-146b levels by ~500-6000 fold rather than the ~ 20 – 70 fold increase seen following IL-1β stimulation, it suggested that these actions of miRNA mimics, including the degradation of IRAK1 mRNA, might be mediated through non-physiological processes at these super-maximal concentrations.
Figure 6.

Role of miRNA-146a and miRNA-146b in the IL-1β signalling pathway. A549 cells were exposed IL-1β (1 ng/ml) and the expression of IRAK1 and TRAF6 protein (a) and mRNA (b) were detected at the indicated time points. In separate studies, A549 cells were transfected with an inhibitor or mimic of miRNA-146a, miRNA-146b or the relevant control miRNA and the effect upon IRAK1 protein expression was determined at 0 h and at 6 h following IL-1β stimulation (c). The results in panel (a) and (c) are representative of a least 3 independent experiments whilst panel (b) gives the mean ± SEM of 3 independent experiments.
Figure 7.
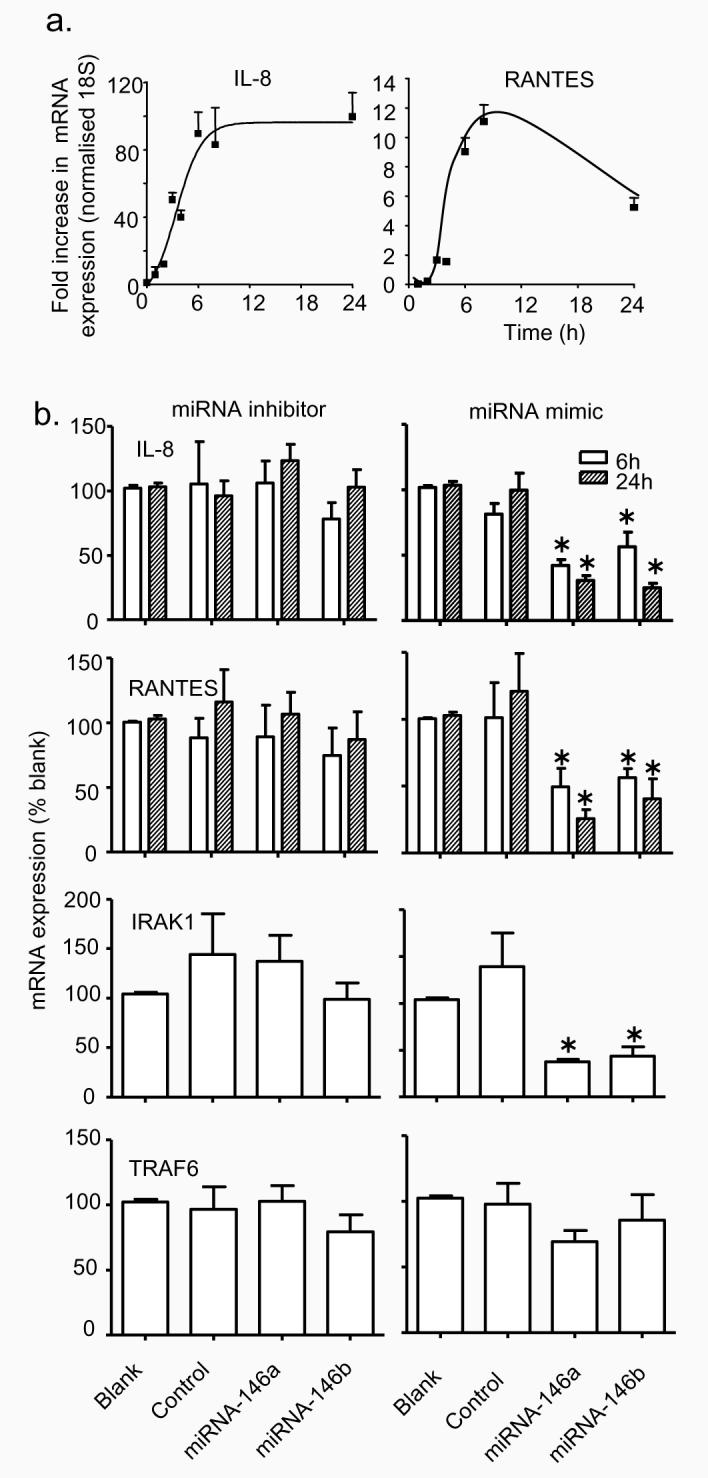
Role of miRNA-146a and miRNA-146b during IL-1β-induced IL-8 and RANTES transcription. A549 cells were stimulated with IL-1β and the expression of IL-8 and RANTES mRNA was determined at the indicated time (a). Alternatively, cells were transfected for 4 h with transfection reagent alone (blank), control inhibitor or mimic (control), miR-146a inhibitor or mimic (miRNA-146a) and miR-146b inhibitor or mimic (miRNA-146b) for 4 h, exposed to 1 ng/ml IL-1β and the expression of IL-8, RANTES, IRAK1 and TRAF6 mRNA was determined at 6 h and/or 24 h. The results are expressed as the mean ± SEM of at least 3 independent experiments. * p < 0.05 versus blanks.
miRNA-146a and miRNA-146b do not target chemokine secretion
Having eliminated an action of miRNA-146a and miRNA-146b upon the IL-1β signalling pathway and chemokine transcription, we proceeded to investigate the possible targeting of proteins involved in IL-8 and RANTES secretion. However, we once again observed no significant reduction in syntaxin-3, synaptotagmin-1 and sec23IP protein expression during the 24 h following IL-1β stimulation (Fig. 8a). Additional evidence that miRNA-146a and miRNA-146b did not act upon secretion, came from studies of the effect of miRNA inhibition and over-expression upon intracellular IL-8 and RANTES levels. Examination of the time course of intracellular protein production showed a time-dependent increase in the IL-8 and RANTES levels that peaked at ~ 24 h and 6 h, respectively (Fig. 8b). If the action of IL-1β-induced miRNA-146a and miRNA-146b were mediated through the down-regulation of proteins involved in secretion, then this would result in the accumulation of intracellular IL-8 and RANTES. Under these circumstances, miRNA mimics would be expected to further block secretion, resulting in increased intracellular IL-8 and RANTES levels whilst miRNA inhibitors should reduce these levels. No significant effect of inhibitor or mimic upon intracellular RANTES levels was observed (Fig. 8c). However, we found that at IL-1β concentrations that induced miRNA-146a and miRNA-146b production, the mimics reduced intracellular expression whilst an inhibitor of miRNA-146a, and to a lesser extend miRNA-146b, increased IL-8 levels (Fig. 8c). In addition to the protein data, this provided further evidence that miRNA-146a and miRNA-146b did not target IL-8 and RANTES secretion.
Figure 8.

Role of miRNA-146a and miRNA-146b during IL-1β-induced IL-8 and RANTES secretion. A549 cells were stimulated with IL-1β (1 ng/ml) and at the indicated times, the intracellular protein expression of syntaxin-3, synaptotagmin-1 or sec23-IP was determined by Western blotting (a) and IL-8 and RANTES by ELISA (b). Alternatively, cells were transfected for 4 h with transfection reagent alone (blank), control inhibitor or mimic, miR-146a inhibitor or mimic and miR-146b inhibitor or mimic for 4 h and then exposed to the indicated IL-1β concentration and the intracellular IL-8 and RANTES protein expression was determined at 6 h. The results are the mean ± SEM of 3 individual samples and are representative of at least 3 independent experiments. * p < 0.05 versus blanks.
Discussion
A number of recent publications have indicated that miRNAs might regulate the inflammatory response associated with adaptive and innate immunity. With the adaptive response, miRNA-155 has been implicated as a positive regulator of cytokine release from B- and T-cells by an uncharacterised mechanism (25,26). Similarly, miRNA-181a has been shown to modulate the inflammatory response in T-cells, although this was shown to be mediated through the down-regulation of multiple phosphatases such as CTLA-4 (27). In the case of the innate immune response, Taganov et al. (29) have suggested that miRNA-146a and miRNA-146b might negatively regulate the activation of the innate immune response through down-regulation of IRAK1 and TRAF6. However, the targets of miRNA-146a and miRNA-146b were identified by over-expression and were not linked to an inflammatory response. For these reasons, we have undertaken studies to elucidate whether changes in miRNA-146a and miRNA-146b expression are functionally linked to the release of inflammatory mediators and to elucidate their mechanism of action. Specifically, we have examined their role during the IL-1β-induced response in the human alveolar A549 lung epithelial cell line.
Examination of the differential expression of 156 miRNAs following IL-1β exposure showed significant reduction in the expression of 6 miRNAs, let-7g, miRNA-26b, miRNA-104, miR-195, miR-296 and miRNA-299 and increased expression of miR-146a and miRNA-146b. The increased expression of miRNA-146a and miRNA-146b is in agreement with the earlier studies in LPS- and TNF-α-stimulated monocytic cell lines although, unlike ourselves, these investigators also reported up-regulation of miRNA-155 and miRNA-132 (28,29). The reasons for these differences are presently uncertain, although this might be related to cell differences, since studies in transgenic mice report that miRNA-155 is selectively expressed in lymphoid and not non-lymphoid cells (26). At present, the functions of let-7g, miRNA-26b, miRNA-104, miR-195, miR-296 and miRNA-299 have not been investigated, although it would be expected that their reduced levels would result in an increase in expression of target proteins.
A role for miR-146a and miRNA-146b in the regulation of the chemokine release was initially suggested from the time- and concentration-dependency of the IL-1β–induced responses. Thus, measurement of the time course of miRNA-146a and miRNA-146b production showed that this correlated with the rapid release of IL-8 and RANTES. Interestingly, although miRNA-146a and miRNA-146b were expressed at comparable levels in untreated cells, IL-1β stimulated a greater increase in absolute levels of miRNA-146a. Furthermore, examination of the concentration dependency showed that miRNA-146a and miRNA-146b expression occurred at high IL-1β concentrations that paralleled RANTES release: at these concentrations IL-8 release was already maximal. From a mechanistic point, miRNA-146a, IL-8 and RANTES are all thought to be regulated by the inflammatory transcription factor, NF-κB, although these observations indicate that IL-8 might be regulated by a high affinity promoter site whilst miRNA-146a and RANTES are regulated by low affinity NF-κB sites (29,41). This conclusion is supported by our studies showing the inhibition of IL-1β-induced miRNA-146a expression in the presence of dexamethasone, a corticosteroid which is known to attenuate the action of multiple pro-inflammatory transcription factors, including NF-κB. The fact that miRNA-146b expression was also prevented indicated that its transcription is also mediated by corticosteroid-sensitive transcription factors.
Subsequent pharmacological studies established a functional link between the increase in miRNA-146a and the negative feedback regulation of the inflammatory response. Thus, inhibition of miRNA-146a was found to increase IL-8 and RANTES release, whilst miRNA-146a over-expression using miRNA mimics attenuated this response. In contrast, inhibiting miRNA-146b had little effect, although a reduction in chemokine release following the administration of miRNA-146b mimic implied that this was probably related to the smaller increase in miRNA-146b expression in response to IL-1β stimulation, rather than lack of biological activity per se. The importance of miRNA-146a rather than miRNA-146b in TIR-mediated negative feedback is also supported by our observations showing robust increases in miRNA-146a but not miRNA-146b expression following activation of human bronchial epithelial and monocytic cells. Crucially, the fact that both miRNA-146a expression and the inhibitor-mediated increases in IL-8 and RANTES release were only seen at high IL-1β concentrations (>0.3 ng/ml), indicates that this negative feedback pathway is important during severe inflammation.
In an attempt to elucidate their mechanism of action, interrogation of the TargetScan database indicated that miRNA-146a and miRNA-146b targeted proteins involved in both the IL-1β signalling pathway (IRAK1 and TRAF6) and IL-8 and RANTES secretion (syntaxin-3, synaptotagmin-1 and sec23 interacting protein). The most recent TargetScan database was chosen since, unlike earlier databases, this predicts targets not only based upon pairing between the miRNA seed region and miRNA recognition elements (MRE) within the mRNA 3′ UTR, but also accounts for factors such as the presence and cooperation between multiple MREs, the spacing between MREs, proximity to the stop codon, position within the 3′ UTR and AU composition (8). However, it is important to remember that the information used to develop this algorithm might be inaccurate or biased since it is based upon measurement of mRNA knockdown following miRNA over-expression.
Examination of the TargetScan database showed that the IL-1β signalling proteins, IRAK1 and TRAF6 contain multiple MREs for miRNA-146a and miRNA-146b. This is a significant observation as this is thought to be an important determinant of miRNA targeting (8) and is supported experimentally by Taganov et al. (29) who showed that ectopic over-expression of both miRNA-146a and miRNA-146b leads to down-regulation of IRAK1 and TRAF6. However, our studies of the IL-1β-induced changes in the protein expression showed that neither miRNA-146a nor miRNA-146b likely target IRAK1 or TRAF6. Thus, IL-1β stimulation had no effect upon TRAF6 protein expression and although we observed a reduction in IRAK1 protein expression, this response was unaffected by exposure to inhibitors and mimics of miRNA-146a and miRNA-146b. Instead, as previously reported, IRAK1 reduction is likely to be mediated through proteosomal degradation following IRAK1 phosphorylation (39) and subsequent ubiquitination (40). This phosphorylation and ubiquitination was indicated by the characteristic increase in the molecular weight of IRAK1 at 1-3 h following IL-1β exposure. Additional evidence that neither miRNA-146a nor miRNA-146b acted upon the IL-1β signalling pathway or IL-8 and RANTES transcription was provided by the observation that IL-1β-induced IL-8 and RANTES mRNA expression was unaffected by inhibitors of miRNA-146a and miRNA-146b. Paradoxically, miRNA-146a and miRNA-146b mimics reduced chemokine expression, although we speculate that this might have been the result of the super-maximal miRNA concentrations that occur during these over-expression studies. Thus, although we observed no changes in IRAK1 mRNA expression following IL-1β stimulation, there was a significant reduction following transfection with miRNA-146a and miRNA-146b mimics. These observations at high miRNA concentrations might also explain the reduction in IRAK1 and TRAF6 mRNA expression observed by Taganov et al. (29) following ecoptic expression of miRNA-146a and miRNA-146b and underlines the problems associated with using over-expression systems to determine miRNA targets and function.
Having eliminated a likely action upon the IL-1β pathway and chemokine transcription, we proceeded to investigate whether miRNA-146a targeted IL-8 and RANTES secretion through down-regulation of syntaxin-3, synaptotagmin-1 or sec23-IP, all predicted by TargetScan to contain miRNA-146a MRE's within their 3′ UTRs. An action of miRNAs upon secretion had previously been suggested from studies of glucose-induced insulin secretion, in which miRNA-375 and miRNA-9 had been shown to down-regulate the exocytotic proteins, myotrophin (42) and granulphilin/Slp4 (43), respectively. However, this mechanism also appears unlikely since we observed no changes in syntaxin-3, synaptotagmin-1 and sec23-IP protein expression following IL-1β stimulation. Furthermore, examination of the effect of miRNA inhibitors and mimics upon intracellular IL-8 concentrations produced the opposite response to what might be expected if IL-1β-induced miRNA-146a acted upon secretion i.e. an increase and decrease in intracellular IL-8 concentrations following miRNA-146a inhibition and over-expression, respectively.
If miRNA-146a does not target either proteins involved in IL-1β signalling or chemokine transcription and secretion, what is the mechanism of action? A process of elimination would suggest that the action of miRNA-146a (and miRNA-146b) is mediated at the translational level. The fact that existing algorithms have failed to identify potential MREs within the IL-8 and RANTES 3′-UTRs implies that miRNA-146a must act either directly upon the translational mechanism or targets translational protein(s) that are subject to rapid turnover or induction. Interestingly, the stability and/or translation of mRNA for many inflammatory mediators, including IL-8 and RANTES, are known to be regulated by interaction between AU-rich elements (ARE) within their 3′ UTR and regulatory elements. These include proteins such as HuR which stabilise mRNAs, and members of the Tristetraprolin (TTP) family which promote mRNA decay (44). Significantly, recent studies on the mechanisms that regulate TNF-α mRNA stability and translation have shown that there is an interaction between components of the ARE and the miRNA-mediated RNAi pathway. Thus, two crucial components of the RISC complex, fragile-X-mental-retardation-related protein 1 (FXR1) and argonaute 2 (Ago2), have been shown to bind to the ARE and mediate TNF-α up-regulation following serum starvation (45). Similarly, miRNA-16 has been shown to block TNF-α translation through binding to complimentary sequences in the ARE by a mechanism that involves members of the TTP and Ago/eiF2C family (13). Another ARE regulatory protein, HuR (14) has been shown to reverse miRNA-122-mediated repression of cationic amino acid transporter 1 (CAT-1) mRNA in response to stress. It might therefore be speculated that the rapid increases in miRNA-146a could regulate the production of inflammatory mediators through a direct interaction or down-regulation of components involved in post-transcriptional regulation by the ARE and miRNA mediated RNAi pathways. Of relevance, a recent in silico survey of the interactions between miRNAs and immune genes unexpectedly found preferential targeting of proteins involved in the regulation of AU-rich elements and miRNA metabolism (46). Furthermore, this report indicated that major targets of miRNAs are transcription factors, cofactors and chromatin modifiers rather than receptors, their ligands or inflammatory mediators (46).
In conclusion, we have identified a novel mechanism for the negative feedback regulation of the inflammation following activation of the innate immune response by demonstrating that IL-1β–induced increases in miRNA-146a expression negatively regulates IL-8 and RANTES release. Importantly, this is only observed at high IL-1β concentrations, which indicates that it might be an important feedback mechanism during severe inflammation. Significantly, these results also demonstrate that changes in miRNA expression are able to regulate acute biological responses and suggest that pharmacological targeting of miRNAs might provide a novel therapeutic approach to the treatment of inflammation. Since rapid increases in miRNA-146a have also been reported following the TIR-mediated activation of monocytes and macrophages, this suggests that this might represent a common pathway for the regulation of inflammation following activation of the innate immune response.
Acknowledgments
Grant Support
Sterghios A. Moschos is supported by BBSRC (BB/C508234/1), Hanna M. Larner-Svensson is supported on a National Heart and Lung Institute PhD studentship, Mark M. Perry, Andrew E. Williams and Mark A Lindsay are supported by the Wellcome Trust (076111).
Footnotes
Disclosures
The authors declare no competing interests.
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
REFERENCES
- 1.Braddock M, Quinn A. Targeting IL-1 in inflammatory disease: new opportunities for therapeutic intervention. Nat.Rev.Drug Discov. 2004;3:330–339. doi: 10.1038/nrd1342. [DOI] [PubMed] [Google Scholar]
- 2.Li X, Qin J. Modulation of Toll-interleukin 1 receptor mediated signaling. J.Mol Med. 2005;83:258–266. doi: 10.1007/s00109-004-0622-4. [DOI] [PubMed] [Google Scholar]
- 3.Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev.Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 4.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 5.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat.Rev.Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 6.Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 7.Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17:438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- 9.Saetrom P, Heale BS, Snove O, Jr., Aagaard L, Alluin J, Rossi JJ. Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res. 2007;35:2333–2342. doi: 10.1093/nar/gkm133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long D, Lee R, Williams P, Chan CY, Ambros V, Ding Y. Potent effect of target structure on microRNA function. Nat.Struct.Mol Biol. 2007;14:287–294. doi: 10.1038/nsmb1226. [DOI] [PubMed] [Google Scholar]
- 11.Pillai RS, Bhattacharyya SN, Filipowicz W. Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 2007;17:118–126. doi: 10.1016/j.tcb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 12.Giraldez AJ, Cinalli RM, Glasner ME, Enright AJ, Thomson JM, Baskerville S, Hammond SM, Bartel DP, Schier AF. MicroRNAs regulate brain morphogenesis in zebrafish. Science. 2005;308:833–838. doi: 10.1126/science.1109020. [DOI] [PubMed] [Google Scholar]
- 13.Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120:623–634. doi: 10.1016/j.cell.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-Mediated Translational Repression in Human Cells Subjected to Stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 15.Kiriakidou M, Tan GS, Lamprinaki S, Planell-Saguer M, Nelson PT, Mourelatos Z. An mRNA m7G cap binding-like motif within human Ago2 represses translation. Cell. 2007;129:1141–1151. doi: 10.1016/j.cell.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 16.Thermann R, Hentze MW. Drosophila miR2 induces pseudo-polysomes and inhibits translation initiation. Nature. 2007;447:875–878. doi: 10.1038/nature05878. [DOI] [PubMed] [Google Scholar]
- 17.Chendrimada TP, Finn KJ, Ji X, Baillat D, Gregory RI, Liebhaber SA, Pasquinelli AE, Shiekhattar R. MicroRNA silencing through RISC recruitment of eIF6. Nature. 2007;447:823–828. doi: 10.1038/nature05841. [DOI] [PubMed] [Google Scholar]
- 18.Petersen CP, Bordeleau ME, Pelletier J, Sharp PA. Short RNAs repress translation after initiation in mammalian cells. Mol Cell. 2006;21:533–542. doi: 10.1016/j.molcel.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 19.Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- 20.Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, Subramaniam A, Propp S, Lollo BA, Freier S, Bennett CF, Bhanot S, Monia BP. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 21.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 22.Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat.Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 23.Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, Wang H, Chen G, Wang Z. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat.Med. 2007;13:486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 24.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, Vetrie D, Okkenhaug K, Enright AJ, Dougan G, Turner M, Bradley A. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 27.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, Klein LO, Davis MM, Chen CZ. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 28.O'connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc.Natl.Acad.Sci.U.S.A. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc.Natl.Acad.Sci.U.S.A. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moschos SA, Williams AE, Perry MM, Birrell MA, Belvisi MG, Lindsay MA. Expression profiling in vivo demonstrates rapid changes in lung microRNA levels following lipopolysaccharide-induced inflammation but not in the anti-inflammatory action of glucocorticoids. BMC.Genomics. 2007;8:240. doi: 10.1186/1471-2164-8-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Moschos SA, Jones SW, Perry MM, Williams AE, Erjefalt JS, Turner JJ, Barnes PJ, Sproat BS, Gait MJ, Lindsay MA. Lung Delivery Studies Using siRNA Conjugated to TAT(48-60) and Penetratin Reveal Peptide Induced Reduction in Gene Expression and Induction of Innate Immunity. Bioconjug.Chem. 2007;18:1450–1459. doi: 10.1021/bc070077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Souza PM, Kankaanranta H, Michael A, Barnes PJ, Giembycz MA, Lindsay MA. Caspase-catalyzed cleavage and activation of Mst1 correlates with eosinophil but not neutrophil apoptosis. Blood. 2002;99:3432–3438. doi: 10.1182/blood.v99.9.3432. [DOI] [PubMed] [Google Scholar]
- 34.Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, Mann M, Karin M. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- 35.Takaesu G, Ninomiya-Tsuji J, Kishida S, Li X, Stark GR, Matsumoto K. Interleukin-1 (IL-1) receptor-associated kinase leads to activation of TAK1 by inducing TAB2 translocation in the IL-1 signaling pathway. Mol.Cell Biol. 2001;21:2475–2484. doi: 10.1128/MCB.21.7.2475-2484.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, Sun Y, Koo S, Perera RJ, Jain R, Dean NM, Freier SM, Bennett CF, Lollo B, Griffey R. MicroRNA-143 regulates adipocyte differentiation. J.Biol.Chem. 2004;279:52361–52365. doi: 10.1074/jbc.C400438200. [DOI] [PubMed] [Google Scholar]
- 37.Hutvagner G, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS.Biol. 2004;2:E98. doi: 10.1371/journal.pbio.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA Targeting Specificity in Mammals: Determinants beyond Seed Pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao Z, Henzel WJ, Gao X. IRAK: a kinase associated with the interleukin-1 receptor. Science. 1996;271:1128–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- 40.Yamin TT, Miller DK. The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphorylation. J.Biol.Chem. 1997;272:21540–21547. doi: 10.1074/jbc.272.34.21540. [DOI] [PubMed] [Google Scholar]
- 41.Henriquet C, Gougat C, Combes A, Lazennec G, Mathieu M. Differential regulation of RANTES and IL-8 expression in lung adenocarcinoma cells. Lung Cancer. 2007;56:167–174. doi: 10.1016/j.lungcan.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- 43.Plaisance V, Abderrahmani A, Perret-Menoud V, Jacquemin P, Lemaigre F, Regazzi R. MicroRNA-9 controls the expression of Granuphilin/Slp4 and the secretory response of insulin-producing cells. J.Biol.Chem. 2006;281:26932–26942. doi: 10.1074/jbc.M601225200. [DOI] [PubMed] [Google Scholar]
- 44.Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2006;33:7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vasudevan S, Steitz JA. AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell. 2007;128:1105–1118. doi: 10.1016/j.cell.2007.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asirvatham AJ, Gregorie CJ, Hu Z, Magner WJ, Tomasi TB. MicroRNA targets in immune genes and the Dicer/Argonaute and ARE machinery components. Mol Immunol. 2007 doi: 10.1016/j.molimm.2007.10.035. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]