

Abstract
An improved understanding of the biological activities of heparin requires structurally defined heparin oligosaccharides. The chemoenzymatic synthesis of heparin oligosaccharides relies on glycosyltransferases that use UDP-sugar nucleotides as donors. Uridine 5′-diphosphoiduronic acid (UDP-IdoA) and uridine 5′-diphosphohexenuronic acid (UDP-HexUA) have been synthesized as potential analogues of uridine 5′-diphosphoglucuronic acid (UDP-GlcA) for enzymatic incorporation into heparin oligosaccharides. Non-natural UDP-IdoA and UDP-HexUA were tested as substrates for various glucuronosyltransferases to better understand enzyme specificity.
Introduction
Glycosaminoglycans (GAGs) are an important class of therapeutic agents that engage in specific interactions with a wide variety of proteins in both the intracellular and extracellular environment.1 Heparin, the most studied GAG, is widely used as an injectable anticoagulant and has the highest negative charge density of any known biological molecule.2 Pharmaceutical grade heparin is commonly derived from mucosal tissues of slaughtered meat animals, such as porcine intestine or bovine lung, and is a polydisperse, polycomponent, and polypharmacologic agent.3 Low molecular weight heparins (LMWHs) are rapidly replacing heparin as the clinical anticoagulant of choice. However, these LMWHs are derived from heparin and are also polydisperse mixtures, making their controlled preparation and analysis difficult. Thus, structurally defined heparin oligosaccharides are still of great interest as potential anticoagulant agents. Fondaparinux (Arixtra, GSK) and Idraparinux (Sanofi-Aventis), homogeneous pentasaccharide analogues of the ATIII binding pentasaccharide, have been chemically synthesized and introduced into the marketplace. These drugs can reduce the side effects associated with the use of heparin, particularly the risk for heparin-induced thrombocytopenia (HIT). Unfortunately, the chemical synthesis of Fondaparinux is both complicated and costly.4 Therefore, the development of new strategies to enable the efficient, high-yield, and stereocontrolled synthesis of homogeneous heparin-like oligosaccharides is of major importance.
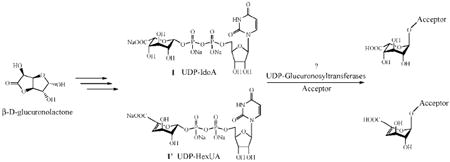
Heparin is composed of alternating (1→4) linked α-l-IdoA or β-d-GlcA and β-d-GlcN units, which are diversely sulfonated. The uronic acid portion of heparin consists of approximately 80–90% of α-l-IdoA units and only 10–20% of β-d-GlcA units. The biosynthesis of heparin involves glycosyltransferases that successively add a N-acetylglucosamine unit (GlcNAc) from UDP-GlcNAc and a glucuronic acid unit (GlcA) from UDP-GlcA. The GlcA residues of the polysaccharide precursor are subsequently C-5 epimerized to form iduronic acid (IdoA) through the action of a C-5 epimerase. C-5 epimerases involved in heparin biosynthesis act on GlcA residues flanked by N-sulfoglucosamine residues.5 This enzyme specificity reduces access to well-defined, unnatural structures. Using the completely controlled, glucuronosyltransferase-based, introduction of IdoA residues might be possible using the unnatural analogue UDP-IdoA. The glucuronosyltransferase-based transfer of UDP-IdoA would allow access to many natural and unnatural sequences.
The availability of well-defined heparin oligosaccharides should provide an improved understanding of the important interactions that occur between heparin and proteins. Synthetic chemists have developed a variety of synthetic methodologies for accessing GAG oligosaccharides.6 However, while these multistep chemical strategies display elegant chemistry, they result in products that are simply too expensive. The chemoenzymatic synthesis of heparin oligosaccharides represents an interesting alternative to the complex chemical syntheses and purification processes. Indeed, enzymatic and chemoenzymatic strategies have been developed in the past few years and proven to be efficient, relying on the unique capacities of enzymes (glycosyltransferases, sulfotransferases, epimerases, etc.) to catalyze regiospecific and stereospecific transformations.7
During the past decade, several unnatural UDP-hexosamines have been synthesized and used as substrates or inhibitors of hexosamine glycosyltransferases.8 UDP-N-trifluoroacetylglucosamine (UDP-GlcNTFA), for example, is accepted as a substrate in place of UDP-GlcNAc by the “core-2” GlcNAc transferase (EC 2.4.1.102) in the biosynthesis of O-linked glycoproteins, and by the GlcNAcT-V transferase (EC 2.4.1.155), a key biosynthetic enzyme controlling the branching pattern of cell surface complex Asn-linked oligosaccharides.9 However, unnatural analogues of UDP-GlcA have not been investigated as potential substrates of glucuronosyltransferases.
In this work, we synthesize UDP-IdoA 1, a potential unnatural substrate of glucuronosyltransferases, which would allow the direct and controlled enzymatic introduction of IdoA units into a growing oligosaccharide. This unnatural substrate might also be useful for the study of the specificity of various glucuronosyltransferases.
Results and Discussion
Several strategies have been developed for the synthesis of UDP-sugar nucleotides.10 Among these, the Khorana–Moffatt procedure,11 involving the coupling of a sugar monophosphate with uridine 5′-monophosphomorpholidate 4-morpholine-N,N′-dicyclohexylcarboxamidine salt (UMP-morpholidate), is still the preferred route. The retrosynthesis of UDP-IdoA is shown in Scheme 1.
Scheme 1.
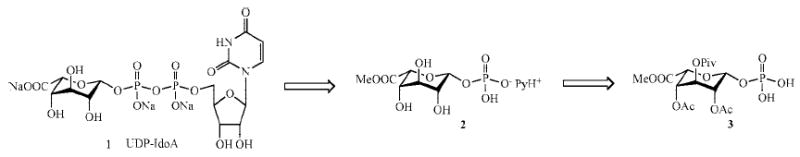
Retrosynthesis of UDP-IdoA
l-IdoA is a rare sugar and is not commercially available. The efficient synthesis of l-IdoA has been a major concern for researchers synthesizing heparin oligosaccharides, and thus a wide variety of strategies have been developed for its preparation. Inversion by nucleophilic substitution at C-5 starting from 3-O-benzyl-1,2-isopropylidene-α-d-glucofuranose12 or d-glucurono-6,3-lactone,13 radical reduction of 5-bromouronate,14 functionalization of Δ4-uronic acid species,15 stereoselective addition on d-xylo-dialose,16 epimerization of d-GlcA derivatives,17 and diastereoselective hydroboration of exoglucals18 have shown good results. Compound 4 (Scheme 2) was first synthesized as described by Ke et al. in 31% overall yield over 5 steps from the inexpensive d-glucurono-6,3-lactone.13
Scheme 2.

Use of a Sulfide Iduronic Acid Type Donor
We initially attempted to prepare the monophosphate 3 through the activation of the anomeric position of compound 4 as a phenyl sulfide.19 According to a procedure developed by Bonnaffé,20 peracetylation of compound 4 was performed in the presence of acetyl chloride, pyridine, and a catalytic amount of dimethylaminopyridine (DMAP) in DCM at −40 °C to limit the potential isomerization of the pyranose ring to the corresponding furanose ring that is known to occur in the presence of pyridine/acetic anhydride. Iduronate donor 5 was then treated with thiophenol in the presence of BF3 · Et2O. The corresponding sulfide 6 was obtained in a modest yield of 37% as a 1/3 α/β mixture after purification on silica gel, but 52% of the starting material could also be recovered and reused. Glycosylation of 6 with dibenzylphosphoric acid in the presence of N-iodosuccinimide (NIS) gave the desired 7 in 91% yield (α/β = 1/4). The β-anomer was isolated by flash chromatography in 70% yield and subjected to hydrogenolysis. During the hydrogenolysis reaction, a β-elimination of the C-4 acetate group was observed and compound 3′ was formed as the major product. Hydrogenolysis in the presence of 2 equiv of NaHCO3 also afforded compound 3′ as the major product. β-Elimination can be acid-catalyzed or base-catalyzed, and thus neither of these reaction conditions were suitable. When we tried to first remove the ester protecting groups using Zemplen conditions, the corresponding methyl glycoside 8 was obtained.
Monophosphate 3 was next approached, using the procedure developed by MacDonald in 1962,21 which involves the use of an anomeric acetate and crystalline phosphoric acid at 60 °C under high vacuum (Scheme 3).
Scheme 3.
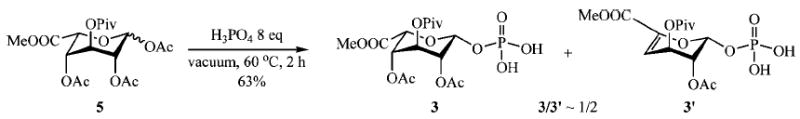
MacDonald’s Phosphorylation of Iduronic Acid Type Donor 5
Iduronate donor 5 was treated with an excess of crystalline phosphoric acid. This strategy gave a mixture of the desired monophosphate 3 and the elimination product 3′ in 1:2 ratio. From a mechanistic point of view, we rationalized the formation of 3′ by a protonation of the C-4 acetyl group by H3PO4 followed by a β-elimination, which is favored in a 1,2-trans axial position, leading to the formation of 3′ and acetic acid. This mechanism is supported by the fact that phosphorylation of methyl 1,2,3,4-tetra-O-acetyl-β-d-glucuronate using the same conditions affords no traces of 3′. Compounds 3 and 3′ were separated on silica gel affording monophosphate 3 as the β-anomer in 21% yield.
In an attempt to increase the yield of the phosphorylation step, another strategy was considered, in which monophosphate 3 would be obtained in two steps from 5 through the activation of the anomeric position as a bromide (Scheme 4). The bromide donor 9 was obtained as the α-anomer 9-α in 95% purity by 1H NMR (see the Supporting Information) and in 89% yield. The desired monophosphate 3 was obtained from the bromide donor 9 in 41% yield by treatment with the tetrabutylammonium salt of phosphoric acid. The reaction was stereospecific and only the β-anomer of 3 was identified (see the Supporting Information, 1H NMR J1,P = 7.5 Hz and J1,2 = 2.0 Hz). Traces of the eliminated monophosphate 3′ could also be observed. Treatment of compound 3 with a catalytic amount of sodium methoxide in methanol afforded the unprotected monophosphate 2, which was converted into its monopyridinium salt by treatment with Amberlite resin (pyridinium form). The methyl ester was kept intact to avoid problems during the coupling reaction. The monophosphate 2 was then reacted with UMP-morpholidate according to the procedure of Khorana and Moffatt. The resulting mixture was then treated with a 2:2:1 mixture of MeOH/H2O/Et3N to remove the methyl ester. Purification was accomplished with a BioGel P2 with 0.25 M NH4HCO3 as eluent, followed by desalting on BioGel P2 with water as eluent. The fractions containing UDP-IdoA were lyophilized and further purified by using a semipreparative SAX-HPLC column (linear gradient from 0 to 1 M NaCl over 50 min.) required for the removal of the uridine diphosphate dimer formed by self-condensation of UMP-morpholidate, which is a classical byproduct of the coupling reaction. After desalting on BioGel P2, the UDP-IdoA 1 target was obtained in 26% yield over 2 steps.
Scheme 4.

Synthesis of UDP-IdoA Using a Bromide Donor
Using the same strategy, the unsaturated analogue of UDP-GlcA and UDP-IdoA, the UDP-HexUA 1′, was obtained in 31% yield over 3 steps from 3′ (Scheme 5).
Scheme 5.

Synthesis of UDP-Hexenuronic Acid 1′
Enzymatic Assays
These unnatural analogues of UDP-GlcA have been tested as substrates for glycosyltransferases isolated from Pasteurella multocida (PmHS1 and PmHS2).7f UDP-IdoA could ultimately serve as a substrate for the direct and specific incorporation of IdoA units in a growing oligosaccharide to enzymatically build the heparin backbone. UDP-HexUA could serve as a potential chain terminator for the preparation of polymers having a defined molecular weight range.
Recently, Sismey-Ragatz et al. reported the enzyme-catalyzed incorporation of unnatural UDP-glucosamine derivatives to different heparosan type acceptors using the Pasteurella multocida heparosan synthases PmHS1 and PmHS2.7f These enzymes both possess two active sites, a glucosaminyltransferase site that transfers UDP-GlcNAc and a glucuronosyltransferase site that transfers UDP-GlcA to an oligosaccharide acceptor. Neither of these glycosyltransferases were able to transfer UDP-HexUA. To build polymers having the backbone of heparin (GlcN-IdoA), UDP-IdoA was used in combination with UDP-GlcNAc, in the presence of PmHS1 or PmHS2, and A-F-AN as acceptor.7f Radiochemical assays suggested that both PmHS1 and PmHS2 coincorporated UDP-IdoA and UDP-GlcNAc, albeit much lower efficiency than with UDP-GlcA (~4% rate of authentic UDP-GlcA). Gel analysis of the PmHS2 polymerization reactions without acceptor demonstrated small amounts of short polymers were generated by using the UDP-IdoA preparation (Figure 1). UDP-GlcA yielded a much greater amount of larger polymers. In another set of experiments, we tested A-F-AN as acceptor and either UDP-GlcA or UDP-IdoA as donor in combination with PmHS2 and monitored the formation of the corresponding tetrasaccharide A-F-AN-UA. MALDI-ToF mass spectrometry allowed the detection of trace amounts of tetrasaccharide with the expected mass when UDP-IdoA was tested (Figure 1, A-F-AN-GlcA observed 1106.55 Da when using UDP-GlcA versus 1106.12 Da when using UDP-IdoA; the mock reaction did not possess this same peak).
Figure 1.

Analysis of GAG polymers by polyacrylamide gel electrophoresis and analysis of the A-F-AN-UA tetrasaccharide by MALDI-ToF. PmHS2 was used in three polymerization reactions containing UDP-GlcNAc and either UDP-GlcA (G), no second sugar (0), or the UDP-IdoA preparation (I). Large molecular mass polymers were formed in abundance with the authentic precursor, but the UDP-IdoA analogue resulted in smaller amounts of short chains (bracketed). The positions of hyaluronan standards are shown in sugar units (18-mer = ~1.6 kDa; 40-mer = ~8 kDa) (note: the light staining band marked with an asterisk (*) is the enzyme). MALDI-ToF spectra: top spectrum = A-F-AN + UDP-GlcA, bottom spectrum = A-F-AN + UDP-IdoA.
While a small portion of the UDP-IdoA incorporated, it remains unclear whether the low levels of incorporation are due to (a) either UDP-IdoA or its elongation products inhibiting the synthase, or (b) the presence of trace UDP-GlcA contaminant, or (c) the formation of UDP-GlcA from UDP-IdoA through epimerization during synthase catalysis.
3H-labeled trisaccharide (GlcN[3H]Ac-GlcA-AnMan) was used as acceptor in the presence of PmHS1 or PmHS2 to help identify the uronic acid residue that is transferred when using the UDP-IdoA preparation (Figure 2), and the reactions were monitored by using PAMN-HPLC.22 3H-labeled trisaccharide was synthesized from a disaccharide (GlcUA-AnMan) by using E. coli derived KfiA and UDP-GlcNAc[3H], where AnMan represents 2,5-anhydromannitol.22
Figure 2.

PAMN-HPLC profiles of PmHS2 modified 3H-labeled trisaccharide. A 3H-labeled trisaccharide with a structure of (GlcN[3H]Ac-GlcUA-AnMan) was incubated with PmHS2 and UDP-GlcA or UDP-IdoA. The product was analyzed by PAMN-HPLC which was eluted with a linear gradient of KH2PO4. Panel A shows the profile of unmodified 3H-trisaccharide. Panel B shows the profile of 3H-trisaccharide modified with PmHS2 in the presence of UDP-GlcA for 2 h. Panel C shows the profile of 3H-labeled trisaccharide modified with PmHS2 in the presence of UDP-IdoA for 2 h. Panel D shows the profile of 3H-labeled trisaccharide modified with PmHS1 in the presence of UDP-IdoA overnight.
When UDP-GlcA was used (positive control), the desired tetrasaccharide was obtained with a very good selectivity. When UDP-IdoA was used as substrate, the conversion was very low, but a small peak corresponding to the tetrasaccharide was obtained. These products were treated with glucuronidase and iduronidase to identify the nature of the uronic acid unit incorporated.23 Both tetrasaccharide products were glucuronidase-sensitive but were iduronidase-insensitive. Thus, the UDP-IdoA could not be incorporated as IdoA into oligosaccharides with use of PmHS1 or PmHS2. While trace amounts of UDP-GlcA contaminant could be present in the synthetic preparation, it could not be detected with NMR or SAX-HPLC. Alternatively, UDP-GlcA might be formed from UDP-IdoA through unexpected and hitherto never previously observed C-5 epimerization during synthase catalysis. A wider library of UDP-GlcA analogues is needed to understand the specificity of these glycosyltransferases. The current study constitutes a preliminary study. Furthermore, it may be possible to incorporate IdoA units in a growing oligosaccharide from UDP-IdoA by modifying the glycosyltransferase to accept such an unnatural substrate. Work is underway to generate a library of glycosyltransferase mutants to test their ability to utilize UDP-IdoA to build IdoA containing oligosaccharides.
In conclusion, two new unnatural analogues of UDP-GlcA, UDP-IdoA 1 and UDP-HexUA 1′, were synthesized and used to study the specificity of different glucuronosyltransferases. UDP-HexUA did not serve as a substrate for the glucuronosyltransferases tested. When UDP-IdoA was used as the substrate, sugar residues were transferred but only GlcA was incorporated into the products formed. This result could be explained by the contamination of synthetic UDP-IdoA with a small amount of UDP-GlcA. Alternatively, UDP-IdoA might be isomerized to UDP-GlcA by the synthase upon transfer by a not known yet understood enzymatic process.
Experimental Section
Methyl 1,2,4-Tri-O-acetyl-3-O-pivaloyl-β-l-idopyranuronate (5)
Methyl 3-O-pivaloyl-l-iduronate (4) (3.1 g, 10.6 mmol) was suspended in anhydrous CH2Cl2 (56 mL) under argon atmosphere at −40 °C (acetone/dry ice) and 2,4-dimethylaminopyridine (130 mg, 1.06 mmol), pyridine (8.66 mL, 106 mmol), and acetyl chloride (4.54 mL, 63.7 mmol) were added. After being stirred at this temperature for 10 h, the mixture was diluted with CH2Cl2 (150 mL) and the resulting organic phase was washed with saturated NaHCO3, water, 1 M HCl, and water. The organic layer was dried over Na2SO4, filtered, and concentrated under diminished pressure. After purification on silica gel with use of a gradient of petroleum ether/ethyl acetate (85/15 to 50/50), methyl 1,2,4-tri-O-acetyl-3-O-pivaloyl-β-l-idopyranuronate (5) (2.88 g, 65%) was obtained. An additional mixture of 5α/5β (302 mg, 7%) was also recovered (combined yield of 5α/5β: 72%). 1H NMR (500 MHz, CDCl3) δ (ppm) 6.03 (1H, d, J = 2.0 Hz, H-1), 5.32 (1H, t, J = 4.1 Hz, H-3), 5.10 (1H, m, H-4), 5.01 (1H, ddd, J = 4.1 Hz, J = 2.0 Hz, J = 0.7 Hz, H-2), 4.68 (1H, d, J = 2.7 Hz, H-5), 3.79 (3H, s, CO2Me), 2.14, 2.12, 2.09 (9H, 3 s, 3 OAc), 1.26 (9H, s, Piv). 13C NMR (125 MHz, CDCl3) δ (ppm) 175.7, 169.4, 169.2, 168.5, 166.8 (Piv, 3 OAc and CO2Me), 89.9 (C-1), 73.2 (C-5), 66.8 (C-3), 66.5 (C-2), 65.6 (C-4), 52.7 (CO2Me), 38.9 (quaternary C of Piv), 27.0 (3 Me of Piv), 20.7, 20.6, 20.5 (3 Me of 3 OAc). HRMS m/z calcd for C18H26O11Na [M + Na]+ 441.1367, found 441.1369.
Methyl 1-Phenylthio-2,4-di-O-acetyl-3-O-pivaloyl-l-idopyranuronate (6)
Methyl 1,2,4-tri-O-acetyl-3-O-pivaloyl-β-L-idopyranuronate (5) (1.0 g, 2.39 mmol) was suspended in anhydrous CH2Cl2 (5 mL) under argon atmosphere at room temperature and freshly activated MS 4Å (200 mg), thiophenol (0.3 mL, 2.91 mmol), and BF3 · Et2O (0.45 mL, 3.59 mmol) were added. After being stirred at this temperature for 1 day, the mixture was diluted with CH2Cl2 (150 mL) and the resulting organic phase was washed with cold water then saturated NaHCO3. The organic layer was dried over Na2SO4, filtered, and concentrated under diminished pressure. After purification on silica gel with a gradient of petroleum ether/ethyl acetate (85/15 to 50/50), methyl 1-phenylthio-2,4-di-O-acetyl-3-O-pivaloyl-l-idopyranuronate (6) (413 mg, α/β = 1/3, 37%) was obtained. Further elution allowed recovery of the starting material 5 (520 mg, 52%) that could be reused.
6α: 1H NMR (500 MHz, CDCl3) δ (ppm) 7.60–7.58 (2H, m, HAr), 7.33–7.31 (3H, m, HAr), 5.13 (1H, t, H-4), 5.08–5.04 (3H, m, H-2, H-3, H-5), 4.46 (1H, d, J = 1.8 Hz, H-1), 3.78 (3H, s, CO2Me), 2.16, 2.09 (6H, 2 s, 2 OAc), 1.21 (9H, s, OPiv). 13C NMR (125 MHz, CDCl3) δ (ppm) 175.1, 169.2, 169.1, 167.1 (Piv, 2 OAc and CO2Me), 133.4 (C-1′), 132.1 (2C-2′), 129.1 (2C-3′), 128.1 (C-4′), 85.1 (C-1), 74.6 (C-5), 67.6 (C-3), 66.1 (C-2), 65.6 (C-4), 52.6 (CO2Me), 38.7 (quaternary C of Piv), 27.0 (3 Me of Piv), 20.6, 20.5 (2 Me of 2 OAc). HRMS m/z calcd for C22H28O9SNa [M + Na]+ 491.1346, found 491.1353.
6β: 1H NMR (500 MHz, CDCl3) δ (ppm) 7.51–7.49 (2H, m, HAr), 7.32–7.27 (3H, m, HAr), 5.68 (1H, d, J = 2.6 Hz, H-1), 5.22 (1H, d, H-5), 5.18 (1H, t, H-3), 5.13, (1H, t, H-2), 4.99 (1H, t, H-4), 3.80 (3H, s, CO2Me), 2.09, 2.08 (6H, 2 s, 2 OAc), 1.30 (9H, s, OPiv). 13C NMR (125 MHz, CDCl3) δ (ppm) 176.1, 169.3, 169.2, 168.2 (Piv, 2 OAc and CO2Me), 134.0 (C-1′), 131.4 (2C-2′), 129.1 (2C-3′), 127.8 (C-4′), 85.5 (C-1), 68.5 (C-5), 67.7 (C-3), 66.9 (C-2), 66.2 (C-4), 52.6 (CO2Me), 38.9 (quaternary C of Piv), 27.0 (3 Me of Piv), 21.0, 20.8 (2 Me of 2 OAc). HRMS m/z calcd for C22H28O9SK [M + K]+ 507.1086, found 507.1087.
Methyl 1-Dibenzylphospho-2,4-di-O-acetyl-3-O-pivaloyl-l-idopyranuronate (7)
Methyl 1-phenylthio-2,4-di-O-acetyl-3-O-pivaloyl-l-idopyranuronate (6) (47 mg, 100 μmol), dibenzyl phosphoric acid (57 mg, 200 μmol), and freshly activated MS 4Å (60 mg) were suspended in anhydrous dichloroethane (1 mL) at room temperature. N-Iodosuccinimide (45 mg, 200 μmol) was added and the mixture was stirred overnight. The molecular sieves was then filtered off and washed with CH2Cl2. The resulting organic phase was washed with saturated NaHCO3, water, saturated Na2S2O3, then water. The organic layer was dried over Na2SO4, filtered, and concentrated under diminished pressure. After purification on silica gel with a gradient of petroleum ether/ethyl acetate (7/3 to 1/1), methyl 1-dibenzylphospho-2,4-di-O-acetyl-3-O-pivaloyl-β-l-idopyranuronate (7β) (45 mg, 73%) and methyl 1-dibenzylphospho-2,4-di-O-acetyl-3-O-pivaloyl-α-l-idopyranuronate (7α) (12 mg, 18%) were obtained. 7β: 1H NMR (500 MHz, CDCl3) δ (ppm) 7.37–7.28 (10H, HAr), 5.84 (1H, dd, J1,P = 6.4 Hz, J1,2 = 0.6 Hz, H-1), 5.15–5.00 (6H, m, H-3, H-4, 2CH2 of OBn), 4.89 (1H, d, J = 2.4 Hz, H-5), 4.84 (1H, m, H-2), 3.70 (3H, s, CO2Me), 2.08, 2.07 (6H, 2 s, 2 OAc), 1.17 (9H, s, Piv). 13C NMR (125 MHz, CDCl3) δ (ppm) 177.3 (d, JC,P = 6.6 Hz, Piv), 169.2 (d, JC,P = 2.6 Hz, OAc), 168.9 (d, JC,P = 2.4 Hz, OAc), 167.3 (CO2Me), 135.3, 135.2 (2d, JC,P = 7.3 Hz and JC,P = 7.3 Hz, 2 quaternary Ar), 128.6, 128.5, 128.4, 128.0, 127.8 (10 CHAr), 94.9 (d, JC,P = 5.6 Hz, C-1), 69.8 (d, JC,P = 5.2 Hz, C-5), 69.5 (d, JC,P = 5.2 Hz, C-3), 67.5 (d, JC,P = 3.9 Hz, C-2), 66.4 (d, JC,P = 2.5 Hz, C-4), 65.9, 65.4 (2d, JC,P = 11.0 Hz and JC,P = 3.7 Hz, 2CH2 OBn), 52.6 (d, JC,P = 2.9 Hz, CO2Me), 38.7 (quaternary C of Piv), 26.8 (d, JC,P = 5.7 Hz, 3 Me of Piv), 20.6, 20.5 (2d, JC,P = 2.8 Hz and JC,P = 2.7 Hz, 2 OAc). HRMS m/z calcd for C30H37O13PNa [M + Na]+ 659.1864, found 659.1860.
Methyl 1-Phospho-2,4-di-O-acetyl-3-O-pivaloyl-β-l-idopyranuronate (3)
Phosphoric acid (30 mg, 0.3 mmol) was treated with a 40 wt % aqueous solution of tetrabutylammonium hydroxide (0.6 mL, 0.9 mmol) at 0 °C in an ice bath. After 10 min the mixture was freeze-dried to afford the teatrabutylammonium phosphate (246 mg, 0.3 mmol). Methyl 1-bromo-2,4-di-O-acetyl-3-O-pivaloyl-β-l-idopyranuronate (9) (101 mg, 0.23 mmol) dissolved in acetonitrile (7 mL), triethylamine (32 μL, 0.23 mmol) dissolved in acetonitrile (4 mL), and molecular sieves 3Å (250 mg) were added. This mixture was heated in refluxing acetonitrile (80 °C) for 30 min. After the mixture had cooled to rt, the molecular sieves was filtered off and washed with acetonitrile. The filtrate was evaporated under diminished pressure to afford a brown oil. After purification on silica gel with a gradient of EtOAc/MeOH (1/0 to 1/1), methyl 1-phospho-2,4-di-O-acetyl-3-O-pivaloyl-β-l-idopyranuronate (3) was obtained. To facilitate NMR assignment, it was then treated with Amberlite (NH4+ form) in methanol to obtain the corresponding ammonium salt of 3 (46 mg, 41%). 1H NMR (500 MHz, MeOD) δ (ppm) 5.66 (1H, dd, J1,P = 7.5 Hz, J1,2 = 2.0 Hz, H-1), 5.14–5.05 (4H, m, H-2, H-3, H-4, H-5), 3.76 (3H, s, CO2Me), 2.09 and 2.04 (6H, 2 s, 2 OAc), 1.25 (9H, s, Piv). 13C NMR (125 MHz, MeOD) δ (ppm) 178.2, 171.0, 170.8, 170.3 (Piv, 2 OAc and CO2Me), 95.1 (d, J1,P = 4.9 Hz, C-1), 69.1 (d, J2,P = 10.3 Hz, C-2), 69.0 (C-4), 68.9 (C-3), 68.2 (C-5), 52.9 (CO2Me), 39.9 (quaternary C of Piv), 27.4 (3 Me of Piv), 20.7, 20.5 (2 Me of 2 OAc). HRMS m/z calcd for C16H24O13P [M – H]− 455.0960, found 455.0956.
Methyl 1-Bromo-2,4-di-O-acetyl-3-O-pivaloyl-α-l-idopyranuronate (9)
Methyl 1,2,4-tri-O-acetyl-3-O-pivaloyl-β-l-idopyranuronate (5) (120 mg, 0.287 mmol) was suspended in anhydrous CH2Cl2 (1 mL) at 0 °C and water (31 μL, 1.72 mmol) followed by PBr3 (46 μL, 0.49 mmol) were added. After being stirred at this temperature for 10 min, the mixture was warmed to rt and vigorously stirred for 3 h. The resulting mixture was then diluted with CH2Cl2 (15 mL) and washed with water and saturated NaHCO3 solution. The organic layer was dried over Na2SO4, filtered, and concentrated under diminished pressure to give the methyl 1-bromo-2,4-di-O-acetyl-3-O-pivaloyl-α-l-idopyranuronate (9α) (112 mg, 89%). This compound was used in the next step without further purification after 1H NMR showed purity higher than 95%. 1H NMR (500 MHz, CDCl3) δ (ppm) 6.46 (1H, br s, H-1), 5.19 (1H, br s, H-4), 5.08–5.06 (2H, m, H-3 and H-2), 4.97 (1H, d, J = 1.7 Hz, H-5), 3.82 (3H, s, CO2Me), 2.12, 2.10 (6H, 2 s, 2 OAc), 1.30 (9H, s, Piv).
Uridine 5′-Diphosphoiduronic Acid (UDP-IdoA) (1)
Methyl 1-phospho-2,4-di-O-acetyl-3-O-pivaloyl-α-l-idopyranuronate (3) (18 mg, 36 μmol) was dissolved in anhydrous MeOH (1 mL), cooled to 0 °C, and treated with a 0.5 M solution of MeONa in MeOH (0.5 mL). After 3 h at rt, the solution was cooled to 0 °C and Amberlite (H+ form, 250 mg) was added. After filtration, MeOH was evaporated and the resulting mixture was redissolved in water before treatment with Amberlite (PyH+ form). The corresponding monopyridinium salt of 2 (14 mg) was obtained after freeze-drying. The monopyridinium salt of methyl 1-phospho-α-l-idopyranuronate (2) (14 mg, 36 μmol) was dissolved in anhydrous pyridine (1 mL) and evaporated under diminished pressure. This operation was repeated 3 times. A solution of 4-morpholine N,N′-dicyclohexylcarboxamidinium uridine 5′-phosphomorpholidate (34 mg, 49 μmol) in anhydrous pyridine (2 mL) was concentrated to dryness in vacuo. This operation was also repeated 3 times. A solution of the 4-morpholine N,N′-dicyclohexylcarboxamidinium uridine 5′-phosphomorpholidate in anhydrous pyridine (1 mL) was then added to the dry monopyridinium salt of methyl 1-phospho-α-l-idopyranuronate (6) followed by concentration in vacuo. The mixture was then redissolved in anhydrous pyridine (800 μL) and stirred at rt for 5 days under argon atmosphere. Pyridine was then evaporated under reduced pressure and the mixture was treated with MeOH/H2O/Et3N 2:2:1 (1.5 mL) for 24 h at RT, in order to deprotect the methyl ester. The mixture was then concentrated under reduced pressure and redissolved in water. Purification was achieved on a P2 BioGel column with a 0.25 M NH4HCO3 solution as eluent. The fractions containing UDP-IdoA (UV at 262 nm) were pooled and lyophilized. Further purification was achieved with use of a semipreparative SAX column (20 × 250 mm), using a linear gradient of NaCl (0 to 1 M over 50 min) at pH 3.5. After desalting on P2 BioGel, UDP-IdoA 1 (6 mg, 9.3 μmol, 26%) was obtained as a white powder. 1H NMR (500 MHz, D2O) δ (ppm) 7.86 (1H, d, J = 8.1 Hz, H-6), 5.95–5.85 (2H, m, H-1′ and H-5), 5.46 (1H, dd, J1″,P = 7.9 Hz, J1,2 = 2.9 Hz, H-1″), 4.55 (1H, d, J = 2.8 Hz, H-5″), 4.30–4.10 (5H, m, 2H-5′, H-4′, H-3′, H-2′), 3.92 (1H, dd, J = 3.9 Hz, J = 2.8 Hz, H-4″), 3.79 (1H, t, J = 4.8 Hz, H-3″), 3.69 (1H, dd, J = 5.1 Hz, J = 2.9 Hz, H-2″). 13C NMR (125 MHz, D2O) δ (ppm) 175.9 (C-6″), 166.3 (d, J = 22.1 Hz, C-4), 151.9 (d, J = 21.9 Hz, C-2), 141.6 (C-6), 102.7 (d, J = 10.1 Hz, C-5), 97.2 (d, J = 6.5 Hz, C-1″), 88.2 (d, J = 6.3 Hz, C-1′), 83.4 (d, J = 9.3 Hz, C-4′), 83.3 (d, J = 9.0 Hz, C-5″), 73.8 (d, J = 21.1 Hz, C-3′), 70.6 (d, J = 7.1 Hz, C-3″), 70.3 (C-4″), 69.7 (C-2″), 66.0 (d, J = 6.1 Hz, C-2′), 64.9 (d, J = 5.8 Hz, C-5′).31P NMR (202.34 MHz, D2O) δ (ppm) − 10.2 (d, J = 20 Hz), −12.2 (d, J = 20 Hz). HRMS m/z, calcd for C15H21N2O18P2 [M – H]− 579.0270, found 579.0255.
Uridine 5′-Diphosphohex-4,5-enuronic Acid (1′)
Following the same procedure as described above, uridine 5′-diphosphohex-4,5-enuronic acid (1′) was obtained (10 mg, 31%). 1H NMR (500 MHz, D2O) δ (ppm) 7.87 (1H, d, J = 8.1 Hz, H-6), 5.93 (1H, d, J = 4.5 Hz, H-1′), 5.90 (1H, d, J = 8.1 Hz, H-5), 5.82 (1H, d, J = 3.9 Hz, H-4″), 5.60 (1H, dd, J1″,P = 8.4 Hz, J 1,2 = 5.3 Hz, H-1″), 4.30 (1H, m, H-2′), 4.21 (1H, m, H-3′), 4.16 (1H, m, H-4′), 4.13–4.10 (2H, m, H-3″, H-5′), 3.84 (1H, t, J = 5.0 Hz, H-2″). 13C NMR (125 MHz, D2O) δ (ppm) 168.9 (C-6″), 166.63 and 166.56 (C-2 and C-4), 144.6 (C-5″), 141.5 (C-6), 107.3 (C-4″), 102.7 (C-5), 95.3 (C-1″), 88.2 (C-1′), 83.4 (C-3′), 83.3 (C-4′), 70.0 (C-2″), 69.7 (C-2′), 66.0 (C-5′), 65.1 (C-3″). 31P NMR (202.34 MHz, D2O) δ (ppm) −9.98 (d, J = 20 Hz), −11.95 (d, J = 20 Hz). HRMS m/z calcd for C15H19N2O17P2 [M – H]− 561.0164, found 561.0158.
Enzymatic Assays. Polymerization Reactions with Pasteurella multocida Heparosan Synthases (PmHS1 and PmHS2)
UDP-sugars were dissolved at 15 mM in 10 mM Tris, pH 7.2, buffer for storage as aliquots at −80 °C. Maltose binding protein fusions of PmHS1 or PmHS2, two isozymes of heparosan synthase, were employed as the catalysts.7f Sugar addition reactions typically contained 50 mM Tris, pH 7.2, 1 mM MnCl2, PmHS1 or PmHS2, various amounts of UDP-GlcNAc, and either (a) UDP-GlcA (the authentic uronic acid) or (b) UDP-IdoA as noted. To measure relative incorporation rates of the two UDP-uronic acids (1 mM), radioactive UDP-[3H]GlcNAc (0.27 μM; 0.2 μCi/reaction) addition into polymer by enzyme (12–15 μg; 24 h at 30 °C) was monitored by paper chromatography.7f For another proof of polymerization, reactions with nonradioactive UDP-sugars (10–25 mM each) and enzyme (46 μg; 22 h at 30 °C) were analyzed on polyacrylamide gels with combined Alcian Blue/Silver staining.7f Single sugar addition reactions with A-F-AN, a synthetic acceptor (0.5–1 mM), and synthase (6–15 μg; 18–60 h at 22–30 °C) were used under similar conditions as polymerization assays, but only a single UDP-uronic acid (0.75–1.5 mM) was employed. Products were analyzed by MALDI-ToF mass spectrometry with ATT matrix in negative ion mode.7f
Enzymatic Synthesis of a Tetrasaccharide
PmHS2 (50 μg) was incubated with trisaccharide (50,000 cpm) in a buffer containing 25 mM Tris (pH 7.5), 10 mM MnCl2, 10 mM MgCl2, and 1 μM UDP-GlcUA or UDP-IdoUA at 37 °C for 2 h or overnight. The presence of anticipated 3H-labeled tetrasaccharide (GlcUA-GlcN[3H]Ac-GlcUA-AnMan) was determined by determining the retention time of the 3H-peak on polyamine-based anion exchange HPLC.22 Briefly, the polyamine II column (YMC) was equilibrated with ddH2O for 10 min, and then eluted with a linear gradient of 0 to 1 M KH2PO4 in 60 min at 0.5 mL/min. 3H-labeled trisaccharide and tetrasaccharide were respectively eluted at around 33 and 83 mM of KH2PO4.
Glucuronidase/Iduronidase Treatment of the Tetrasaccharide
The resulting 3H-labeled tetrasaccharide was purified by PAMN-HPLC and desalted by dialyzing against water, using MWCO 1000 membrane. The 3H-labeled tetrasaccharide was digested with either β-glucuronidase or α-iduronidase as described previously.23 The digested samples were reanalyzed by PAMN-HPLC to determine if the tetrasaccharide was susceptible to the digestion with either β-glucuronidase or α-iduronidase.
Supplemental Material
Acknowledgments
This work was supported by the National Institute of Health (NIH HL062244 to R.J.L.). The authors would like to thank Nigel J. Otto for the preparation of the heparosan synthases, Dr. Zhenqing Zhang for his help in acquiring NMR data, and Dr. Dmitri Zagorevski for HRMS analyses.
Footnotes
Supporting Information Available
1H and 13C NMR data of compounds 1, 1′, 3–7, and 9 and 31P NMR, COSY, and HMQC of compounds 1 and 1′. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Casu B. Chapter 1, Structure and active domains of heparin. In: Garg HG, Linhardt RJ, Hales CA, editors. Chemistry and Biology of Heparin and Heparan Sulfate. Elsevier; Oxford, UK: 2005. [Google Scholar]
- 2.Linhardt RJ, Dordick JS, Deangelis PL, Liu J. Semin Thromb Hemostasis. 2007;33(5):453–465. doi: 10.1055/s-2007-982076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linhardt RJ, Gunay NS. Semin Thromb Hemostasis. 1999;3:5–16. [PubMed] [Google Scholar]
- 4.Sinay P, Jacquinet J-C. Carbohydr Res. 1984;132:C5–C9. [Google Scholar]
- 5.Kusche M, Hannesson HH, Lindahl U. Biochem J. 1991;275:151–158. doi: 10.1042/bj2750151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Karst NA, Linhardt RJ. Curr Med Chem. 2003;10:1993–2031. doi: 10.2174/0929867033456891. [DOI] [PubMed] [Google Scholar]; (b) Avci FY, Karst NA, Linhardt RJ. Curr Pharm Res. 2003;9:2323–2335. doi: 10.2174/1381612033453929. [DOI] [PubMed] [Google Scholar]; (c) Van Boeckel CAA, Petitou M. Angew Chem Int Ed Engl. 1993;32:1671–1690. [Google Scholar]; (d) Seeberger PH, Haase W-C. Chem Rev. 2000;100:4349–4393. doi: 10.1021/cr9903104. [DOI] [PubMed] [Google Scholar]; (e) Ojeda R, Terentí O, de Paz J-L, Martín-Lomas M. Glycoconj J. 2004;21:179–195. doi: 10.1023/B:GLYC.0000045091.18392.a8. [DOI] [PubMed] [Google Scholar]; (f) Prabhu A, Venot A, Boons G-J. Org Lett. 2003;5(26):4975–4978. doi: 10.1021/ol0359261. [DOI] [PubMed] [Google Scholar]; (g) Yeung BKS, Chong PYC, Petillo PA. J Carbohydr Chem. 2002;21(7–9):799–865. [Google Scholar]
- 7.(a) Linhardt RJ, Weïwer M. Comprehensive Glycoscience From chemistry to systems biology. Vol. 1. Elsevier Science; Boston, MA: 2007. pp. 713–746. [Google Scholar]; (b) Kuberan B, Lech MZ, Beeler DL, Wu ZL, Rosenberg RD. Nat Biotechnol. 2003;21(11):1343–1346. doi: 10.1038/nbt885. [DOI] [PubMed] [Google Scholar]; (c) Muñoz EM, Xu D, Avci F, Kemp MM, Liu J, Linhardt RJ. Biochem Biophys Res Commun. 2006;339:597–602. doi: 10.1016/j.bbrc.2005.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen J, Avci FY, Muñoz EM, McDowell LM, Chen M, Pedersen LC, Zhang L, Linhardt RJ, Liu J. J Biol Chem. 2005;280(52):42817–42825. doi: 10.1074/jbc.M504338200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lindahl U, Li J-P, Kusche-Gullberg M, Salmivirta M, Alaranta S, Veromaa T, Emeis J, Roberts I, Taylor C, Oreste P, Zoppetti G, Naggi A, Torri G, Casu B. J Med Chem. 2005;48:349–352. doi: 10.1021/jm049812m. [DOI] [PubMed] [Google Scholar]; (f) Sismey-Ragatz AE, Green DE, Otto NJ, Rejzek M, Field RA, DeAngelis PL. J Biol Chem. 2007;282(39):28321–28327. doi: 10.1074/jbc.M701599200. [DOI] [PubMed] [Google Scholar]
- 8.(a) Busca P, Martin OR. Tetrahedron Lett. 2004;45:4433–4436. [Google Scholar]; (b) Lazarevic D, Thiem J. Carbohydr Res. 2002;337:2187–2194. doi: 10.1016/s0008-6215(02)00183-0. [DOI] [PubMed] [Google Scholar]; (c) Chang R, Vo T-T, Finney NS. Carbohydr Res. 2004;341:1998–2004. doi: 10.1016/j.carres.2006.05.008. [DOI] [PubMed] [Google Scholar]; (d) Busca P, Piller V, Piller F, Martin OR. Bioorg Med Chem Lett. 2003;13:1853–1856. doi: 10.1016/s0960-894x(03)00287-7. [DOI] [PubMed] [Google Scholar]; (e) Gross BJ, Kraybill BC, Walker S. J Am Chem Soc. 2005;127:14588–14589. doi: 10.1021/ja0555217. [DOI] [PubMed] [Google Scholar]; (f) Losey HC, Jiang J, Biggins JB, Oberthür M, Ye X-Y, Dong SD, Kahne D, Thorson JS, Walsh CT. Chem Biol. 2002;9:1305–1314. doi: 10.1016/s1074-5521(02)00270-3. [DOI] [PubMed] [Google Scholar]
- 9.Sala RF, MacKinnon SL, Palcic MM, Tanner ME. Carbohydr Res. 1998;306:127–136. doi: 10.1016/s0008-6215(97)10033-7. [DOI] [PubMed] [Google Scholar]
- 10.(a) Palmacci ER, Plante OJ, Seeberger PH. Eur J Org Chem. 2002:595–606. [Google Scholar]; (b) Arlt M, Hindsgaul O. J Org Chem. 1995;60:14–15. [Google Scholar]; (c) Hanessian S, Lu P-P, Ishida H. J Am Chem Soc. 1998;120:13296–13300. [Google Scholar]; (d) Ernst C, Klaffke W. Tetrahedron Lett. 2001;42:2973–2975. [Google Scholar]
- 11.Roseman S, Distler JJ, Moffatt JG, Khorana HG. J Am Chem Soc. 1961;83:659–663. [Google Scholar]
- 12.(a) Barroca N, Jacquinet J-C. Carbohydr Res. 2000;329:667–679. doi: 10.1016/s0008-6215(00)00234-2. [DOI] [PubMed] [Google Scholar]; (b) Van Boeckel CAA, Beetz T, Vos JN, De Jong AJM, Van Aelst SF, Van den Bosch RH, Mertens JMR, Van der Vlugt FA. J Carbohydr Chem. 1985;4:293–321. [Google Scholar]
- 13.Ke W, Whitfield DM, Gill M, Larocque S, Yu S-H. Tetrahedron Lett. 2003;44:7767–7770. [Google Scholar]
- 14.(a) Chiba T, Sinay P. Carbohydr Res. 1986;151:379–389. [Google Scholar]; (b) Yu HN, Furukawa J-I, Ikeda T, Wong C-H. Org Lett. 2004;6(5):723–726. doi: 10.1021/ol036390m. [DOI] [PubMed] [Google Scholar]
- 15.Bazin HG, Kerns RJ, Linhardt RJ. Tetrahedron Lett. 1997;38:923–926. [Google Scholar]
- 16.Lubineau A, Gavard O, Alais J, Bonnaffe D. Tetrahedron Lett. 2000;41:307–311. [Google Scholar]
- 17.(a) Vlahov IR, Linhardt RJ. Tetrahedron Lett. 1995;36:8379–8382. [Google Scholar]; (b) Schell P, Orgueira HA, Roehrig S, Seeberger PH. Tetrahedron Lett. 2001;42:3811–3814. [Google Scholar]
- 18.(a) Chiba T, Jacquinet J-C, Sinay P, Petitou M, Choay J. Carbohydr Res. 1988;174:253–264. [Google Scholar]; (b) Rochepeau-Jobron L, Jacquinet J-C. Carbohydr Res. 1997;303:395–406. doi: 10.1016/s0008-6215(98)00298-5. [DOI] [PubMed] [Google Scholar]
- 19.Deng S, Gangadharmath U, Chang C-WT. J Org Chem. 2006;71:5179–5185. doi: 10.1021/jo060374w. [DOI] [PubMed] [Google Scholar]
- 20.Gavard O, Hersant Y, Alais J, Duverger V, Dilhas A, Bascou A, Bonnaffé D. Eur J Org Chem. 2003:3603–3620. [Google Scholar]
- 21.MacDonald DL. J Org Chem. 1962;27:1107–1109. [Google Scholar]
- 22.Chen M, Bridge A, Liu J. Biochemistry. 2006;45:12358–12365. doi: 10.1021/bi060844g. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Duncan MB, Carrick K, Pope RM, Liu J. Glycobiology. 2003;13:785–794. doi: 10.1093/glycob/cwg101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.