

Abstract
A variety of binding events in biological systems are mediated by multivalent interactions between oligosaccharides and saccharide receptors present on pathogens and cell surfaces. In particular, given the important role of multivalent interaction between proteins and carbohydrates in the initial step of pathogen recognition, many glycosylated molecules and polymers have been synthesized in order to mimic the carbohydrate ligands and to inhibit the binding of the pathogen to its target. In this work, we extend our evaluation of the impact of the architecture of well-defined glycopolypeptides on the inhibition of binding of the cholera toxin B pentamer (CT B5) subunit. Here we report the production of two families of α-helical glycopolypeptides which were synthesized via a combination of protein engineering and chemical methods. The presentation of pendant saccharides on the polypeptide backbones, as well as their valencies, can be well controlled via these methods. Control of the backbone conformation, introduced in this report, is also possible via these strategies. The polypeptides and glycopolypeptides were characterized via SDS-PAGE analysis, 1H NMR, and MALDI-TOF mass spectrometry. Their conformation and hydrodynamic volume were characterized by circular dichroic (CD) spectroscopy and gel permeation chromatography (GPC), respectively. The binding of CT B5 by these glycopolypeptides was evaluated via direct enzyme-linked immunosorbent assay (DELA). The effects of spacing and conformation were elucidated by comparison of the binding exhibited by helical glycopolypeptides with that of random-coil glycopolypeptides.
Introduction
In biological systems, multivalent interactions play unique roles owing to their greater avidity and specificity versus monovalent interactions. Moreover, multivalent interactions can also induce reorganization of receptors on cell surfaces, which can enhance receptor binding selectivity.1-4 Such multivalent interactions, especially those between proteins and carbohydrates, occur in a broad range of biological contexts, involving cell surface receptors, enzymes, G-protein-coupled receptors, ion channels, lectins, and toxins found in cells and pathogens.5-13 The study of multivalent interactions is therefore very important for producing effective inhibitors of surface—surface interactions as well as effectors of biological processes that could be important in the development of vaccines or vaccine adjuvants.9,14-16 A more detailed understanding of specific cellular activities, such as cell communication,8 inflammation,13 cell signaling,17 fertilization,6,18 and selective recognition of antigens by antibodies,19,20 has been elucidated by the studies of multivalent interactions.
Protein—carbohydrate multivalent interactions also occur in the initial step of host infection by a variety of pathogens, such as the AB5 family of bacterial toxins (including heat labile enterotoxin, cholera toxin, shiga-like toxins, and pertussis toxin), which contain one enzymatic A subunit as a lethal factor and five B subunits that mediate binding of the toxin to the cell membrane.10,21 The specific multivalent interactions between the B pentamer and specific gangliosides expressed on the host cell surface, which enable toxin entry into the host cells, have been mimicked in high-affinity multivalent inhibitors and offer potential therapeutic strategies.14,15,22,23 Moreover, the binding of synthetic inhibitors with CT B5 can be achieved with simple monosaccharides and does not require the penetration of cell membranes, which allows saccharide ligands of a variety of identities and/or electrostatic charge to be easily appended on the multivalent backbone for investigations of inhibition enhancement.24 Therefore, CT B5 has been used as a model lectin for studies of multivalent interactions.
Many types of scaffolds have been used for multivalent displays of ligands, including one-dimensional linear small molecule scaffolds, rigid or flexible linear polymeric scaffolds,25 dendrimers,26 nanoparticle surfaces,27 monolayer surfaces,28 and liposomes.29 These and other previous reports have illustrated that various architectural features such as scaffold conformation, valency, linker arm, and ligand density can all significantly affect binding.2,28,30 For example, multivalent enhancement factors ranging from 10 to 106 have been reported for the binding of multivalent sialic acid-functionalized macromolecules to the hemagglutinin receptors on the surface of the influenza virus, depending on the degree of valency and the type of scaffold.31 In contrast, a series of sialosides with chemically defined divalent to tetravalent structures have been synthesized, and their relative binding affinities for sialoadhesin (Siglec 1) have shown poor correlation with valency; the tetrasialoside with the highest valency exhibits the poorest binding perhaps due to the steric restriction caused by the close spacing of the ligands.32 It has been reported by Cloninger and co-workers that the binding activities and the degree of protein clustering induced by dendrimeric glycopolymers can be attenuated by the ligand types and densities.30 More recently, Sampson and co-workers have reported multivalent fertilin β oligopeptides, which exhibit variations in the inhibition of fertilization depending on differences in scaffold length and ligand density.6 The nature of the linker arm between scaffolds and saccharide ligands can also have an impact on multivalent binding. Two bivalent Pk trisaccharide ligands reported by Toone and co-workers have been suggested to bind shiga-like toxin in different modes and with different enhancements based on differences in the physicochemical nature of the linkers.33 It has been shown that the increasing of length and hydrophobicity of linkers of pendant ligands can improve the CT B5 inhibition of a series of poly-(glutamic acid)-based glycopolymers,34 as had been previously suggested in investigations of small molecule35 and dendrimeric inhibitors.36
Very useful information has been obtained from these studies to guide the design of new multivalent ligands. An increased level of detail to guide ligand design, however, could be facilitated via the production of increasingly well-defined scaffolds. Most of the previously reported scaffolds are heterogeneous with respect to valencies, spacing, ligand densities, or scaffold length and have not offered opportunities to alter polymer conformation, which have been barriers to the detailed interpretation of the factors that affect multivalent interactions. Moreover, some of the scaffolds, such as liposomes and monolayers, can be difficult to stabilize, so in many cases only the average densities of the functional groups can be controlled.28 Protein engineering methods with subsequent chemical coupling of ligands provide significant advantages for the manipulation of polypeptide backbone conformation and functional group placement. We have previously reported a family of random-coil glycopolypeptides synthesized via such strategies as potential inhibitors of CT B5.34,37 The scaffolds were equipped with modified galactopyranosides, as the natural CT B5 ligand on the cell surface (the GM1 ganglioside (Gal β1-3GalNAc β1-4(Neu5Acα2-3)Galβ1-4Glc-ceramide)) binds through its terminal galactose. Modification of scaffolds with simple monosaccharides has thus provided a facile and useful strategy for production of multivalent inhibitors.38
In the work reported here, we report the synthesis and characterization of two families of α-helical glycopolypeptides designed to display galactopyranosides at nominally 35 and 17 Å between two adjacent pendant saccharide ligands. The crystal structure of the complex of CT B5 and GM1 reveals that the distance between two adjacent sugar-recognizing binding sites on CT B5 is ∼35 Å, so the production of these scaffolds has afforded additional opportunities to probe the sensitivity of inhibition to the presentation of saccharides at these distances.24 The polypeptides and glycopolypeptides were characterized via multiple methods, including 1H NMR, circular dichroic spectroscopy (CD), gel permeation chromatography (GPC), and DELA. Comparison of the CT B5 inhibition of these α-helical glycopolypeptides with those of previously reported random-coil glycopolypeptides37 has also provided assessment of the contribution of backbone conformation on the binding event. Opportunities for the design of additional glycopolypeptides with tunable inhibition are suggested on the basis of these investigations.
Materials and Methods
Materials
2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and Fmoc-6-aminohexanoic acid (Fmoc-Ahx-OH) were purchased from EMD Biosciences Inc. (San Diego, CA). 3,3′,5,5′-Tetramethylbenzidine (TMB) was purchased from Pierce (Rockford, IL). Isopropyl β-D-thiogalactopyranoside (IPTG) and C96 Maxisorp microtiter plates were purchased from Fisher Scientific (Pittsburgh, PA). Ganglioside GD1b, cholera toxin B subunit horseradish peroxidase conjugate (CT B5-HRP), β-D-galactosylamine, β-D-galactose, dimethyl sulfoxide (DMSO), diisopropylethylamine (DIEA), and all other reagents were obtained from Sigma-Aldrich (St. Louis, MO) and used without any further purification.
Protein Expression and Purification
The plasmids pET28b-JS1-A3, pET28b-JS1-A6, and pET19b-RF1-B6 were used to transform chemically competent cells of E. coli strain BL21(DE3)-pLysS for expression of polypeptides 17-H-3, 17-H-6, and 35-H-6, respectively.39 The sequences and expected molecular weights (which were confirmed via MALDI-TOF analysis of the purified polypeptides) of these three polypeptides are listed in Table 1. The expression was conducted via standard methods employing chemical induction (final concentration of IPTG of 0.4 mM) of cultures of the appropriate expression host, grown in 500 mL 2 x YT media with ampicillin (100 μg/mL) and chloramphenicol (34μg/mL) for 35-H-6 or kanamycin (25 μg/mL) and chloramphenicol (34 μg/ mL) for 17-H-3 and 17-H-6.39,40 Cells were harvested 4 h later via centrifugation (10 000 rpm, 20 min), and the cell pellets were resuspended in buffer B (8 M urea, 100 mM NaH2PO4, 10 mM Tris‚·Cl, pH = 8.0), with 4 mL of buffer B for 1 g of cell pellet, and frozen at −20 °C.
Table 1.
Polypeptides Employed in These Studies
| polypeptide sequences | MW (Da) | approx spacing (Å) | |
|---|---|---|---|
| 17-H-3a | [AAAQEAAAAQAAAQAEAAQAAQ]3 | 8875b | 17 |
| Cap 17-H-3c | 17-H-3 | 17 | |
| 17-H-6 | [AAAQEAAAAQAAAQAEAAQAAQ]6 | 14770 | 17 |
| Cap17-H-6 | 17-H-6 | 17 | |
| 35-H-6 | [AAAQAAQAQAAAEAAAQAAQAQ]6 | 14159 | 35 |
| Cap 35-H-6 | 35-H-6 | 35 |
x-H-y: x represents the approximate distance between the adjacent glutamic acids, H represents helical conformation, and y represents the number of repeats of the monomeric peptide sequence.
The given molecular weight of polypeptides are theoretical; purified polypeptides show these expected masses in MALDI-TOF analysis.
Cap x-H-y represents the N-(ϵ-aminocaproyl)-β-D-galactosylamine-functionalized glutamic acid residues in the glycopolypeptides.
The cell suspensions then were lysed via sonication, and the cell lysates were collected via centrifugation (15 000 rpm, 20 min). The target polypeptides were purified from the cell lysate via immobilized Ni-NTA affinity chromatography with stepwise pH gradient elution under denaturing conditions. Eluted polypeptides were dialyzed against deionized water at 4 °C for 3 days (3 changes of water per day), and the dialysates were lyophilized to yield 15–20 mg of the purified target polypeptides per liter of culture. The protein purification was monitored with sodium dodecyl sulfate—polyacrylamide gel electrophoresis (SDS-PAGE) and visualized via Coomassie blue staining.
Synthesis of N-(ϵ-Aminocaproyl)-β-D-galactosylamine
The N-(ϵ-caproyl)-β-D-galactosylamine was prepared using previously described methods.34 Fmoc-Ahx-OH (1.4 mmol, 500 mg), HBTU (1.4 mmol, 550 mg), and DIEA (7 mmol, 1.3 mL) were dissolved in 2.5 mL of DMSO and preactivated for 5 min, and the predissolved β-D-galactosylamine solution (1.4 mmol, 250 mg in 5 mL of DMSO) was added into the reaction and stirred at room temperature overnight. The reaction solution was mixed with ∼200mL of H2O and then and washed with DCM three times to remove the byproducts and DMSO. The aqueous phase was collected and lyophilized. For deprotection of the Fmoc group, the obtained yellow powder was dissolved in piperidine/DMF (20:80) and stirred at room temperature for 2 h, followed by precipitation into cold ethyl ether. After centrifugation, the precipitate was dissolved in H2O and lyophilized (∼340 mg of product, ∼83% yield).
Glycosylation of Polypeptides
The polypeptides were dissolved in DMSO at a concentration of 1 mg/mL. A 5-fold excess (based on moles of carboxylic acid groups in the polypeptides) of N-(ϵ-aminocaproyl)-β-D-galactosylamine, a 2-fold excess of HBTU, and a 5-fold excess of DIEA were added into the reaction solution every 3 h (3 times) to ensure completion of the reaction. The reaction solutions were stirred at room temperature for 9 h. The reaction mixtures were dialyzed against 1 M NaCl (4 changes) for 1 day, then against H2O (4 changes) for another day, and then lyophilized.
General Characterization
Electrospray mass spectrometry and NMR were used to characterize the N-(ϵ-aminocaproyl)-β-D-galactosylamine. The purity of the polypeptides and glycopolypeptides was monitored via SDS-PAGE, the composition was confirmed via NMR and amino acid analysis, and the molecular weight of the polypeptide and glycopolypeptides was confirmed via MALDI-TOF mass spectrometry. The secondary structure of the polypeptides and glycopolypeptides was characterized via circular dichroic (CD) spectroscopy. Concentrations of the samples of polypeptides and glycopolypeptides were determined via amino acid analysis (University of Iowa, Iowa City, IA).
Circular Dichroic Spectroscopy (CD)
Circular dichroic spectra were recorded on a Jasco J-810 spectropolarimeter (Jasco Inc, Easton, MD) equipped with a Jasco PTC-424S temperature controller. Background scans of buffers (PBS pH 7.4) were recorded and subtracted automatically from the sample scans. Samples were made at a concentration of approximately 1–2 μM via dilution. The stock concentrations were confirmed via amino acid analysis. The samples (400 μL) were loaded into a 1 mm path length quartz cuvette. Data points for the wavelength-dependent CD spectra were recorded at every nanometer with a 1 nm bandwidth and a 10 s averaging time for each data point. Data points for the temperature scans were recorded at 222 nm, at 1 °C intervals with an equilibration time of 1 min. The molar ellipticity, [θ]MRE (deg cm2/dmol), was calculated via use of the concentration, molecular weight of the samples, and cell path length.
Gel Permeation Chromatography (GPC)
The relative hydrodynamic volumes of this family of polypeptides and glycopolypeptides were examined via gel permeation chromatography employing a Waters GPC (Milford, MA). 50 μL of filtered 1 mg/mL samples in PBS buffer (pH 7.4) or PBS with 20% acetonitrile was injected and separated on a Waters Ultrahydrogel linear column (7.8 × 300 mm) followed by a Ultrahydrogel 250 column (7.8 × 300 mm). The absorbance at 214 nm was detected with a Waters 2996 photodiode array detector.
Direct Enzyme-Linked Assay (DELA)
A direct enzyme-linked immunosorbent assay was carried out in 96-well microplates as previously reported.41 Microplates were incubated at 37 °C for 16 h with 100 μL of 2 μg/mL ganglioside GD1b PBS solution (pH 7.4) in each well and then blocked with 1% BSA—PBS solution for 30 min. The plates were washed with 0.05% Tween 20 in PBS and then used for the inhibition assays. For the inhibition assay, a series of glycopolypeptide samples of various concentrations (determined by amino acid analysis) were prepared and mixed with the cholera toxin B subunit horseradish peroxidase conjugate (CT B5-HRP) in sample buffer (0.1% BSA, 0.05% Tween 20, PBS) to obtain a final concentration of CT B5-HRP of 6 ng/mL. The glycopolypeptides (or monovalent galactose as a control) were incubated with the CT B5-HRP at room temperature for 2 h; 100 μL of the sample mixture was then added into wells of the prepared plates and incubated for 30 min at room temperature. Unbound toxin was removed via washing with 0.05% Tween 20 in PBS, and the remaining CT B5-HRP bound to GD1b was developed with TMB solution. The reaction was quenched with 2 M H2SO4, and the amounts of CT B5-HRP were monitored with absorbance measurements at 450 nm. IC50 values were calculated via fitting of the data by nonlinear regression.42
Results
Synthesis of Glycopolypeptides
The coupling reaction of N-(ϵ-aminocaproyl)-β-D-galactosylamine to the polypeptides is shown in Figure 1. The six-carbon linker arm of the galactopyranoside ligands was chosen on the basis of previous studies of the contribution of the hydrophobic linker arms to the binding of CT B5 as well as studies of N-(ϵ-aminocaproyl)-β-D-galactosylamine-modified PGA glycopolypeptides that indicated improved inhibition of CT B5 in a DELA format.34,35 The nominal distance between adjacent saccharide ligands in these helical glycopolypeptides was estimated on the basis of energy minimization calculations, as previously described.43 The polypep-tide sequences, their molecular weights, and the approximate distance between glutamic acid residues are listed in Table 1. Amide bond formation was carried out under basic condition with HBTU activation. To approach full substitution of the glycopolypeptide, fresh saccharide ligands, HBTU, and DIEA were added in three cycles. The excess caproyl-functionalized galactosylamine and other small molecules were removed from the reaction mixture via dialysis against 1 M NaCl and then H2O, yielding pure glycopolypeptides with high yield. The purity of polypeptides and glycopolypeptides was determined with HPLC and SDS-PAGE (Supporting Information, Figure S1). The composition, molecular mass, and degree of substitution were characterized via 1H NMR and MALDI-TOF (Supporting Information, Figures S2—S4); the tabulated results of these experiments are shown in Table 2.
Figure 1.

Glycosylation of α-helical polypeptides with N-(ϵ-aminocaproyl)-β-D-galactosylamine.
Table 2.
Estimated Degrees of Substitution of Synthesized Glycopolypeptides
| glyco- polypeptide |
no. of reactive -COOH (including C-termini) |
no. of saccharides determined from NMR |
mol mass (Da) determined from MALDI |
no. of saccharides calcd from MALDI |
av no. | degree of substitution (%) |
|---|---|---|---|---|---|---|
| Cap 17-H-3 | 7 | 6.7 | 10673 | 6.5 | 6.6 ± 0.1 | 94 ± 2 |
| Cap 17-H-6 | 13 | 11.5 | 17808 | 11.7 | 11.6 ± 0.1 | 90 ± 1 |
| Cap 35-H-6 | 7 | 7.3 | 16017 | 6.3 | 6.8 ± 0.7 | 97 ± 10 |
Secondary Structure Characterization
CD experiments were conducted to study any potential changes in polypeptide conformation upon glycosylation; the studies were conducted in PBS buffer at pH 7.4 to mimic the buffer conditions employed for the DELA. The spectra of all polypeptides and glycopolypeptides at 4 °C are shown in Figure 2. As shown in the figure, each polypeptide and glycopolypeptide exhibits a spectrum with two minima at 208 and 222 nm and a maximum at ∼195 nm, consistent with the expected spectrum of a helical polypeptide. The mean residue ellipticity ([θ]MRE) values of samples were calculated on the basis of concentrations determined via amino acid analysis and according to standard equations. Because the α-helical polypeptide chains contain nonalanine-rich N- and C-tags, which have lower helical propensities than the alanine-rich sequences, the helicities of the repeated alanine-rich region are predicted to be higher than the overall apparent fractional helicities. As shown in Figure 2, the intensities of the spectra of the glycopolypeptides are generally slightly lower than those of the corresponding polypeptides, indicating that the glycopolypeptides have slightly lower helicities than the corresponding polypeptides. Standard methods and equations for correlating [θ]MRE and the helicity of the macromolecules have been reported previously43,44 (Supporting Information) and were used to calculate the maximum fractional helicities of the polypeptides and glycopolypeptides. Polypep-tides 17-H-3, 17-H-6, and 35-H-6 show fractional helicities of 71%, 78%, and 68%, while glycopolypeptides Cap 17-H-3, Cap 17-H-6, and Cap 35-H-6 exhibit fractional helicities of 57%, 71%, and 52%, respectively. Variations in the helicities of the polypeptides have been observed with differences in sample preparation, with errors generally less than 15% (although near 25% in isolated cases). The greater loss in helicity of the Cap 17-H-3 after glycosylation, relative to that observed for the Cap 17-H-6, may be due to the shorter chain length. The greater loss of helicity for the Cap 35-H-6 compared with Cap 17-H-6 may result from interruption of stabilizing salt bridges, between glutamic acid and glutamine at i and i + 4 positions, after glycosylation45,46
Figure 2.

CD spectra of polypeptides and glycopolypeptides PBS buffer (pH 7.4) at 4 °C: (A) 17-H-3 and Cap 17-H-3; (B) 17-H-6 and Cap 17-H-6; (C) 35-H-6 and Cap 35-H-6.
Thermal Stability
The thermal stability of polypeptides and glycopolypeptides with increasing temperature was studied via CD to examine the effect of glycosylation. Upon heating from 4 to 80 °C, the minima at 208 and 222 nm decrease in absolute intensity, with the appearance of a single minimum near 198 nm at elevated temperature for all samples (Supporting Information, Figure S5). The observed results are consistent with the adoption of a nonhelical conformation by the macromolecules upon heating. The thermal transition is fully reversible upon heating and cooling for both the polypeptides and glycopolypeptides (Supporting Information, Figure S6).
The existence of an isodichroic point (at ∼203 nm) in the CD spectra of all polypeptides and glycopolypeptides suggests the existence of a two-state transition.47,48 The [θ]222 values as a function of temperature, for the polypeptides and glycopolypeptides, and the fitted curves (fits based on a two-state function) are shown in Figure 3. As shown in the figure, both polypeptides and glycopolypeptides undergo a thermal transition over a reasonably broad temperature range, consistent with data reported for other alanine-rich peptides.49-51 Given the apparent two-state nature of the transition, assessment of the reduction in helicity (via measurement of the [θ]222) as a function of temperature can provide a measure of the melting temperature and enthalpy of the transition (Supporting Information). By fitting these data to the standard two-state equation,52,53 the transition temperature (Tm) and van’t Hoff enthalpy values (ΔHm) of the polypeptides and glycopolypeptides can be obtained; these values are listed in Table 3. The glycopolypeptides show transitions that are broader than those of the unmodified polypeptides, suggesting that the transition is less cooperative. In addition, the glycopolypeptides show higher transition temperatures than the corresponding polypeptides, suggesting that they may be more thermally stable despite their lower fractional helicities, although these differences may result from the increased breadth of the transition. The ΔHm values are only slightly greater in the glycopolypeptides versus the polypeptides, which may support this speculation; on the whole, the thermal properties of the polypeptides and glycopolypeptides are not widely dissimilar, indicating that the helical polypeptides can be employed as templates for the multivalent display of these and potentially other ligands.
Figure 3.

Plots of [θ]222 vs temperature and fitting curves based on a two-state function for polypeptides and glycopolypeptides: (A) 17-H-3 and Cap 17-H-3; (B) 17-H-6 and Cap 17-H-6; (C) 35-H-6 and Cap 35-H-6.
Table 3.
van’t Hoff Enthalpy (ΔHm) and Melting Temperature (Tm) for the Polypeptides and Glycopolypeptides
| 17-H-3 | Cap 17-H-3 | 17-H-6 | Cap 17-H-6 | 35-H-6 | Cap 35-H-6 | |
|---|---|---|---|---|---|---|
| ΔHm (kcal/mol)a,b | 20.3 ± 1.9 | 19.4 ± 1.8 | 22.2 ± 1.0 | 29.2 ± 4.8 | 18.5 ± 1.8 | 20.0 ± 2.0 |
| Tm (°C)c | 33.6 ± 2.3 | 38.4 ± 0.7 | 34.9 ± 0.1 | 47.8 ± 3.1 | 35.2 ± 1.4 | 38.4 ± 1.5 |
Reported errors are based on the calculated error from duplicate measurements.
The ΔHm values for all the polypeptides and the relative glycopolypeptides are not statistically different based on the results of ANOVA, Turkey’s test (p > 0.05).
The statistical comparison of Tm values using ANOVA (Turkey’s test) showed that the Cap 17-H-3 and Cap 35-H-6 do not have significantly different Tm values than their corresponding polypeptides backbones, while the Tm of Cap 17-H-6 is statistically different than that of 17-H-6 (p < 0.05) with unknown origins.
Gel Permeation Chromatography (GPC)
In our investigations of random-coil glycopolypeptides, a correlation of hydrodynamic volume and inhibition was observed.37 GPC experiments were therefore conducted to study the hydrodynamic volume of the α-helical polypeptides and glycopolypeptides, for comparison with the glycosylated random-coil polypeptides of chemical and recombinant origins reported in our previous studies.37 In these experiments, the absorbance at 214 nm was monitored as a function of elution time for the polypeptides and glycopolypeptides in PBS buffer or PBS buffer containing 20% acetonitrile; representative GPC traces for the helical polypeptides and glycopolypeptides in PBS are shown in Figure 4. All helical glycopolypeptides have a slightly longer elution time than their corresponding polypeptides, suggesting that the glycopolypeptides have smaller hydrodynamic volumes. The GPC traces of 35-H-6 and Cap 35-H-6 are shown in Figure 4 as an example. In the unmodified polypeptides, the chains may be more extended due to the negatively charged glutamic acid residues, despite the fact that the Debye length under these conditions (7.8 Å)54 is shorter than the expected distance between glutamic acid residues in chains of 100% helicity. Neutralization of charge by coupling with saccharide ligands yields smaller hydrodynamic volumes for the glycopolypeptides although they are of higher molecular mass. These results are similar to the GPC results for PGA and random-coil polypep-tides;37 this similarity may result from the incomplete helicity of the helical polypeptides. In addition, a comparison of the elution times for the random-coil and helical glycopolypeptides in PBS is shown in Figure 5. As illustrated in the figure, the Cap 35-H-6 shows longer elution times than Cap PGA 86 and Cap 35-RC-6, indicating the relatively small hydrodynamic volume of the helical polypeptides. Given the similarity of the molecular masses of the Cap 35-RC-6 and Cap 35-H-6, if these α-helical polypeptides were rigid, they should exhibit a greater radius of gyration (RG) value than the random-coil polypep-tides.37 The fact that these α-helical polypeptides show an apparently lower hydrodynamic volume than random-coil polypeptides may be due to their flexibility, despite their significant helicity.55 Nevertheless, comparisons of hydrodynamic volume and inhibition enhancement of helical glycopolypeptides with those of the glycosylated random-coil glycopolypeptides permit the effects of backbone conformation on inhibition to be evaluated.
Figure 4.
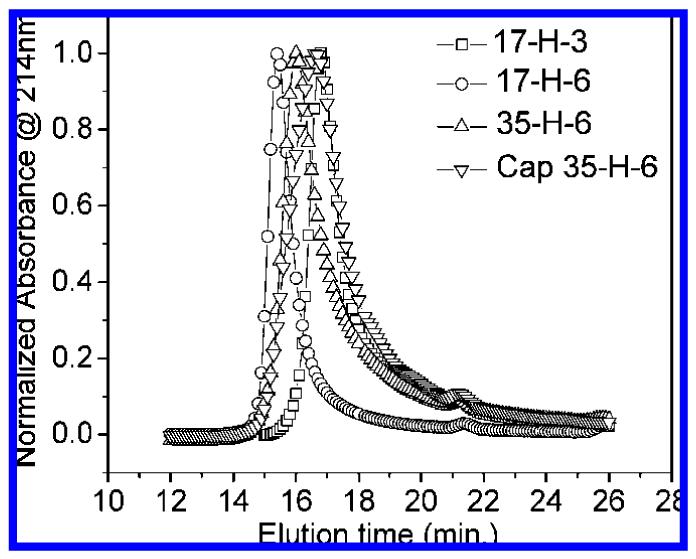
Elution times for polypeptides 17-H-3, 17-H-6, and 35-H-6 and glycopolypeptide Cap 35-H-6 in PBS. Plots are shown as normalized absorbance at 214 nm vs elution time.
Figure 5.
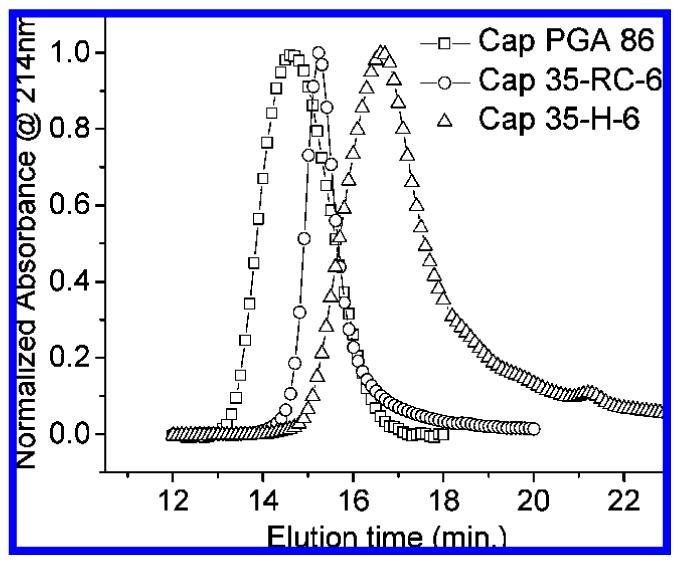
Elution times of Cap PGA 86, Cap 35-RC-6, and Cap 35-H-6 in PBS. Plots are shown as normalized absorbance at 214 nm vs elution time.
CT B5 Binding Assay
The CT B5 inhibition of these glycopolypeptides were examined via the DELA protocol reported by Minke et al.41 The inhibition curves for all the glycopolypeptides are shown in Figure 6. The concentration of samples of glycopolypeptides in the DELA assay ranged from 0 to 350 μM on polypeptide basis (0 to 3 mM on saccharide basis), whereas the concentration of monovalent galactose was varied from 0 to 150 mM. The data were fit via nonlinear regression42 with the corresponding results shown in Table 4. The IC50 value obtained for monovalent galactose in these assays was 54 mM, while the IC50 values (given in saccharide concentration) for the glycopolypeptides were 3.34 mM (Cap 17-H-3), 725 μM (Cap 17-H-6), and 160 μ M (Cap 35-H-6). The inhibition enhancements for the glycopolypeptides over the monovalent galactose are 16-fold, 74-fold, and 340-fold, respectively. In control experiments of the unmodified polypep-tides, some nonspecific inhibition was observed at high concentrations for the 17-H-3 and 17-H-6 (greater than 250 μM polypeptide (1.75 mM saccharide) for 17-H-3, and 100 μM polypeptide (1.3 mM saccharide) for 17-H-6); 35-H-6 showed no nonspecific inhibition at these concentrations.
Figure 6.
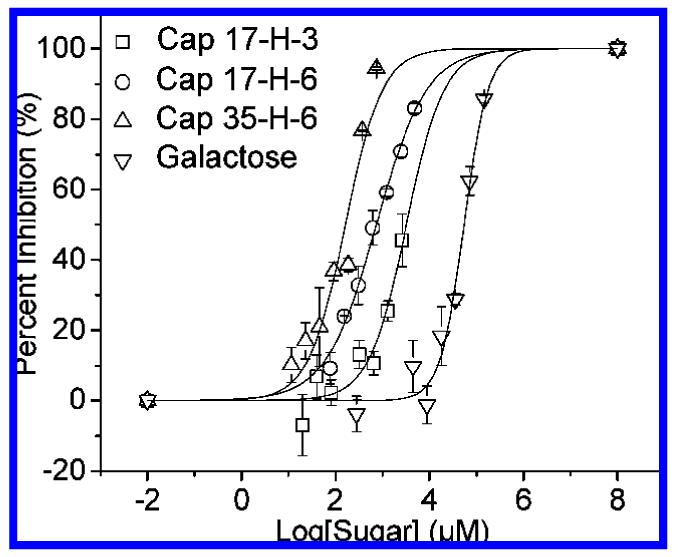
DELA inhibition data for inhibition of CT B5 by glycopolypeptides and galactose. Curves shown are fits to the data by a nonlinear regression method.
Table 4.
IC50 Values for Glycopolypeptides and Galactose As Determined via DELA and Inhibition Enhancement of Glycopolypeptides over Galactose
| sample | IC50 (μM) | inhibition enhancement over galactosea |
|---|---|---|
| Cap 17-H-3 | 3340 ± 650 | 16 ± 3 |
| Cap 17-H-6 | 725 ± 50 | 74 ± 3 |
| Cap 35-H-6 | 160 ± 26 | 340 ± 30 |
| galactose | 53900 ± 8920 |
The statistical comparison of the IC50 values using ANOVA (Turkey’s test) showed that the inhibition enhancement of Cap 35-H-6 was statistically significant as compared with the other two glycopolypeptides (p < 0.05).
Discussion
Design of Glycopolypeptides
Protein engineering strategies were applied to produce a family of α-helical polypeptides as the backbones of multivalent saccharide ligands with well-defined spacing and conformation. Alanine- and glutamine-rich polypeptides were chosen owing to their high helicity and reduced tendency for aggregation,39,43 while the glutamic acid residues were employed for specified placement of saccharide residues in the chain through standard amide bond formation strategies. The inclusion of the glutamic acid residues at various positions and in various numbers permitted the manipulation of spacing and valency of these glycopolypeptides. The production of these scaffolds was targeted so that the inhibition of CT B5 by this family of α-helical glycopolypeptides could be compared with that of previously studied random-coil glycopolypeptides and so that backbone effects on multivalent binding could be estimated.37
This distance between adjacent carbohydrate binding sites of CT B5 is estimated to be 35 Å from the crystal structure of the complex of CT B5 and its native receptor GM1 ganglioside-Galβ1-3GalNAcβ1-4(Neu5Acα2-3)Gal-β1-4Glc-ceramide.24 Additionally, between the carbohydrate binding sites on the adjacent subunits, there is no significant steric hindrance, which allows the saccharide ligands presented on the scaffolds access to those carbohydrate binding sites.56 As shown in Table 1, 35-H-6 was designed with an adjacent functional group spacing of 35 Å, while 17-H-3 and 17-H-6 were designed with a spacing of 17 Å; these values were estimated on the basis of energy minimization calculations.39,43 They are commensurate with the distances between saccharide receptors in the CT B5 and therefore permit study of the influence of saccharide spacing on the binding for these helical glycopolypeptides.
The distance between the C4 carbon of the terminal galactose and the C4 carbon of the glucose of the GM1 has been estimated to be ∼16 Å, which provides an estimate of the optimal length of a linker arm required to permit unimpeded interaction of the pendant saccharides in the binding pocket of CT B5.34 Favorable hydrophobic interaction between an alkyl linker arm and hydrophobic amino acids in the binding pocket of CT B5 has also been indicated to improve the binding affinity of saccharide ligands, based on comparison of the inhibition by a series of 3,5-substituted phenyl galactosides.35 The favorable effect of the alkyl linker arm has also been indicated in studies of CT B5 inhibition by our previously reported PGA-derived glycopolypeptides and recombinant glycopolypeptides; glycopolypeptides with caproyl-linked galactopyranosides showed higher inhibition enhancement than those without the six-carbon linker.34,37 The caproyl-linked galactopyranoside was therefore applied in this family of glycopolypeptides; the distance between the α-carbon on the glutamic acid residue and the C4 carbon of the pendant galactose is ∼18.7 Å when this linker is used, which is commensurate with the measured distance between relevant atoms in the GM1 ligand.
Conformational Properties and Effects of Glycosylation
Both unmodified polypeptides and glycopolypeptides show α-helical secondary structures, as illustrated in the CD results (Figure 2). The fractional helicities of these macromolecules range from approximately 50% to 80% at 4 °C; the helicity of the alanine-rich segments of these sequences would be correspondingly greater (60% to nearly 100%). At room temperature (the conditions of the DELA experiment), the alanine-rich segments of these macromolecules retain fractional helicities ranging from approximately 45% to 80%. Glycopolypeptides exhibited lower maximum helicities than the corresponding polypeptides, with losses in helicity ranging from 4% to 24%. Such a loss in secondary structure upon glycosylation is consistent with previously reported conformation changes upon glycosylation as reported by de Jongh et al.57 In these studies, fructosylated β-lactoglobulin exhibited a 5% reduction in β-sheet character upon comparison to unmodified β-lactoglobulin, as assessed via CD.57 The change in secondary structure observed for the helical glycopolypeptides may result from the influence of molecular weight, hydrogen bonding, and steric factors in the folding of these glycopolypeptides.58,59 The retention of greater helicity of the Cap 17-H-6 relative to that of Cap 17-H-3 may due to the longer chain length of the 17-H-6, which may stabilize the helical conformation and decrease the effect of glycosylation on the secondary structure.59 Compared with 35-H-6, Cap 35-H-6 has ∼24% lower helicity, which may result from the reduction of favorable Glu-Gln interactions that have been shown to stabilize the helical conformation of alanine-rich peptides.45,46
The melting temperatures observed here for the α-helical glycopolypeptides are greater than those reported for other shorter alanine-rich peptides,50 which is expected given their greater length, as mentioned above. The Tm values for the glycopolypeptides, obtained from the CD melting curves, are greater than the values for the corresponding polypeptides, which may suggest that the secondary structure of the glycopolypeptides is stabilized against thermal denaturation via glycosylation. The apparent increase in thermal stabilization observed as an increased Tm may also simply result from lower cooperativity during the folding and unfolding process, which may be caused by the steric effects of bulky saccharide groups on the side chain of glycopolypeptides and that complicates estimates of Tm. Specifically, the Tm values of Cap 17-H-3 and Cap 35-H-6 (each carrying 6–7 saccharides) increased by 4.8 and 3.2 °C relative to those of the unmodified polypeptides, respectively. The increase in the Tm of the Cap 17-H-6 (with 12–13 saccharides), in contrast, was significantly higher ( ∼12.9 °C), despite the similarity in its length to that of Cap 35-H-6. The observed increase in Tm values is also comparable with what has been reported for glycosylated β-lactoglobulin. Although the β-sheet content of β-lactoglobulin was reduced upon glycosylation, the glycosylated protein showed slightly increased thermal stability than β-lactoglobulin based on CD and DSC studies; the Tm was observed to increase from 68 to 77 °C for β-lactoglobulin containing seven fructose residues.57 Similar effects of glycosylation on protein thermal stability have been reported for α-chymotrypsin glycosylated with lactose or dextran, with the glycosylated α-chymotrypsin (with 7–8 lactose per chain) showing an increase in Tm of 7 °C (from 46 to 53 °C).60 Although no specific molecular mechanisms have been determined for the improved thermal stability in these cases, glycosylation has been widely shown to increase protein thermal stability61,62 and has therefore become an often-explored method for modulating protein thermal stabilities as well as other properties such as the kinetics of protein unfolding and the formation of insoluble protein aggeragates.60,63,64 Similarly, we cannot assign any molecular mechanisms to the origins of apparently increased thermal stability in the glycopolypeptides or assess if this change in folding reflects any significant change in chain rigidity at room temperature. Evaluation of the chain rigidity at room temperature via small-angle scattering methods is currently underway.
The ΔHm values obtained from the CD melting curves are similar for this family of alanine-rich polypeptides and glycopolypeptides, suggesting a similar enthalpy of unfolding for the cooperative unit.65 The ΔHm values measured for the conformational transition of these alanine-rich polypeptides and glycopolypeptides are slightly higher than those reported for shorter alanine-rich peptides (∼11 kcal/mol),66 which may be expected owing the higher molecular weight of the polypeptides here. In contrast, the measured values are smaller than that of collagen-like polypeptides in which folding involves multiple polypeptide chains,67 consistent with the monomeric nature of the transition and the low cooperativity indicated by the breadth of the transition for the helical macromolecules.
Although there are slight differences between the helicities of the polypeptides and glycopolypeptides, both sets of macromolecules exhibit helical conformations of reasonably comparable stability, suggesting that the glutamic acid residues do not play a significant role in stabilizing the α-helical structure and that glycosylation does not disrupt the polypeptide backbone conformation. This type of scaffold, designed on the basis of secondary structure preferences of natural amino acids, therefore represents a useful approach in the production of more complicated, yet defined, multivalent ligands with desired backbone conformation.
Effects of Saccharide Spacing on CT B5 Inhibition
An important goal of this work was to determine the effects, on CT B5 inhibition, of varying the distance between adjacent saccharide ligands along a helical polypeptide backbone. As shown in Table 4, Cap 17-H-3 and Cap 17-H-6 exhibit 16- and 74-fold enhancement in inhibition, while Cap 35-H-6 shows a 340-fold enhancement in binding, clearly indicating that higher inhibition enhancement can be obtained if the distance between the adjacent saccharide ligands along the backbone better matches the spacing between carbohydrate binding sites of the lectin target. The significant reduction in inhibition enhancement for the Cap 17-H-3 and Cap 17-H-6 relative to Cap 35-H-6 (p < 0.05), despite the fact that Cap 17-H-6 displays the same number of saccharides at an approximate 35 Å spacing as does Cap 35-H-6, likely results from unfavorable steric hindrance that reduces the avidity of multisite binding. The origins of the binding avidity of the 17-H-X scaffolds are not fully clear but may reflect a combination of multivalent binding and some statistical effects, given the higher density of saccharide ligands relative to the receptors and the flexibility of the glycopolypep-tide chain. Statistical effects, which increase the local concentration of the saccharide ligands in the vicinity of the receptors, have been commonly observed in synthetic glycopolymer inhibitors and effectors.26,30,68 As the structure of the Cap 17-H-X glycopolypeptides in solution is yet unknown, it is possible that the observed binding results from multivalent binding between alternating saccharides (with minimal statistical effects) and that this binding is attenuated by the steric hindrance of the additional saccharides. Synthesis of glycopolypeptides with saccharide spacing that is fully mismatched (e.g., 25 Å) would provide additional insight into the origins of the binding avidity and is being explored. Compared with Cap 17-H-3 and Cap 17-H-6, the higher-avidity Cap 35-H-6 has a lower saccharide density (and a lower valency compared with Cap 17-H-6) but an adjacent saccharide spacing designed to match the distance between two adjacent carbohydrate binding domains on CT B5. In this case, the multivalent effect almost certainly dominates the binding event and is suggested by these data to result in greater inhibitory potency.
In addition to saccharide spacing, other factors, such as valency, scaffold length, and molecular weight, may also affect the multivalent binding. The difference in inhibition enhancements for the Cap 17-H-3 and Cap 17-H-6 (16-fold and 74-fold, respectively), albeit small, likely reflects the higher valency of the Cap 17-H-6 which would improve binding on the basis of statistical effects and may also reflect the higher molecular mass and scaffold length of the 17-H-6, effects that have been previously indicated to be favorable for multivalent binding.26 A detailed comparison of the relative importance of valency, scaffold length, and molecular mass will require comparisons with additional glycopolypeptides. In addition, more detailed characterization of the binding event via isothermal titration calorimetry will permit a quantitative evaluation of the number of binding sites occupied and the enthalpic and entropic contributions to the binding event. These results provide initial evidence that the binding avidity can be manipulated in helical scaffolds via structural modulation of architecture, which has not been previously accessible synthetically.26
Backbone Conformation Effects on CT B5 Inhibition
As illustrated above, the presentation of saccharide residues on the helical scaffold improves inhibition enhancement, with the Cap 35-H-6 showing a 340-fold improvement in inhibition in comparison to the 162-fold improvement of the Cap 35-RC-6. Taken alone, these data suggest that improvements in inhibition are to be gained by altering polymer conformation, perhaps as a result of entropic effects, despite the less than 100% helicity of the helical glycopolypeptides. In our previous studies, random-coil glycopolypeptide-based inhibitors with smaller hydrodynamic volume consistently exhibited poorer inhibition than those with greater hydrodynamic volume, independent of molecular mass.34,37 On the basis of the GPC data of Figure 5, we therefore would have predicted that Cap 35-H-6 should show the lowest inhibition of the glycopolypeptides produced to date; expected trends are highlighted by the trend line for the data in Figure 7. As shown in the figure, the observed improvements in inhibition for the Cap 35-H-6 are markedly greater than anticipated on the basis of its size; such improvements are not observed for either of the helical glycopolypeptides with the higher saccharide densities, suggesting the need for optimized spacing of adjacent saccharides along with optimization of chain conformation/flexibility. Additional GPC experiments conducted in PBS containing 20% acetonitrile showed much more similar elution times for the Cap 35-H-6 and Cap 35-RC-6, suggesting the possibility of complicating enthalpic effects in the GPC experiment, although inhibition data could not be collected under these alternate solution conditions. Nevertheless, the combination of the inhibition and GPC data suggest opportunities for substantial improvements in inhibition by these glycopolypeptides with the production of helical scaffolds of greater hydrodynamic volume.
Figure 7.
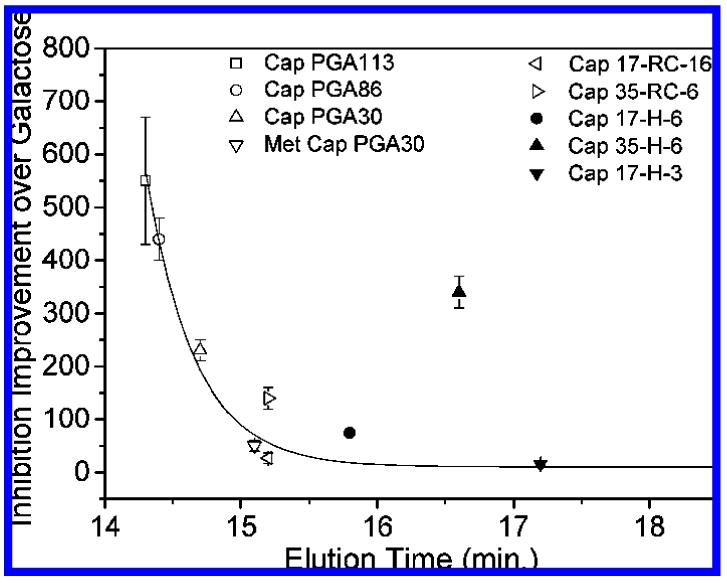
Correlation of inhibition improvement and hydrodynamic volume in PBS (as assessed via elution time in GPC) for random-coil glycopolypeptides (open)37 and α-helical glycopolypeptides (solid).
This inhibition improvement in the α-helical glycopolypeptides could be due to multiple factors. The accessibility of the saccharide ligands to the CT B5 binding sites may simply be improved, owing to the higher rigidity of the helical polypeptides that could improve control over the placement and accessibility of multiple saccharide ligands to the CT B5 binding pocket. In addition, conformational entropy effects may play a role in these observed improvements in inhibition, as the helical chains may be expected to experience a lower loss in conformational entropy upon binding, when compared to the highly flexible glycine-rich random-coil glycopolypeptides.37 The impact of the conformational entropy differences in these glycopolypeptides is likely to be small, however, given the <100% helicity of the helical scaffolds at room temperature (45–80%) and the only 2-fold difference of inhibition enhancements of the helical versus random-coil glycopolypeptides. Isothermal titration calorimetry experiments will help elucidate the origins of avidity differences in these macromolecules.
It is notable that the inhibition improvements of this family of helical glycopolypeptides is comparable with that of most synthetic multivalent glycopolymers (101–103-fold improvements), such as linear synthetic glycopolymers or dendrimers with high valency,36,70–77 despite the relatively small size of the glycopolypeptides. Moreover, compared with globular proteins and linear glycopolymers with similar valencies (100–101-fold improvements),72,73 these helical glycopolypeptides show substantially improved inhibitory potency. Although the inhibition enhancements of these helical glycopolypeptides are not comparable with those of the best pentavalent multivalent molecules reported with architectures specific for pentameric lectin receptors,15,16,22 the modulation of inhibition by manipulation of polymer architecture offers opportunities to describe multivalent interactions on the basis of macromolecular structure, which will serve as the basis for designing new architectures with improved binding avidity. The expansion of these glycopolymeric design principles will also be of significant importance in the manipulation of binding events over larger length scales for which small molecule inhibitors may not be as well-suited.78
Conclusions
The combination of protein engineering and chemical coupling methods was applied to synthesize two families of alanine-rich α-helical glycopolypeptides with well-defined spacing, valency, and conformation. CD experiments were used to monitor the conformation change after glycosylation and showed that glycosylation did not change helical conformation or thermal stabilities significantly. The inhibition of CT B5 by these glycopolypeptides was examined via DELA assay. All three multivalent α-helical glycopolypeptides showed inhibition enhancements over that of monovalent galactose, which likely arise from both multivalent effects and statistical effects. In this binding process, the importance of controlling spacing between adjacent saccharides was suggested by the improved inhibition of Cap 35-H-6 relative to that of Cap 17-H-6 and Cap 17-H-3. The fact that the helical glycopolypeptides exhibit greater enhancements in CT B5 inhibition relative to comparably sized random-coil glycopolypeptides suggests that the α-helical conformation can exert favorable effects on multivalent binding, although the relative contribution of entropic and enthalpic effects is currently unknown. Protein engineering strategies for the production for glycopolymers of improved and specified interactions with multivalent targets show significant promise; accordingly, the design of new inhibitors of controlled architecture, employing the insights of these studies, is underway.
Supplementary Material
Acknowledgment
This work was funded in part by the National Aeronautics and Space Administration (NA68-01923) and the National Center for Research Resources, a component of the National Institutes of Health (1-P20-RR017716-01 and 1-P20-RR015588 (instrument facilities)). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH. Ronak Maheshwari is thanked for helpful discussions and figure preparation. Brian Polizzotti, Ying Wang, and Ayben Top are also thanked for helpful suggestions and protocols.
References and Notes
- (1).Brewer CF, Miceli MC, Baum LG. Curr. Opin. Struct. Biol. 2002;12(5):616–623. doi: 10.1016/s0959-440x(02)00364-0. [DOI] [PubMed] [Google Scholar]
- (2).Kiessling LL, Gestwicki JE, Strong LE. Angew. Chem., Int. Ed. 2006;45(15):2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lee YC, Lee RT. Acc. Chem. Res. 1995;28(8):321–7. [Google Scholar]
- (4).Simanek EE, McGarvey GJ, Jablonowski JA, Wong CH. Chem. Rev. 1998;98(2):833–862. doi: 10.1021/cr940226i. [DOI] [PubMed] [Google Scholar]
- (5).Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson JD. Molecular Biology of the Cell. 4th Garland Publishing; New York: 2002. [Google Scholar]
- (6).Baessler KA, Lee Y, Roberts KS, Facompre N, Sampson NS. Chem. Biol. (Cambridge, MA) 2006;13(3):251–259. doi: 10.1016/j.chembiol.2005.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bunnell SC, Singer AL, Hong DI, Jacque BH, Jordan MS, Seminario MC, Barr VA, Koretzky GA, Samelson LE. Mol. Cell. Biol. 2006;26(19):7155–7166. doi: 10.1128/MCB.00507-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cho BK, Kieke MC, Boder ET, Wittrup KD, Kranz DM. J. Immunol. Methods. 1998;220(12):179–188. doi: 10.1016/s0022-1759(98)00158-6. [DOI] [PubMed] [Google Scholar]
- (9).Choi SK. Synthetic Multivalent Molecules—Concepts and Biomedical Applications. Wiley-Interscience; New York: 2003. [Google Scholar]
- (10).Fan E, Merritt EA, Verlinde CLMJ, Hol WGJ. Curr. Opin. Struct. Biol. 2000;10(6):680–686. doi: 10.1016/s0959-440x(00)00152-4. [DOI] [PubMed] [Google Scholar]
- (11).Lasky LA. Annu. Rev. Biochem. 1995;64:113–39. doi: 10.1146/annurev.bi.64.070195.000553. [DOI] [PubMed] [Google Scholar]
- (12).Merritt EA, Hol WGJ. Curr. Opin. Struct. Biol. 1995;5(2):165–71. doi: 10.1016/0959-440x(95)80071-9. [DOI] [PubMed] [Google Scholar]
- (13).Mowery P, Yang ZQ, Gordon EJ, Dwir O, Spencer AG, Alon R, Kiessling LL. Chem. Biol. 2004;11(5):725–732. doi: 10.1016/j.chembiol.2004.03.027. [DOI] [PubMed] [Google Scholar]
- (14).Solomon D, Kitov Pavel, I., Paszkiewicz E, Grant Gordon, A., Sadowska Joanna, M., Bundle David, R. Org. Lett. 2005;7:4369–72. doi: 10.1021/ol051529+. [DOI] [PubMed] [Google Scholar]
- (15).Zhang Z, Merritt EA, Ahn M, Roach C, Hou Z, Verlinde CLMJ, Hol WGJ, Fan E. J. Am. Chem. Soc. 2002;124:12991–12998. doi: 10.1021/ja027584k. [DOI] [PubMed] [Google Scholar]
- (16).Kitov PI, Sadowska JM, Mulvey G, Armstrong GD, Ling H, Pannu NS, Read RJ, Bundle DR. Nature (London) 2000;403(6770):669–672. doi: 10.1038/35001095. [DOI] [PubMed] [Google Scholar]
- (17).Chiu GNC, Edwards LA, Kapanen AI, Malinen MM, Dragowska WH, Warburton C, Chikh GG, Fang KYY, Tan S, Sy J, Tucker C, Waterhouse DN, Klasa R, Bally MB. Mol. Cancer Ther. 2007;6(3):844–855. doi: 10.1158/1535-7163.MCT-06-0159. [DOI] [PubMed] [Google Scholar]
- (18).Konkar S, Gupta S, Sampson NS. Bioorg. Med. Chem. Lett. 2004;14(6):1381–1384. doi: 10.1016/j.bmcl.2003.09.097. [DOI] [PubMed] [Google Scholar]
- (19).Sharkey RM, Cardillo TM, Rossi EA, Chang CH, Karacay H, McBride WJ, Hansen HJ, Horak ID, Goldenberg DM. Nat. Med. (N.Y.) 2005;11(11):1250–1255. doi: 10.1038/nm1322. [DOI] [PubMed] [Google Scholar]
- (20).Perez L, Ayala M, Pimentel G, Bell H, Canaan-Haden L, Bequet M, Gonzalez LJ, Miranda M, Ravelo R, Roque L, Acevedo B, Oliva JP, Gavilondo JV. Biotechnol. Appl. Biochem. 2006;43(1):39–48. doi: 10.1042/BA20050073. [DOI] [PubMed] [Google Scholar]
- (21).Fan E, O’Neal CJ, Mitchell DD, Robien MA, Zhang Z, Pickens JC, Tan XJ, Korotkov K, Roach C, Krumm B, Verlinde CLMJ, Merritt EA, Hol WGJ. Int. J. Med. Microbiol. 2004;294(4):217–223. doi: 10.1016/j.ijmm.2004.07.002. [DOI] [PubMed] [Google Scholar]
- (22).Fan E, Zhang Z, Minke WE, Hou Z, Verlinde CLMJ, Hol WGJ. J. Am. Chem. Soc. 2000;122:2663–2664. [Google Scholar]
- (23).Williams SJ, Davies GJ. Trends Biotechnol. 2001;19(9):356–362. doi: 10.1016/s0167-7799(01)01699-7. [DOI] [PubMed] [Google Scholar]
- (24).Pickens JC, Mitchell DD, Liu J, Tan X, Zhang Z, Verlinde CLMJ, Hol WGJ, Fan E. Chem. Biol. 2004;11:1205–1215. doi: 10.1016/j.chembiol.2004.06.008. [DOI] [PubMed] [Google Scholar]
- (25).Kiessling LL, Gestwicki JE, Strong LE. Curr. Opin. Chem. Biol. 2000;4(6):696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- (26).Cloninger MJ. Curr. Opin. Chem. Biol. 2002;6(6):742–748. doi: 10.1016/s1367-5931(02)00400-3. [DOI] [PubMed] [Google Scholar]
- (27).Lin CC, Yeh YC, Yang CY, Chen CL, Chen GF, Chen CC, Wu YC. J. Am. Chem. Soc. 2002;124:3508–3509. doi: 10.1021/ja0200903. [DOI] [PubMed] [Google Scholar]
- (28).Lee Y, Sampson NS. Curr. Opin. Struct. Biol. 2006;16(4):544–550. doi: 10.1016/j.sbi.2006.05.015. [DOI] [PubMed] [Google Scholar]
- (29).Spevak W, Foxall C, Charych DH, Dasgupta F, Nagy JO. J. Med. Chem. 1996;39:1018–20. doi: 10.1021/jm950914+. [DOI] [PubMed] [Google Scholar]
- (30).Wolfenden ML, Cloninger MJ. Bioconjug. Chem. 2006;17:958–966. doi: 10.1021/bc060107x. [DOI] [PubMed] [Google Scholar]
- (31).Kiessling LL, Pohl NL. Chem. Biol. 1996;3(2):71–7. doi: 10.1016/s1074-5521(96)90280-x. [DOI] [PubMed] [Google Scholar]
- (32).Kalovidouris SA, Blixt O, Nelson A, Vidal S, Turnbull WB, Paulson JC, Stoddart JF. J. Org. Chem. 2003;68:8485–8493. doi: 10.1021/jo030203g. [DOI] [PubMed] [Google Scholar]
- (33).Lundquist JJ, Debenham SD, Toone EJ. J. Org. Chem. 2000;65:8245–8250. doi: 10.1021/jo000943e. [DOI] [PubMed] [Google Scholar]
- (34).Polizzotti BD, Kiick KL. Biomacromolecules. 2006;7:483–490. doi: 10.1021/bm050672n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mitchell DD, Pickens JC, Korotkov K, Fan E, Hol WGJ. Bioorg. Med. Chem. 2004;12(5):907–920. doi: 10.1016/j.bmc.2003.12.019. [DOI] [PubMed] [Google Scholar]
- (36).Vrasidas I, De Mol NJ, Liskamp RMJ, Pieters RJ. Eur. J. Org. Chem. 2001;(24):4685–4692. [Google Scholar]
- (37).Polizzotti BD, Maheshwari R, Vinkenborg J, Kiick KL. Macromolecules. 2007;40:7103–7110. doi: 10.1021/ma070725o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Masserini M, Freire E, Palestini P, Calappi E, Tettamanti G. Biochemistry (Moscow) 1992;31(8):2422–6. doi: 10.1021/bi00123a030. [DOI] [PubMed] [Google Scholar]
- (39).Farmer RS, Argust LM, Sharp JD, Kiick KL. Macromolecules. 2006;39:162–170. doi: 10.1021/ma051534t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Frederick M, Ausubel RB, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Short Protocols in Molecular Biology. 4th Wiley John & Sons; New York: 1999. [Google Scholar]
- (41).Minke WE, Roach C, Hol WGJ, Verlinde CLMJ. Biochemistry (Moscow) 1999;38(18):5684–5692. doi: 10.1021/bi982649a. [DOI] [PubMed] [Google Scholar]
- (42).Bowen WP, Jerman JC. Trends Pharmacol. Sci. 1995;16(12):413–7. doi: 10.1016/s0165-6147(00)89091-4. [DOI] [PubMed] [Google Scholar]
- (43).Farmer RS, Kiick KL. Biomacromolecules. 2005;6:1531–1539. doi: 10.1021/bm049216+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Miller JS, Kennedy RJ, Kemp DS. J. Am. Chem. Soc. 2002;124:945–962. doi: 10.1021/ja011726d. [DOI] [PubMed] [Google Scholar]
- (45).Scholtz JM, Qian H, Robbins VH, Baldwin RL. Biochemistry (Moscow) 1993;32:9668–76. doi: 10.1021/bi00088a019. [DOI] [PubMed] [Google Scholar]
- (46).Lyu PC, Liff MI, Marky LA, Kallenbach NR. Science (N.Y.) 1990;250(4981):669–73. doi: 10.1126/science.2237416. [DOI] [PubMed] [Google Scholar]
- (47).Kohn WD, Monera OD, Kay CM, Hodges RS. J. Biol. Chem. 1995;270(43):25495–506. doi: 10.1074/jbc.270.43.25495. [DOI] [PubMed] [Google Scholar]
- (48).Krylov D, Mikhailenko I, Vinson C. EMBO J. 1994;13(12):2849–61. doi: 10.1002/j.1460-2075.1994.tb06579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kennedy RJ, Tsang KY, Kemp DS. J. Am. Chem. Soc. 2002;124:934–944. doi: 10.1021/ja016285c. [DOI] [PubMed] [Google Scholar]
- (50).Miller JS, Kennedy RJ, Kemp DS. J. Am. Chem. Soc. 2002;124:945–962. doi: 10.1021/ja011726d. [DOI] [PubMed] [Google Scholar]
- (51).Farmer RS, Top A, Argust LM, Liu S, Kiick KL.Pharm. Res 2007. DOI: 10.1007/s11095-007-9344-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Allen DL, Pielak GJ. Protein Sci. 1998;7(5):1262–1263. doi: 10.1002/pro.5560070524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Brumano MH, Rogana E, Swaisgood HE. Arch. Biochem. Biophys. 2000;382(1):57–62. doi: 10.1006/abbi.2000.1983. [DOI] [PubMed] [Google Scholar]
- (54).Israelachvili JN. Intermolecular and Surface Forces: With Applications to Colloidal and Biological Systems. Academic Press; New York: 1992. [Google Scholar]
- (55).Creighton TE. Proteins Structures and Molecular Properties. 2nd W. H. Freeman; New York: 1993. [Google Scholar]
- (56).Merritt EA, Sarfaty S, van den Akker F, L’Hoir CL, Martial JA, Hol WGJ. Protein Sci. 1994;3(2):166–75. doi: 10.1002/pro.5560030202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Broersen K, Voragen Alphons, G. J., Hamer Rob, J., De Jongh Harmen, H. J. Biotechnol. Bioeng. 2004;86(1):78–87. doi: 10.1002/bit.20030. [DOI] [PubMed] [Google Scholar]
- (58).O’Neil KT, DeGrado WF. Science (N.Y.) 1990;250(4981):646–51. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- (59).Bertozzi CR, Hoeprich PD, Jr, Bednarski MD. J. Org. Chem. 1992;57:6092–4. [Google Scholar]
- (60).Sola RJ, Al-Azzam W, Griebenow K. Biotechnol. Bioeng. 2006;94(6):1072–1079. doi: 10.1002/bit.20933. [DOI] [PubMed] [Google Scholar]
- (61).Swanwick RS, Daines AM, Tey LH, Flitsch SL, Allemann RK. ChemBioChem. 2005;6(8):1338–1340. doi: 10.1002/cbic.200500103. [DOI] [PubMed] [Google Scholar]
- (62).Davis BG, Jones JB. Synlett. 1999;(9):1495–1507. [Google Scholar]
- (63).Ricardo J. Sola, Griebenow K. FEBS Lett. 2006;580(6):1685–90. doi: 10.1016/j.febslet.2006.02.001. [DOI] [PubMed] [Google Scholar]
- (64).Vegarud G, Christensen TB. Biotechnol. Bioeng. 1975;17(9):1391–7. doi: 10.1002/bit.260170918. [DOI] [PubMed] [Google Scholar]
- (65).MacCallum JL, Moghaddam MS, Chan HS, Tieleman DP. Proc. Natl. Acad. Sci. U.S.A. 2007;104(15):6206–6210. doi: 10.1073/pnas.0605859104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Scholtz JM, Marqusee S, Baldwin RL, York EJ, Stewart JM, Santoro M, Bolen DW. Proc. Natl. Acad. Sci. U.S.A. 1991;88(7):2854–8. doi: 10.1073/pnas.88.7.2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Persikov AV, Xu Y, Brodsky B. Protein Sci. 2004;13(4):893–902. doi: 10.1110/ps.03501704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Wolfenden ML, Cloninger MJ. J. Am. Chem. Soc. 2005;127:12168–12169. doi: 10.1021/ja053008n. [DOI] [PubMed] [Google Scholar]
- (69).Mammen M, Chio SK, Whitesides GM. Angew. Chem., Int. Ed. 1998;37(20):2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- (70).Kitov PI, Shimizu H, Homans SW, Bundle DR. J. Am. Chem. Soc. 2003;125:3284–3294. doi: 10.1021/ja0258529. [DOI] [PubMed] [Google Scholar]
- (71).Merritt EA, Zhang Z, Pickens JC, Ahn M, Hol WGJ, Fan E. J. Am. Chem. Soc. 2002;124:8818–8824. doi: 10.1021/ja0202560. [DOI] [PubMed] [Google Scholar]
- (72).Lundquist JJ, Toone EJ. Chem. Rev. 2002;102:555–578. doi: 10.1021/cr000418f. [DOI] [PubMed] [Google Scholar]
- (73).Gestwicki JE, Strong LE, Kiessling LL. Chem. Biol. 2000;7(8):583–591. doi: 10.1016/s1074-5521(00)00002-8. [DOI] [PubMed] [Google Scholar]
- (74).Mangold SL, Cloninger MJ. Org. Biomol. Chem. 2006;4(12):2458–2465. doi: 10.1039/b600066e. [DOI] [PubMed] [Google Scholar]
- (75).Schlick KH, Udelhoven RA, Strohmeyer GC, Cloninger MJ. Mol. Pharmaceutics. 2005;2:295–301. doi: 10.1021/mp050014h. [DOI] [PubMed] [Google Scholar]
- (76).Appeldoorn CCM, Joosten JAF, el Maate FA, Dobrindt U, Hacker J, Liskamp RMJ, Khan AS, Pieters RJ. Tetrahedron: Asymmetry. 2005;16(2):361–372. [Google Scholar]
- (77).Yamaguchi S, Nishida Y, Sasaki K, Kambara M, Kim CL, Ishiguro N, Nagatsuka T, Uzawa H, Horiuchi M. Biochem. Biophys. Res. Commun. 2006;349(2):485–491. doi: 10.1016/j.bbrc.2006.08.072. [DOI] [PubMed] [Google Scholar]
- (78).Kiessling LL, Gestwicki JE, Strong LE. Angew. Chem., Int. Ed. 2006;45(15):2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.