

Abstract
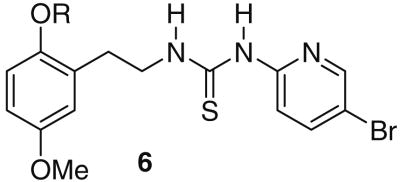
Several novel thiourea derivatives of the NNRTI HI-236 substituted at the C-2 oxygen of the phenyl ring have been synthesized and evaluated for their inhibitory activity against HIV-1 (IIIB) replication in MT-2 cell cultures. The compounds were synthesized in order to fine-tune the activity of HI-236 as well as to gain insight into spatial characteristics in the pocket pertaining to the positional choice of tether in the design of [NRTI]-tether-[HI-236] bifunctional inhibitors. Two of the thiourea derivatives bearing a butynyl (6c) or hydroxyethyl tether (6n) were endowed with improved anti-HIV activity compared to HI-236. NNRTI activity was confirmed by a cell-free RT assay on six of the derivatives in which 6c returned an IC50 of 3.8 nM compared to 28 nM for HI-236, establishing it as an improved lead for HI-236. The structure-activity profile is discussed in terms of potential interactions in the NNRTI pocket as suggested by a docking model using AutoDock, which have a bearing on the bifunctional drug design.
Keywords: NNRTI, HI-236, AutoDock, Thiourea, HIV, Bifunctional inhibitors
1. Introduction
The HIV-1 reverse-transcriptase enzyme is responsible for converting the genomic single-stranded RNA of HIV into a double-stranded DNA and continues to be a major target for anti-HIV drug discovery.1 Inhibitors fall into two distinct classes as nucleoside analogues (NRTIs) and non-nucleoside analogues (NNRTIs), and their modes of action are distinct and well documented.2 Thus, while NRTIs act as competitive substrates at the substrate-binding site, the NNRTIs work allosterically in an adjacent pocket, interfering with reverse transcription by altering the conformational mobility of RT, which results in non-competitive inhibition. To date, more than 30 structurally diverse NNRTIs have been identified, which have been comprehensively reviewed by several workers.3 NNRTIs are vulnerable to HIV's high mutation rate, and to circumvent this, they are currently used in combination with NRTIs. An alternative strategy that has been investigated by a number of groups over the last decade or so has been to combine an NRTI and an NNRTI into a single bifunctional molecule.4 Such inhibitors may be classified into two types: cleavable5 and non-cleavable.6 The former involve NRTI–linker–NNRTI systems that are designed to release the two drugs into the cytoplasm via enzymatic hydrolysis in the hope of promoting synergistic inhibitory action. Conversely, the latter, based on an original suggestion by Nanni7 and coworkers, are designed to promote inhibition at both sites cooperatively through the individual drugs in the same entity, on the basis that the NRTI and NNRTI drug–target sites are in close proximity of 10–15 Å.8 The latter, ambitious strategy has produced some inventive combinations, but true bifunctional character has yet to be comprehensively established. In the design of such bifunctional drugs, it is important to identify attachment points on each drug as well as to choose an appropriate tether regarding size and functional character.
We have been interested in developing non-cleavable bifunctional NRTI/NNRTI compounds involving d4T as the NRTI and the PETT (phenethylthiazolyl) derivative HI-236 as the NNRTI. These compounds were selected in view of their excellent profiles as antiviral agents, and it was decided to attach the linker on the NRTI side of the molecule to C-5 of the pyrimidine base in view of anticipated low interference with base pairing.9 On the HI-236 side, it was decided to use the C-2 phenolic oxygen in view of studies conducted by Uckun10 regarding the binding mode of HI-236 in the RT pocket. Our recently published11 first prototype, [d4T]-butyne-[HI-236], returned an EC50 of 250 nM against HIV-1 (IIIB) in MT-2 cell culture using an MTT assay. This result compares favourably with Ladurée's bifunctional,12 also based on d4T but conjugated to a PETT derivative through a glycylsuccinyl cleavable linker, which turned out to be inactive (Fig. 1).
Figure 1.

Structures of two recent bifunctional inhibitors based on PETT compounds with d4 T.
PETT (phenethylthiazolyl) derivatives such as those shown in Figure 1 were first introduced by the Eli Lilly group in 1995.13 Changing the thiazole ring to a pyridine ring as N-(5-bromo-2-pyridinyl)-N′-(2-pyridylethyl)-thiourea (named as Trovirdine), resulted in enhanced activity and a detailed structural analysis of it carried out by the Uckun group revealed abundant sterically usable space surrounding the pyridyl group as well as the ethyl linker.14,15 They postulated that an efficient use of this space would lead to more potent anti-HIV agents with higher affinity for the NNRTI binding pocket, and this resulted in several more potent derivatives including the highly potent HI-236,10,15 with a much superior inhibition profile in which the pyridyl ring of trovirdine was replaced by a 2,5-dimethoxyphenyl group, Figure 2.
Figure 2.

Structures of HI-236 and Trovirdine.
Structural studies around HI-236 revealed, as with other PETT derivatives, that the thiourea portion interacts in Wing 1 towards the front of the pocket via H-bonding with K101.10,15 Conversely, the C-5-methoxy group forges cooperative C–Hπ interactions with the highly conserved W229, while the phenyl ring interacts with Y188. The methoxy group at C-2 was found to lie underneath the ethyl linker and was considered to occupy some of the vacant space near Y188 in Wing 2, an issue which assumes a greater weight of importance for drug-resistant strains where there is an increase in pocket size due to mutated residues of smaller size such as Y181C and Y188L. The realization of the [d4T]-butynyl-[HI-236] bifunctional prototype mentioned previously11 prompted a study of the influence of the nature of the C-2 tether of HI-236 on its biological activity. In this paper, we present the synthesis and biological activity of a small library of HI-236 derivatives involving substitution of the C-2 phenolic methyl group, the objective being not only to shed light on NNRTI attachment at the C-2 oxygen for the bifunctional design, but also to probe the possibility of further binding in the region of the pocket just mentioned in an attempt to improve the activity of HI-236 even further. Tethers attached to the C-2 phenolic oxygen were chosen to probe length, functionality and polarity. Bearing in mind that a Sonogashira connection16 between C-5 of d4T and a terminal alkyne was used for the bifunctional connection chemistry, some, but not all of the derivatives studied were selected with an alkyne terminus.
2. Chemistry
Synthesis of the tethered derivatives utilized phenolic amine 1 as a key intermediate that was readily available in multigram quantities using chemistry reported by Glennon17 via a three-step sequence from commercially available 2-hydroxy-5-methoxybenzaldehyde, Scheme 1. In our case, it was found it easier to isolate 1 as an N-Boc carbamate 2. Yields for the various steps in the sequence to carbamate 2 were high and spectroscopic data was in accordance with literature values for all of the known compounds, Scheme 1.
Scheme 1.

Reagents and conditions: (i) BnBr, K2CO3, EtOH, reflux, 16 h; 95%; (ii) CH3NO2, NH4OAc, 70 °C, 14 h; 75%; (iii) LiAlH4, THF, reflux, 4 h; (iv) (Boc)2O, Et3N, CH3CN, rt, overnight; 76% (2 steps); (v) H2, Pd/C, EtOH, rt, 5 h; 66%.
Thereafter, following benzyl group deprotection to 3, the various O-alkylations at the C-2 phenolic oxygen could be carried out with introduction of the thiourea moiety last for each case. A more convergent approach involving converting amine 1 first into the thiourea derivative, debenzylating and then carrying out the C-2 phenolic alkylation reaction to generate a small library of targets was deemed to be unattractive in view of the anticipated incompatibility of the thiourea functionality with the alkylation chemistry.
Alkylation of carbamate 3 (Scheme 2 and Table 1) to afford a library of alkylated N-Boc derivatives 4 proceeded efficiently with bromide or tosylate electrophiles using K2CO3 in acetonitrile at reflux for around 20 h and proved to be superior to a Mitsunobu reaction with the corresponding alcohol. Yields of chromatographed products were generally in excess of eighty percent (see Section 4). In most cases, the electrophile was readily available either commercially or via a one-step derivatisation.
Scheme 2.

Reagents and conditions: (i) K2CO3, CH3CN, 80 °C, 20 h or NaH, DME, 80 °C, 20 h with ROTs.
Table 1.
O-Alkylation of 3 to afford 4 as precursors of C-2 substituted HI-236 derivatives.
| R | No. | R | No. | R | No. | R | No. |
|---|---|---|---|---|---|---|---|
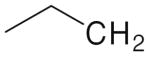 |
4a | 4e | 4i | 4m | |||
| 4b | 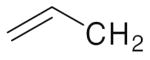 |
4f | 4j | 4n | |||
| 4c | Bn | 4g | 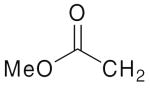 |
4k | 4o | ||
| 4d | 4h | 4l |
The exceptions came for the alkynyl tethers used to probe the question of the influence of tether length and aqueous solubility of the bifunctional compounds. These involved alkynyl PEG (ethylene glycol) chains and were synthesized by the following sequence shown in Scheme 3 in which the tether was developed divergently. Thus, standard phenolic alkylation of a monobenzylated glycol bromide afforded the alkylated product, which was committed to a two-step sequence involving hydrogenolytic debenzylation followed by tosylation to afford tosylates 5a and 5b. Nucleophilic displacement with propargyloxide anion then furnished the products 4i and 4j. The latter18 turned out to be superior to hydroxyl-group alkylation with propargyl bromide. Similarly, alcohols 4n and 4o were obtained from hydrogenolysis of their benzyl ethers obtained from alkylation of 3 as described.
Scheme 3.

Reagents and conditions: (i) BnPEGBr (n = 0 or 1), K2CO3, CH3CN, 80 °C, 20 h or NaH, DME, 80 °C, 20 h; (ii) H2, Pd/C, EtOH, rt, 18 h (iii) TsCl, Et3N, DMAP, CH2Cl2, 0 °C–rt, 20 h (iv) propargyl alcohol, NaH, THF, 70 °C, 5 h.
Table 1 summarises the various products of C-2 phenolic alkylation of Boc carbamate 3.
All derivatives 4a–o returned acceptable NMR spectra together with acceptable combustion analysis data (solids) and/or HRMS mass spectral data. Notable in the NMR were the triad of signals for the three aromatic protons in the 1H NMR spectrum integrating correctly against the N-Boc tert-butyl singlet. The 13C NMR spectra returned the correct number of carbon singlets in each case.
Finally, synthesis of the target thioureas 6 was accomplished via a Boc-deprotection, condensation sequence shown in Scheme 4.
Scheme 4.

Reagents and conditions: (i) CF3COOH, CH2Cl2, 0 °C, 1 h and then DIEA, 0 °C, 10 min, followed by 8, THF, rt, 2 h.
Thus, each derivative 4 was reacted with trifluoroacetic acid (TFA) in DCM at 0 °C for 1–3 h until TLC revealed complete deprotection to a polar amine spot. In view of the amine's anticipated water solubility, the final step was conducted without using an aqueous work-up. Thus, following complete removal of all volatiles including any excess TFA, the residue was redissolved in THF, Hünig's base (EtN(i-Pr)2) was added to liberate the free amine and the thiocarbonyl reagent 7 added according to the original procedure. As described by the Eli Lilly group,13 the latter could be readily prepared by reacting 2-amino-5-bromopyridine with 1,1′-thiocarbonyldiimidazole in acetonitrile at room temperature for 12 h to afford a precipitate that was used without purification. Attempted recrystallization of 7 from methanol resulted in a product with imidazole substituted by methoxy (Scheme 5).
Scheme 5.

Reagents and conditions: (I) 2-amino-5-bromopyridine, CH3CN, rt, 120.
Condensation between the amine and 7 could be realized in DMF at 100 °C as described by Bell and coworkers13 to afford the targets 6a–o in an overall yield of around 30% for the two steps. However, later on, it was discovered that condensation could be carried out under much milder conditions in THF or DMF at room temperature to increase the two-step yield to around 60%. All of the targets were isolated by silica-gel column chromatography as crystalline solids that were crystallized to a constant, sharp melting point to return acceptable combustion analysis data. A full spectroscopic analysis using 1H and 13C NMR spectroscopy was also carried out on each derivative to return acceptable data. Notably, the aromatic region in the 1H NMR spectra provided convenient markers for the aromatic and heteroaromatic rings to demonstrate that condensation had taken place. The thiourea N–H's could be discerned downfield as two separate resonances. The thiourea thiocarbonyl carbon could be identified using 13C NMR, resonating at around 179 ppm. The retention of bromine in the pyridine ring was confirmed by 13C NMR (δC–Br ∼112.6 ppm) as well as microanalytical and IR data. Table 2 summarises yields for the two-step sequence of 4–6 in Scheme 4.
Table 2.
Yields for converting 4–6.
| Compound | Yield | Compound | Yield | Compound | Yield | Compound | Yield |
|---|---|---|---|---|---|---|---|
| 6a | 65 | 6e | 60 | 6i | 62 | 6m | 53 |
| 6b | 17a | 6f | 67 | 6j | 60 | 6n | 65 |
| 6c | 25a | 6g | 37 | 6k | 50 | 6o | 51 |
| 6d | 69 | 6h | 58 | 6l | 68 |
DMF at 100 °C; the others involved THF at rt.
3. Biological results, modeling, and discussion
The inhibitory activity of compounds 6a–o was measured against HIV-1 (IIIB) replication in MT-2 cell culture using an MTT assay.19 HI-236 was included as a reference compound. The results are shown in Table 3.
Table 3.
Antiviral activity and cytotoxicity assaya results for 6a–o against HIV-1 (IIIB) in MT-2 cell culture.
| R | EC50b (μM) | CC50c (μM) | T.Id | R | EC50b (μM) | CC50c (μM) | T.Id |
|---|---|---|---|---|---|---|---|
| CH3 HI-236 | 0.048 | 1.0 | 21 |
|
2.0 | 5.8 | 3 |
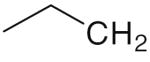 6a 6a
|
0.095 | 0.1 | 1 |
|
0.095 | 9.5 | 100 |
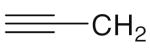 6b 6b
|
0.052 | 0.1 | 2 |
|
0.39 | 6.8 | 17 |
|
|
0.026 | 1 | 38 |
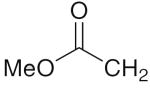 6k 6k
|
12 | 48 | 4 |
|
|
0.25 | 10.0 | 40 |
|
0.12 | 5.5 | 46 |
|
|
0.27 | 20.0 | 74 |
|
0.05 | 3.8 | 76 |
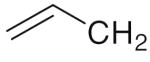 6f 6f
|
0.20 | 11.0 | 55 |
|
0.012 | 1.0 | 83 |
| Bn 6g | 0.22 | 4.0 | 18 |
|
0.098 | 8.5 | 94 |
Ref. 19; MOI was 0.1.
Effective concentration that inhibits viral-mediated T-cell death by 50%, determined by averaging samples of each concentration in triplicate.
Concentration that kills 50% of the T-cells, also determined by averaging triplicate samples.
In vitro therapeutic index (CC50/EC50).
In order to lend support that these HI-236 derivatives act as NNRTIs, six of the derivatives (6c, 6d, 6i, 6k, 6n, and 6o) covering the most (6n) to the least active (6k) were subjected to an in vitro steady-state RT inhibition assay, the results from which are shown in Table 4. The results present strong support for all of the derivatives in this study to be acting as NNRTIs since they have no possibility of being phosphorylated for NRTI activity against RT. This is also in keeping with the published findings on HI-236. The activities ranged from 3.8 to 100 nM, and as expected were superior to those from the cell-culture results except for 6n, which was slightly lower. Such improvements likely reflect the lipophilicity of the derivatives and their poor solubility in the aqueous cell-culture medium. Notably, the ester 6k, which was effectively inactive in cell culture (12 μM), revealed a 120-fold improvement to 100 nM in the RT assay, possibly due to ester hydrolysis in the cell-culture medium.20 Importantly, the alkyne 6c returned a value of 3.8 nM in the RT assay, which was more active compared to that of the alcohol 6n (19 nM), in spite of the reverse being true in cell culture (26 nM vs 12 nM, respectively). The most active derivative 6c was then evaluated in cell culture against the Y181C resistant strain and retained activity much better than HI-236 (8-fold decrease against 25 for HI-236).
Table 4.
Comparison of cell culture versus in vitro RT inhibition (nM) for selected derivatives of 6.
 | ||||
|---|---|---|---|---|
| R | Antiviral activity EC50 (nM) | RT inhibition assay IC50 (nM) | Antiviral activity Y181C EC50 (nM) | Cellular Toxicity Y181C CC50 (TI) (nM) |
| Me (HI-236) | 48 | 28 | 1200 | 18,000 (15) |
|
|
26 | 3.8 | <200 | 4000 (>20) |
|
|
250 | 27 | ||
|
|
95 | 41 | ||
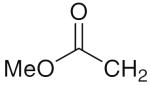 6k 6k
|
12,000 | 100 | ||
|
|
12 | 19 | ||
|
|
98 | 24 | ||
Regarding a general comparison of all derivatives, the results from the cell culture shown in Table 3 were taken to reveal important trends within classes of different tether (polar vs lipophilic). Four of the compounds returned activities equal to HI-236 (6b and 6m) or greater (6c, 2-fold increase; 6n, 4-fold increase). In order to assist with interpretation of the results, docking studies were carried out using AutoDock 3.0521 based on the recently published HIV-1 RT protein crystal structure of N-(5-chloro-2-pyridinyl)-N′-[2-(4-ethoxy-3-fluoro-2-pyridinyl)ethyl]-thiourea as template.22 The conformation of the PETT derivatives studied was set to accommodate the well documented14,22,23 intramolecular hydrogen bond between the hydrogen of the thiourea nitrogen attached to the alkyl side and the nitrogen of the pyridinyl ring to form a flat six-membered pseudo-ring. This protocol resulted in a strong preference for one low energy conformation which dominated 153 of the 250 possible conformations. HI-236 was docked first, and we were able to establish parity of result with that obtained by Uckun. The two structures are shown in Figure 3.
Figure 3.
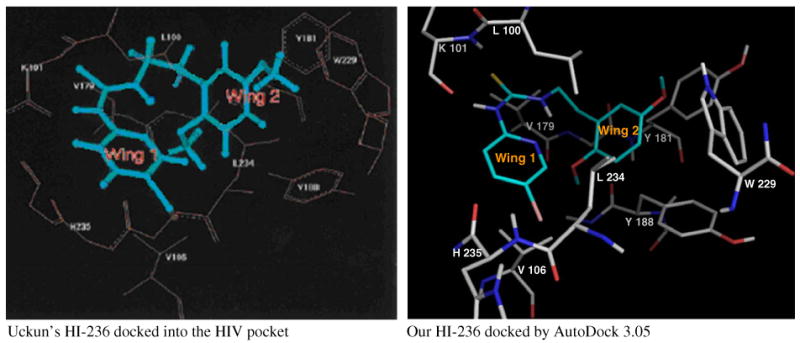
HI-236 docked into the HIV pocket.
Thus, as established by Uckun, the inhibitor adopts the characteristic butterfly-shaped orientation with the thiourea moiety embedded in Wing 1 in which hydrogen bonding between the thiourea hydrogen on the N-pyridyl side and the carbonyl oxygen of K101 is clearly visible. Wing 2 accommodates the phenyl ring of HI-236 close to the Y181 and Y188 region with the 5-methoxy group interacting with W229. The 2-alkoxy substituent positions into a well-containing residues Y181, Y188, F227, V106, and V179. This feature had significance for the aims of this study. The only difference between our structure and that of Uckun's was the orientation of W229 relative to Y181. In Uckun's model, the six-membered ring of the indole of W229 overlapped Tyr181, whereas the model obtained from our calculations had the five-membered ring of the indole overlapping Tyr181. This could have been due to differences in the static protein structure used in our docking calculations as compared with the structure of the HIV-1 RT binding pocket used in the studies by Uckun. Attention was then focused on docking two of the derivatives, chosen as the ester 6k and the alcohol 6o. The parity of 6k and 6o with our docked HI-236 in terms of overall topology in the pocket was first investigated, as illustrated in Figure 4 for 6k. The elongation of the 2-alkoxy substituent can be seen but the positioning of the rest of the PETT structure essentially stayed the same, revealing that EC50 differences are likely to be based on interactions in the Wing 2 region as postulated. Alcohol 6o gave a similar result.
Figure 4.
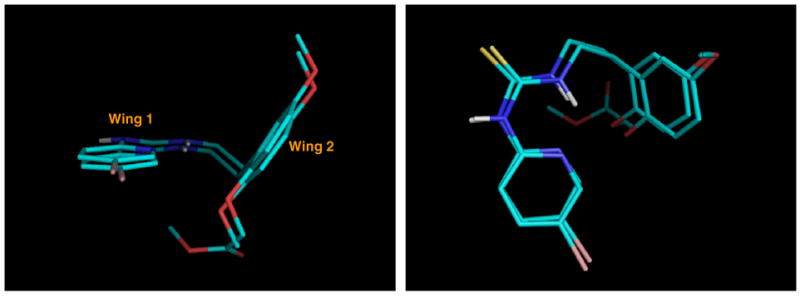
The similarities in orientation between the AutoDock models of HI-236 and 6k.
Examination of the way in which the 2-alkoxy substituents of 6k and 6o positioned in the hydrophobic region of Wing 2 as shown in Figures 5 and 6 taken from each end of the pocket for both cases, gave insights into the possible interactions responsible for fluctuation of biological activity.
Figure 5.
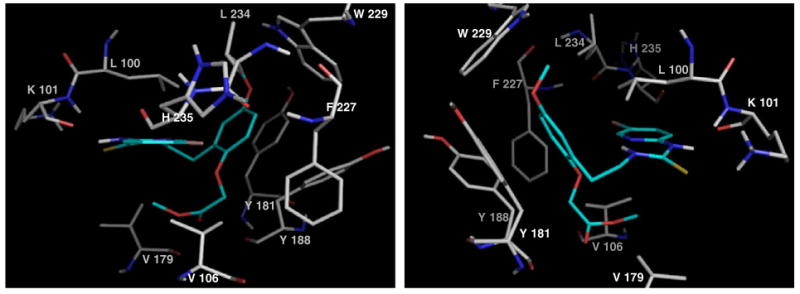
Docking of ester 6k.
Figure 6.
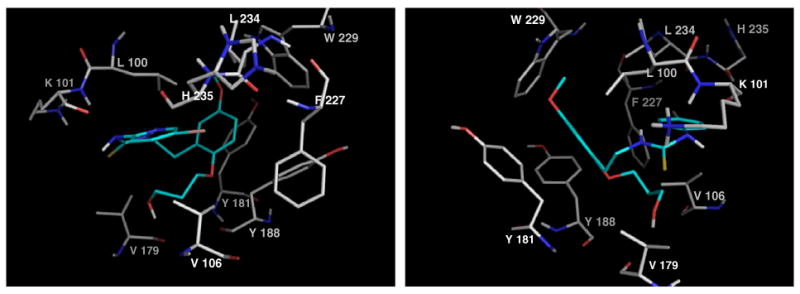
Docking of alcohol 6o.
Both structures suggest the possibility of interaction with V109 or, to a lesser extent V179, towards the floor of the pocket. A cooperative hydrogen bond for 6o would nicely explain the much enhanced activity of alcohol 6n (EC50 = 0.012 μM) with a shorter (two-carbon) chain, in which presumably the distance between hydrogen-bond donor and acceptor is optimal. 6n returned an activity four times higher than HI-236 in cell culture, and almost twice that in the RT assay and thus presents itself as an interesting candidate for the problematic V106A mutation if the hydrogen bonding postulate is correct.
Similarly, for the alkyl and alkynyl compounds 6a–f, the results show that the hydrophobic pocket is accommodating but with the only significant cooperative interaction being with the triple bond. In this regard, the butynyl chain of 6c, rather than the propargyl (6b) or pentynyl substituents (6d) appears to be optimal, with 6c indicating a 2-fold increase in activity compared to HI-236 in cell culture and 7-fold in the RT assay. Although this classical force field does not include an explicit π–π term, the charge distribution on the aromatic rings mimics this fundamentally quantum effect surprisingly well. Therefore we suggest that the increased activity of 6c is an optimal cooperative π–π face-to-face interaction between the aromatic residues of Y181, Y188, and the triple bond. In addition, a much lower fold reduction in activity for 6c (8×) was observed against the Y181C24 mutant strain compared to that of HI-236 (25×), (see Table 4). The PEG derivatives predictably returned lower activities, but not overly so given their bulk. Thus 6j, with 10 atoms in the substituent, returned an activity of 0.39 μM as only 8-fold less potent than HI-236. By comparison, the benzyl derivative 6h is too bulky to be satisfactorily accommodated. Finally, the nitrile derivative 6m indicated some level of cooperation in the pocket being as active as HI-236, in spite of its five-atom substituent. Once again, π–π and/or hydrogen bonding possibilities appear to be likely explanations.
In summary, it is important to note that of the 15 compounds tested in cell culture, two (6c and 6n) were more active than HI-236 and two (6b and 6m) were as active, while this picture improved in the RT assay in which of the six derivatives tested, two (6c and 6n) were more active and two were as active as HI-236 (6d and 6o). Such results endorse the conclusions drawn from the study by Uckun15 that the PETT derivatives have unused available pocket volume with good potential for drug-development, and have identified tethered butynyl derivative 6c as an advanced lead. Further fine-tuning is worthwhile pursuing on developing side chains that can cope with mutated residues contained in resistant strains,25 and the modelling results suggest that this might be possible to achieve by adding further substitution at the C-3 position of the aromatic ring ortho to the C-2 O-tether. In addition, the study has generated important insights regarding the choice of the C-2 oxygen as the attachment point for the tether in the bifunctional compounds, and the likelihood of a tether at this position providing a route from the pocket to the NRTI binding site. In this regard, a comprehensive study of elongated alkylated bifunctional double-drugs in order to shed light on the origin of biological activity for the prototype in Figure 111 will be communicated in a forthcoming paper.
4. Experimental
4.1. Docking aspects
The binding conformations of HI-236 (1) and its ester (6k) and alcohol (6o) derivatives bound to HIV-1 Reverse Transcriptase (RT) were modelled using AutoDock 3.0521 based on the published HIV-1 RT protein crystal structure of N-(5-chloro-2-pyridinyl)-N′-[2-(4-ethoxy-3-fluoro-2-pyridinyl)ethyl]-thiourea.22 Each ligand was built in Gaussview,26 and optimised using Gaussian 98 to relax bond lengths and angles that were not varied in the docking simulation. Polar hydrogen atoms were added to the protein and Kollman united-atom partial charges assigned using the AutoDockTools package. For each ligand, Gasteiger-Marsili27 partial charges were assigned, as implemented in AutoDockTools. Autodock 3.05 utilises an empirical scoring function21 to calculate binding free energies, which incorporates five energy terms, including a Lennard–Jones 12-6 term and a directional 12-10 hydrogen bond term, for which the default parameters distributed with AutoDockTools were used. Electrostatic interactions were calculated using a distance-dependent dielectric constant.28 Atomic solvation parameters and fragmental volumes were assigned using the AddSol utility, from which the desolvation contribution to the binding free energy is calculated. A 61 × 61 × 61 grid map was used in all docking calculations, with a grid spacing of 0.375 Å. Given the known location of the NNRTI binding site, the grid was centred on the coordinates for the equivalent atom of PETT-lig corresponding to the default selected root atom of each ligand investigated. Docked conformations were generated using the Lamarckian genetic algorithm (LGA) with an initial population size of 150 structures. Translation, quaternion and torsional step sizes were set to 2 Å, 5.0° and 5.0° respectively for HI-236 and 0.1 Å, 1.0° and 1.0° for ligands 9k and 9o. Further parameters were set to their default values. A total of 250 runs were performed for each ligand, and the resulting conformations clustered using a root mean-squared deviation criterion of 0.5 Å in x, y, z positional coordinates. Bond rotation from the pyridinyl ring across to the thiourea moiety was disallowed, and the conformation was set to accommodate the well documented14,21,22 intramolecular hydrogen bond between the hydrogen of one of the thiourea nitrogens and the nitrogen of the pyridinyl ring to form a flat six-membered pseudo-ring. This protocol resulted in a strong preference for one low energy conformation which dominated 153 of the 250 possible conformations. Although using a static protein structure in the docking simulation and limiting the rotation of certain bonds, the binding conformation of HI-236 in this study compared well with that obtained by Uckun.15
4.2. General procedures for synthesis
Microanalyses were obtained with a Fisons EA 110 CHN Elemental Analyser. Infrared (IR) absorptions were measured on a Perkin-Elmer Spectrum One FT-IR spectrometer. 1H NMR spectra were recorded on a Varian Mercury Spectrometer at 300 MHz and a Varian Unity Spectrometer at 400 MHz with Me4Si as internal standard. 13C NMR spectra were recorded at 75 MHz on a Varian Mercury Spectrometer or at 100 MHz on a Varian Unity Spectrometer with Me4Si as internal standard. High resolution mass spectra were recorded on a VG70 SEQ micromass spectrometer. Melting points were determined using a Reichert-Jung Thermovar hot-stage microscope and are uncorrected. Analytical thin-layer chromatography (TLC) was performed on silica-gel 60 F254 (Merck). Column chromatography was performed with Merck silica-gel 60 (70–230 mesh). HI-23610 was synthesized by the same sequence as for derivatives 6 and returned acceptable NMR data.
4.3. Procedure for tosylates 5a and 5b
The phenol 3 was O-alkylated with the appropriate PEGbromide as its monobenzyl ether according to the procedure below for formation of the alkylated derivatives 4. The alkylated products were subjected to hydrogenolysis in ethanol using Pd–C (10 mol%) at atmospheric pressure for 18 h. Following filtration through Celite, the alcohols were purified by column chromatography in yields in excess of 80% before being tosylated with p-toluenesulfonyl chloride (1.5 equiv) in dichloromethane using triethylamine (2 equiv) and DMAP (cat). Following a conventional work-up, the product was isolated by column chromatography. Solid products were generally recrystallized from ethyl acetate/hexane mixtures.
4.3.1. N-(tert-Butoxycarbonyl)-2-[5-methoxy-2-(2-p-toluenesulfonyloxyethoxy)phenyl]ethylamine (5a)
76% Yield as a colourless oil; IR (CHCl3) νmax 3690, 3451, 1706, 1367, 1164 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.75 (2H, d, J = 8.2 Hz), 7.28 (2H, d, J = 8.2 Hz), 6.62 (3H, m), 4.77 (1H, br s), 4.30 (2H, t, J = 4.7 Hz), 4.05 (2H, t, J = 4.7 Hz), 3.68 (3H, s), 3.22 (2H, q, J = 6.8 Hz), 2.65 (2H, t, J = 6.8 Hz), 2.38 (3H, s), 1.38 (9H, s); 13C NMR (100 MHz, CDCl3) δ: 155.9, 154.1, 150.2, 145.0, 132.9, 129.9, 129.3, 127.8, 116.7, 112.0, 111.9, 78.8, 68.4, 66.4, 55.5, 40.5, 30.9, 28.4, 21.5; EI-HRMS: m/z; found: 465.18130 (M+). C23H31NO7S (M+) requires 465.18212.
4.3.2. N-(tert-Butoxycarbonyl)-2-{5-methoxy-2-[2-(2-p-toluenesulfonyloxyethoxy)ethoxy]phenyl}ethylamine (5b)
83% Yield as a colourless oil; IR (CHCl3) νmax 3693, 3453, 1706, 1367, 1168 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.76 (2H, d, J = 7.9 Hz), 7.28 (2H, d, J = 7.9 Hz), 6.73 (1H, d, J = 8.8 Hz), 6.68 (2H, m), 4.80 (1H, br s), 4.17 (2H, t, J = 4.7 Hz), 3.98 (2H, t, J = 4.7 Hz), 3.75 (4H, m), 3.73 (3H, s), 3.29 (2H, q, J = 6.7 Hz), 2.74 (2H, t, J = 6.7 Hz), 2.39 (3H, s), 1.39 (9H, s); 13C NMR (100 MHz, CDCl3) δ: 155.9, 153.9, 150.9, 144.8, 133.1, 129.8, 129.3, 127.9, 116.6, 113.0, 112.0, 78.8, 70.0, 69.3, 68.8, 68.3, 55.6, 40.6, 31.0, 28.4, 21.5; EI-HRMS: m/z; found: 509.20675 (M+). C25H35NO8S (M+) requires 509.20834.
4.4. Procedure for the N-Boc O-alkylated derivatives 4
The alkylating agent as a tosylate or bromide (2.0 mmol) in dry acetonitrile (5 mL) was added dropwise over 1 h to a refluxing and stirring mixture of the phenol 3 (1.0 mmol) and anhydrous potassium carbonate (4.0 mmol) in dry acetonitrile (15 mL). The reaction was refluxed for 20 h. The mixture was filtered, the acetonitrile evaporated, and the residue subjected to silica-gel column chromatography (10% EtOAc/pet ether) to afford the product generally as a colourless solid. Entries 4n and 4o were derived via hydrogenolysis of their corresponding benzyl ethers, while entries 4i and 4j were obtained via substitution of tosylates 5a and 5b respectively using propargyloxide ion in refluxing THF (5 h). Yields cited in the text refer to the final step in each case.
4.4.1. N-(tert-Butoxycarbonyl)-2-(5-methoxy-2-propoxyphenyl)ethylamine (4a)
95% Yield, mp 52–54 °C; IR (CHCl3) νmax 3684, 3452, 3002, 2977, 1706, 1502 cm−1; 1H NMR (400 MHz, CDCl3) δ: 6.76 (1H, d, J = 9.2 Hz), 6.70 (2H, m), 4.78 (1H, br s), 3.88 (2H, t, J = 6.4 Hz), 3.75 (3H, s), 3.35 (2H, q, J = 6.6 Hz), 2.78 (2H, t, J = 6.6 Hz), 1.80 (2H, m), 1.42 (9H, s), 1.04 (3H, t, J = 7.4 Hz); 13C NMR (100 MHz, CDCl3) δ: 155.9, 153.5, 151.3, 129.5, 116.8, 112.3, 112.0, 79.8, 70.2, 55.7, 40.9, 30.9, 28.4, 22.8, 10.7; EI-HRMS: m/z; found: 309.19401 (M+). C17H27NO4 (M+) requires 309.19400. Anal. Found: C, 66.13; H, 8.80; N, 3.91. C17H27NO4 requires; C, 65.99; H, 8.80; N, 4.53.
4.4.2. N-(tert-Butoxycarbonyl)-2-(5-methoxy-2-propargyloxyphenyl)ethylamine (4b)
75% Yield, mp 49–50 °C; IR (CHCl3): νmax 3691, 3454, 3308, 3022, 2124, 1707, 1501 cm−1; 1H NMR (300 MHz, CDCl3): δ 6.90 (1H, d, J = 9.3 Hz), 6.72 (2H, m), 4.65 (3H, d, J = 2.4 Hz), 3.75 (3H, s), 3.35 (2H, q, J = 6.8 Hz), 2.79 (2H, t, J = 6.8 Hz), 2.47 (1H, t, J = 2.4 Hz), 1.42 (9H, s); 13C NMR (75 MHz, CDCl3): δ 155.9, 154.3, 149.8, 129.6, 116.7, 113.6, 112.0, 79.0, 79.0, 56.7, 55.6, 40.6, 30.9, 28.4; EI-HRMS: m/z; found: 305.16244 (M+). C17H23NO4 (M+) requires 305.16271. Anal. Found: C, 66.90; H, 7.54; N, 4.54. C17H23NO4 requires C, 66.86; H, 7.59; N, 4.59.
4.4.3. N-(tert-Butoxycarbonyl)-2-[2-(3-butynyl-1-oxy)-5-methoxyphenyl]ethylamine (4c)
61% Yield, mp 76–77 °C; IR (CHCl3): νmax 3455, 3309, 2413, 1707, 1602 cm−1; 1H NMR (300 MHz, CDCl3): δ 6.75 (3H, m), 4.70 (1H, br s), 4.05 (2H, t, J = 6.8 Hz), 3.75 (3H, s), 3.36 (2H, q, J = 6.6 Hz), 2.79 (2H, t, J = 6.6 Hz), 2.66 (2H, dt, J = 2.7, 6.8 Hz), 2.03 (1H, t, J = 2.7 Hz), 1.42 (9H, s); 13C NMR (75 MHz, CDCl3): δ 155.9, 153.9, 150.6, 129.3, 116.8, 112.8, 112.0, 80.7, 78.9, 69.8, 66.8, 55.6, 40.7, 30.9, 28.4, 19.7; EI-HRMS: m/z; found: 319.17756 (M+). C18H25NO4 (M+) requires 319.17836. Anal. Found: C, 67.10; H, 7.66; N, 3.67. C18H25NO4 requires C, 67.89; H, 7.89; N, 4.39.
4.4.4. N-(tert-Butoxycarbonyl)-2-[5-methoxy-2-(4-pentynyl-1-oxy)phenyl]ethylamine (4d)
93% Yield; mp 69–71 °C; IR (CHCl3) νmax 3696, 3452, 3307, 2980, 2344, 1707, 1502, 1218, 1164 cm−1; 1H NMR (300 MHz, CDCl3) δ: 6.79 (1H, d, J = 9.6 Hz), 6.70 (2H, m), 4.72 (1H, br s), 4.02 (2H, t, J = 6.5 Hz), 3.75 (3H, s), 3.34 (2H, q, J = 6.5 Hz), 2.78 (2H, t, J = 6.5 Hz), 2.41 (2H, td, J = 2.7, 6.5 Hz), 1.99 (2H, m), 1.96 (1H, t, J = 2.7 Hz), 1.43 (9H, s); 13C NMR (75 MHz, CDCl3) δ: 155.9, 153.7, 151.0, 129.0, 116.7, 112.5, 112.0, 83.4, 79.3, 68.9, 66.9, 55.7, 40.8, 30.9, 28.4, 28.4, 15.4. Anal. Found: C, 68.44; H, 8.16; N, 4.20. C19H27NO4 requires; C, 68.53; H, 8.28; N, 3.87.
4.4.5. N-(tert-Butoxycarbonyl)-2-[2-(2-butynyl-1-oxy)-5-methoxyphenyl]ethylamine (4e)
92% Yield; mp 53–55 °C; IR (CHCl3) νmax 3681, 3449, 3014, 2246, 1703, 1501, 1205 cm−1; 1H NMR (300 MHz, CDCl3) δ: 6.90 (1H, d, J = 9.6 Hz), 6.71 (2H, m), 4.65 (1H, br s), 4.60 (2H, q, J = 2.3 Hz), 3.76 (3H, s), 3.35 (2H, q, J = 6.0 Hz), 2.79 (2H, t, J = 6.0 Hz), 1.83 (3H, t, J = 2.3 Hz), 1.43 (9H, s); 13C NMR (75 MHz, CDCl3) δ: 155.9, 154.0, 150.1, 129.4, 116.5, 113.6, 111.9, 83.3, 78.6, 74.5, 57.3, 55.6, 40.6, 30.6, 28.4, 3.6. Anal. Found: C, 67.39; H, 7.97; N, 4.12. C18H25NO4 requires C, 67.69; H, 7.89; N, 4.23.
4.4.6. N-(tert-Butoxycarbonyl)-2-(2-allyloxy-5-methoxyphenyl)ethylamine (4f)
99% Yield; mp 54–56 °C; IR (CHCl3) νmax 3682, 3449, 3201, 1502, 1703, 1210 cm−1; 1H NMR (300 MHz, CDCl3) δ: 6.78 (1H, d, J = 9.0 Hz), 6.71 (2H, m), 6.05 (1H, ddt, J = 5.1 Hz, 10.5 Hz, 17.3 Hz), 5.39 (1H, dq, J = 1.6 Hz, 17.3 Hz), 5.26 (1H, dq, J = 1.6 Hz, 10.5 Hz), 4.71 (1H, br s), 4.50 (2H, dt, J = 1.6 Hz, 5.1 Hz), 3.76 (3H, s), 3.40 (2H, q, J = 6.5 Hz), 2.80 (2H, t, J = 6.5 Hz), 1.43 (9H, s); 13C NMR (75 MHz, CDCl3) δ: 156.0, 153.7, 150.8, 133.6, 129.2, 117.1, 116.7, 112.9, 112.0, 79.0, 69.5, 55.7, 40.8, 30.9, 28.4. Anal. Found: C, 66.41; H, 8.21; N, 4.40. C17H25NO4 requires C, 66.43; H, 8.20; N, 4.56.
4.4.7. N-(tert-Butoxycarbonyl)-2-(2-benzyloxy-5-methoxyphenyl)ethylamine (4g)
89% Yield; mp 103-104°C; IR (CHCl3): νmax 3691, 3453, 1707, 1503 cm−1; 1H NMR (CDCl3, 400 MHz): 7.41 (5H, m), 6.84 (1H, d, J = 8.1 Hz), 6.71 (2H, m), 5.03 (2H, s), 4.70 (1H, br s), 3.76 (3H, s), 3.37 (2H, q, J = 6.4 Hz), 2.83 (2H, t, J = 6.4 Hz), 1.42 (9H, s); 13C NMR (CDCl3, 100 MHz) δ: 155.9, 153.9, 151.0, 137.4, 128.6, 128.0, 127.9, 127.3, 116.8, 113.0, 112.0, 79.0, 70.9, 55.7 (OCH3), 40.8 (C-1), 30.9 (C-2), 27.8 (OC(CH3)3); EI-HRMS: m/z; found: 301.13383 [(M+–tert-butyl) + H]. C21H27NO4 requires 301.13409 [(M+ – tert-butyl) + H]. Anal. Found: C, 70.50; H, 7.62; N, 3.86. C21H27NO4 requires C, 70.56; H, 7.61; N 3.92.
4.4.8. N-(tert-Butoxycarbonyl)-2-[2-(2-benzyloxyethyl-1-oxy)-5-methoxyphenyl]ethylamine (4h)
85% Yield; mp 56–58 °C; IR (CHCl3) νmax 3673, 3449, 3000, 2398, 1701, 1501 cm−1; 1H NMR (300 MHz, CDCl3) δ: 7.40 (5H, m), 6.79 (1H, d, J = 8.8 Hz), 6.70 (2H, m), 4.78 (1H, br s), 4.63 (2H, s), 4.10 (2H, t, J = 4.8 Hz), 3.82 (2H, t, J = 4.8 Hz), 3.75 (3H, s), 3.37 (2H, q, J = 6.5 Hz), 2.80 (2H, t, J = 6.5 Hz), 1.42 (9H, s); 13C NMR (75 MHz, CDCl3) δ: 155.9, 153.7, 151.0, 138.1, 129.2, 128.4, 127.7, 127.6, 116.6, 112.9, 111.9, 78.7, 73.2, 68.7, 68.4, 55.5, 40.7, 30.9, 28.4; EI-HRMS: m/z; found: 401.22044 (M+). C23H31NO5 (M+) requires 401.22022. Anal. Found: C, 68.90; H, 7.80; N, 3.37. C23H31NO5 requires C, 68.80; H, 7.78; N, 3.49.
4.4.9. N-(tert-Butoxycarbonyl)-2-[5-methoxy-2-(2-propargyloxyethoxy)phenyl]ethylamine (4i)
74% Yield from tosylate 5a with propargyloxide; colourless oil; IR (CHCl3): νmax 3674, 3452, 3307, 2121, 1703 cm−1; 1H NMR (300 MHz, CDCl3): δ 6.78 (1H, d, J = 7.8 Hz), 6.69 (2H, m), 4.79 (1H, br s), 4.26 (2H, d, J = 2.3 Hz), 4.10 (2H, m), 3.88 (2H, m), 3.75 (3H, s), 3.35 (2H, q, J = 6.7 Hz), 2.80 (2H, t, J = 6.7 Hz), 2.45 (1H, t, J = 2.3 Hz), 1.42 (9H, s); 13C NMR (75 MHz, CDCl3) δ: 156.0, 153.9, 150.9, 129.4, 116.7, 113.0, 112.0, 79.6, 78.9, 74.7, 68.4, 68.3, 58.5, 55.7, 40.8, 31.0, 28.4; EI-HRMS: m/z; found: 349.18937 (M+). C19H27NO5 (M+) requires 349.18892.
4.4.10. N-(tert-Butoxycarbonyl)-2-{5-methoxy-2-[2-(2-propargyloxyethoxy)ethoxy] phenyl}ethylamine (4j)
91% Yield from tosylate 5b with propargyloxide; colourlesss oil; IR (CHCl3): νmax 3693, 3607, 3453, 3308, 3012, 2980, 2934, 2120, 1706, 1502; 1H NMR (400 MHz, CDCl3): δ 6.74 (1H, d, J = 8.6 Hz), 6.66 (2H, m), 4.90 (1H, br s), 4.17 (2H, d, J = 2.4 Hz), 4.05 (2H, t, J = 4.9 Hz), 3.80 (2H, t, J = 4.9 Hz), 3.71 (3H, s), 3.70 (4H, m), 3.31 (2H, q, J = 6.7 Hz), 2.76 (2H, t, J = 6.7 Hz), 2.42 (1H, t, J = 2.4 Hz), 1.39 (9H, s); 13C NMR (100 MHz, CDCl3): δ 156.0, 153.8, 151.0, 129.4, 116.6, 113.1, 112.0, 79.6, 78.7, 74.6, 70.5, 69.8, 69.1, 68.5, 58.3, 55.6, 40.7, 31.0, 28.4; EI-HRMS: m/z; found: 393.21448 (M+). C21H31NO6 (M+) requires 393.21514.
4.4.11. N-(tert-Butoxycarbonyl)-2-(2-methoxycarbonylmethyloxy-5-methoxyphenyl)ethylamine (4k)
98% Yield; mp 66–68 °C; IR (CHCl3) νmax 3681, 3449, 1758, 1704, 1501, 1227 cm−1; 1H NMR (300 MHz, CDCl3): δ 6.67 (3H, m), 4.84 (1H, br s), 4.61 (2H, s), 3.78 (3H, s), 3.74 (3H, s), 3.40 (2H, q, J = 6.5 Hz), 2.90 (2H, t, J = 6.5 Hz), 1.41 (9H, s); 13C NMR (75 MHz, CDCl3) δ: 169.6, 156.0, 154.3, 150.1, 129.5, 116.9, 112.4, 112.0, 78.8, 66.0, 55.6, 52.1, 40.7, 30.9, 28.4. Anal. Found: C, 60.37; H, 7.36; N, 3.91. C17H25NO6 requires C, 60.16; H, 7.42; N; 4.13.
4.4.12. N-(tert-Butoxycarbonyl)-2-(2-cyanomethoxy-5-methoxyphenyl)ethylamine (4l)
96% Yield; mp 98–102 °C; IR (CHCl3) νmax 3681, 3449, 2240, 1703, 1501, 1226 cm−1; 1H NMR (300 MHz, CDCl3) δ: 6.89 (1H, d, J = 9.6 Hz), 6.74 (2H, m), 4.73 (2H, s), 4.61 (1H, br s), 3.76 (3H, s), 3.35 (2H, q, J = 6.8 Hz), 2.80 (2H, t, J = 6.8 Hz), 1.43 (9H, s); 13C NMR (75 MHz, CDCl3) δ: 155.9, 155.4, 148.9, 130.1, 117.1, 115.4, 114.0, 112.2, 79.2, 55.6, 54.9, 40.5, 31.4, 28.4. Anal. Found: C, 62.87; H, 7.09; N, 8.30. C16H22N2O4 requires C, 62.73; H, 7.24; N, 9.14.
4.4.13. N-(tert-Butoxycarbonyl)-2-[2-(3-cyanopropyl-1-oxy)-5-methoxyphenyl]ethylamine (4m)
96% Yield; mp 69–71 °C; IR (CHCl3) νmax 3681, 3449, 3014, 2246, 1703 cm−1; 1H NMR (300 MHz, CDCl3) δ: 6.74 (3H, m), 4.73 (1H, br s), 4.03 (2H, t, J = 5.8 Hz), 3.75 (3H, s), 3.32 (2H, q, J = 6.8 Hz), 2.77 (2H, t, J = 6.8 Hz), 2.60 (2H, t, J = 7.0 Hz), 2.14 (2H, m), 1.42 (9H, s); 13C NMR (75 MHz, CDCl3) δ: 155.9, 154.0, 150.6, 128.9, 119.3, 116.8, 112.5, 112.0, 79.5, 66.3, 55.7, 40.8, 31.2, 28.4, 25.7, 14.4; EI-HRMS: m/z; found: 334.1924 (M+). C18H26N2O4 (M+) requires 334.1893. Anal. Found: C, 63.62; H, 7.82; N, 8.06. C18H26N2O4 requires C, 64.65; H, 7.84; N, 8.38.
4.4.14. N-(tert-Butoxycarbonyl)-2-[2-(2-hydroxyethoxy)-5-methoxyphenyl]ethylamine (4n)
93% Yield via hydrogenolysis of 4h; mp 90–91 °C; IR (CHCl3) νmax 3681, 3615, 3449, 3021, 2405, 1700, 1501, 1165 cm−1; 1H NMR (400 MHz, CDCl3) δ: 6.74 (3H, m), 4.87 (1H, br s), 4.00 (4H, m), 3.88 (1H, br s), 3.75 (3H, s), 3.31 (2H, q, J = 6.7 Hz), 2.78 (2H, t, J = 6.7 Hz), 1.42 (9H, s); 13C NMR (100 MHz, CDCl3) δ: 156.5, 153.9, 151.4, 128.9, 117.1, 112.6, 112.0, 79.7, 70.6, 61.6, 55.9, 41.0, 32.6, 28.6; EI-HRMS: m/z; found: 311.17278 (M+). C16H25NO5 (M+) requires 311.17327. Anal. Found: C, 61.70; H, 8.03; N, 4.14. C16H25NO5 requires C, 61.72; H, 8.09; N, 4.50.
4.4.15. N-(tert-Butoxycarbonyl)-2-[2-(3-hydroxypropyl-1-oxy)-5-methoxyphenyl]ethylamine (4o)
100% and 95% Yields in the alkylation and hydrogenolysis steps respectively; mp 44–46 °C; IR (CHCl3) νmax 3681, 3623, 3449, 2399, 1700, 1505, 1205 cm−1; 1H NMR (300 MHz, CDCl3) δ: 6.80 (1H, d, J = 9.6 Hz), 6.70 (2H, m), 4.78 (1H, br s), 4.06 (2H, t, J = 5.9 Hz), 3.87 (2H, t, J = 5.9 Hz), 3.75 (3H, s), 3.34 (2H, q, J = 6.6 Hz), 2.76 (2H, t, J = 6.6 H), 2.37 (1H, br s), 2.03 (2H, quin, J = 5.9 Hz), 1.41 (9H, s); 13C NMR (100 MHz, CDCl3) δ: 155.9, 153.6, 151.1, 128.9, 116.8, 112.4, 112.0, 79.0, 66.9, 65.9, 55.7, 40.7, 31.0, 29.9, 28.4. Anal. Found: C, 62.97; H, 8.35; N, 4.13. C17H27NO5 requires C, 62.75; H, 8.36; N, 4.30.
4.5. Procedure for the synthesis of thioureas 6a–o
Trifluoroacetic acid (0.2 mL) was added to a solution of the N-Boc carbamate 4 (1 mmol) in CH2Cl2 (2 mL) at 0 °C, and the solution stirred for 2 h. Diisopropylethylamine (0.4 mL) was added, the solvent evaporated in vacuo and the crude amine dried under vacuum for 1 h. Thiocarbonyl reagent 7 (1.3 mmol) was added to the crude amine in DMF (5 mL) and the mixture stirred at 100 °C for 16 h. The mixture was then poured into ice-cold water (5 mL) and stirred for 30 min. The precipitate formed was filtered and washed with cold water (2× 5 mL) or alternatively extracted into ethyl acetate and the residue purified by column chromatography using ethyl acetate/light petroleum mixtures as eluent. Alternatively, the condensation reaction with 7 could be carried out in THF (5 mL) at room temperature for 20 h. Following evaporation of solvent, the residue was subjected directly to column chromatography to afford thioureas 6.
4.5.1. N-(5-Bromo-2-pyridinyl)-N′-[2-(5-methoxy-2-propyl-1-oxyphenyl)ethyl]-thiourea (6a)
Using THF/rt, (65%); mp 162–163 °C; IR (CHCl3) νmax 3696, 3413 (NH), 3167, 2963 (C–H), 1512, 1472 (C=S), 1212 (C–N) cm−1; 1H NMR (400 MHz, CDCl3) δ: 11.18 (1H, br s, NH), 8.95 (1H, br s, NH), 8.08 (1H, d, J = 2.4 Hz), 7.67 (1H, dd, J = 2.4 Hz, 8.8 Hz), 6.81 (1H, d, J = 3.2 Hz), 6.78 (1H, d, J = 8.8 Hz), 6.74 (1H, dd, J = 3.2, 8.8 Hz), 6.72 (1H, d, J = 8.8 Hz), 4.02 (2H, m), 3.87 (2H, t, J = 6.4 Hz), 3.75 (3H, s), 2.99 (2H, t, J = 6.4 Hz), 1.80 (2H, m), 1.04 (3H, t, J = 7.4 Hz); 13C NMR (100 MHz, CDCl3) δ: 179.0 (C=S), 153.3, 151.7, 151.5, 146.8, 141.1, 128.5, 117.7, 113.2, 112.6, 112.2, 111.5, 70.2, 55.6, 45.8, 30.0, 22.8, 10.7. Anal. Found: C, 50.77; H, 5.21; N, 9.50; S, 7.14%. C18H22BrN3O2S requires C, 50.95; H, 5.23; N, 9.90; S, 7.55.
4.5.2. N-(5-Bromo-2-pyridinyl)-N′-[2-(5-methoxy-2-propargyloxyphenyl)ethyl]-thiourea (6b)
Using DMF/100 °C, (17%); mp 121–122°C; IR (CHCl3) νmax 3691, 3416 (NH), 3307 (≡CH), 3174, 1602, 1137 cm−1; 1H NMR (400 MHz, CDCl3) δ: 11.13 (1H, br s, NH), 8.29 (1H, br s, NH), 8.12 (1H, d, J = 2.4 Hz), 7.69 (1H, dd, J = 2.4, 8.8 Hz), 6.95 (1H, d, J = 8.8 Hz), 6.82 (1H, d, J = 3.1 Hz), 6.76 (1H, dd, J = 3.1, 8.8 Hz), 6.59 (1H, d, J = 8.8 Hz), 4.67 (1H, d, 1H, J = 2.2 Hz), 4.01 (1H, q, J = 6.7 Hz), 3.76 (3H, s), 3.01 (2H, t, J = 6.7 Hz), 2.47 (1H, t, J = 2.2 Hz); 13C NMR (75 MHz, CDCl3) δ: 179.1 (C=S), 154.2, 151.7, 150.0, 146.7, 141.1, 129.2, 117.6, 113.5, 113.3, 112.6, 111.5, 79.0, 75.3, 56.8, 55.5, 45.7, 29.8. Anal. Found: C, 51.23; H, 4.50; N, 8.62%. C18H18BrN3O2S requires C, 51.44; H, 4.32; N, 10.00.
4.5.3. N-(5-Bromo-2-pyridinyl)-N′-{2-[2-(3-butynyl-1-oxy)-5-methoxyphenyl]ethyl}-thiourea (6c)
Using DMF/100 °C, (25%); mp 155–156 °C; IR (CHCl3) νmax 3691, 3415, 3308 (≡CH), 3176, 3048, 2360 (C≡C), 1591, 1511, 1138 cm−1; 1H NMR (300 MHz, CDCl3) δ: 11.13 (1H, br s, NH), 8.44 (1H, br s, NH), 8.10 (1H, d, J = 2.4 Hz), 7.68 (1H, dd, J = 2.4, 8.7 Hz), 6.76 (3H, m), 6.61 (1H, d, J = 8.7 Hz), 4.03 (4H, m), 3.75 (3H, s), 3.00 (2H, t, J = 6.6 Hz), 2.68 (2H, td, J = 2.6, 6.9 Hz), 2.04 (1H, t, J = 2.6 Hz); 13C NMR (75δMHz, CDCl3) δ: 179.2 (C=S), 153.8, 151.6, 150.9, 146.9, 141.2, 128.9, 117.8, 113.1, 112.8, 112.7, 111.6, 80.8, 69.9, 66.9, 55.6, 45.8, 30.0, 19.8. Anal. Found: C, 52.01; H, 4.46; N, 9.00%. C19H20BrN3O2S requires C, 52.54; H, 4.64; N, 9.67.
4.5.4. N-(5-Bromo-2-pyridinyl)-N′-{2-[5-methoxy-2-(4-pentynyl-1-oxy)phenyl]ethyl}-thiourea (6d)
Using THF/rt, (69%); mp 140–141 °C; IR (CHCl3) νmax 3684, 3415 (NH), 3308 (≡CH), 3152, 3018, 2956, 2245 (C≡C), 1512, 1468 (C=S), 1226 (C–N) cm−1; 1H NMR (400 MHz, CDCl3) δ: 11.18 (1H, br s, NH), 8.83 (1H, br s, NH), 8.10 (1H, d, J = 2.4 Hz), 7.68 (1H, dd, J = 2.4 Hz, 8.8 Hz), 6.81 (1H, d, J = 9.0 Hz), 6.81 (1H, d, J = 3.0 Hz), 6.75 (1H, dd, J = 3.0, 9.0 Hz), 6.70 (1H, d, J = 8.8 Hz), 4.01 (4H, m), 3.75 (3H, s), 2.98 (2H, t, J = 6.8 Hz), 2.41 (2H, td, J = 2.4, 6.8 Hz), 2.02 (2H, m), 1.96 (1H, t, J = 2.4 Hz); 13C NMR (100 MHz, CDCl3) δ: 179.1 (C=S), 153.5, 151.7, 151.2, 146.7, 141.1, 128.5, 117.7, 113.2, 112.6, 112.3, 111.5, 83.5, 68.9, 66.9, 55.6, 45.7, 30.0, 28.4, 15.4. Anal. Found: C, 53.54; H, 4.78; N, 7.81; S, 5.62%. C20H22BrN3O2S requires C, 53.58; H, 4.95; N, 9.37; S, 7.15.
4.5.5. N-(5-Bromo-2-pyridinyl)-N′-{2-[2-(2-butynyl-1-oxy)-5-methoxyphenyl]ethyl}-thiourea (6e)
Using THF/rt, (60%); mp 149–150 °C; IR (CHCl3) νmax 3690, 3410 (NH), 3018, 2239 (C≡C), 1510, 1469 (C=S), 1207 (C–N) cm−1; 1H NMR (400 MHz, CDCl3) δ: 11.12 (1H, br s, NH), 8.58 (1H, br s, NH), 8.11 (1H, d, J = 2.4 Hz), 7.68 (1H, dd, J = 2.4 Hz, 8.8 Hz), 6.93 (1H, d, J = 8.8 Hz), 6.81 (1H, d, J = 3.2 Hz), 6.75 (1H, dd, J = 3.2 Hz, 8.8 Hz), 6.65 (1H, d, J = 8.8 Hz), 4.61 (2H, q, J = 2.4 Hz), 4.01 (2H, m), 3.75 (3H, s), 3.00 (2H, t, J = 6.8 Hz), 1.83 (3H, t, J = 2.4 Hz); 13C NMR (100 MHz, CDCl3) δ: 179.1 (C=S), 153.9, 151.6, 150.4, 146.8, 141.1, 129.1, 117.5, 113.6, 113.1, 112.6, 111.6, 83.4, 74.5, 57.5, 55.6, 45.8, 29.8, 3.7. Anal. Found: C, 52.51; H, 4.53; N, 8.95; S, 6.31%. C19H20BrN3O2S requires C, 52.54; H, 4.64; N, 9.67; S, 7.38.
4.5.6. N-(5-Bromo-2-pyridinyl)-N′-[2-(2-allyloxy-5-methoxyphenyl)ethyl]-thiourea (6f)
Using THF/rt, (67%); mp 153–154 °C; IR (CHCl3) νmax 3681, 3413 (N–H), 3041, 1509 (C=C), 1422 (C=S), 1212 (C–N) cm−1; 1H NMR (400 MHz, CDCl3) δ: 11.11 (1H, br s, NH), 8.13 (1H, br s, NH), 8.10 (1H, d, J = 2.4 Hz), 7.69 (1H, dd, J = 2.4, 8.7 Hz), 6.82 (1H, d, J = 2.9 Hz), 6.80 (1H, d, J = 9.0 Hz), 6.74 (1H, dd, J = 2.9, 9.0 Hz), 6.55 (1H, d, J = 8.7 Hz), 6.05 (1H, ddt, J = 5.1, 10.5, 17.3 Hz), 5.39 (1H, dq, J = 1.6, 17.3 Hz), 5.26 (1H, dq, J = 1.6, 10.5 Hz), 4.49 (2H, dt, J = 1.6, 5.1 Hz), 4.01 (2H, m), 3.76 (3H, s), 3.01 (2H, t, J = 6.6 Hz); 13C NMR (100 MHz, CDCl3) δ: 179.1 (C=S), 153.5, 151.5, 151.0, 146.9, 141.1, 133.6, 128.7, 117.7, 117.0, 113.0, 112.8, 112.6, 111.5, 69.5, 55.6, 45.8, 30.0. Anal. Found: C, 51.32; H, 4.62; N, 9.56%. C18H20BrN3O2S requires C, 51.18; H, 4.78; N, 9.95.
4.5.7. N-(5-Bromo-2-pyridinyl)-N′-[2-(2-Benzyloxy-5-methoxyphenyl)ethyl]-thiourea (6g)
Using THF/rt, (37%); mp 141–142 °C as a yellow solid; IR (CHCl3) νmax 3691, 3416, 1602, 1505, 1138 cm−1; 1H NMR (300 MHz, CDCl3) δ: 11.18 (1H, br s, NH), 8.80 (1H, br s, NH), 8.03 (1H, d, J = 2.6 Hz), 7.65 (1H, dd, J = 2.6, 8.7 Hz), 7.37 (5H, m, ArH), 6.86 (1H, d, J = 9.0 Hz), 6.84 (1H, d, J = 3.2 Hz), 6.74 (1H, dd, J = 3.2, 9.0 Hz), 6.67 (1H, d, J = 8.7 Hz), 5.02 (2H, s), 4.03 (2H, m), 3.75 (3H, s), 3.03 (2H, t, J = 6.6 Hz); 13C NMR (75 MHz, CDCl3) δ: 179.0 (C=S), 153.6, 151.6, 151.1, 146.7, 141.1, 137.3, 128.8, 128.5, 127.8, 127.2, 117.7, 113.2, 112.9, 112.6, 111.5, 70.7, 55.6, 45.7, 30.0; HRMS (EI) m/z; found: 471.0594 (M+), C22H22BrN3O2S (M+) requires 471.0616. Anal. Found: C, 55.48; H, 4.68; N, 8.59; S, 6.17%. C22H22BrN3O2S requires C, 55.94; H, 4.69; N, 8.90; S, 6.79.
4.5.8. N-(5-Bromo-2-pyridinyl)]-N′-{2-[2-(2-Benzyloxyethyl-1-oxy)-5-methoxyphenyl]ethyl}-thiourea (6h)
Using THF/rt, (58% yield); mp 110–111 °C; IR (CHCl3) νmax 3692, 3416 (NH), 3175, 3016, 1506, 1475 (C=S) cm−1; 1H NMR (400 MHz, CDCl3) δ: 11.09 (1H, br s, NH), 8.38 (1H, br s, NH), 8.04 (1 H, d, J = 2.4 Hz), 7.66 (1H, dd, J = 2.4 Hz, 8.8 Hz), 7.31 (5H, m, ArH), 6.81 (1H, d, J = 8.8 Hz), 6.80 (1H, d, J = 3.2 Hz), 6.74 (1H, dd, J = 3.2 Hz, 8.8 Hz), 6.58 (1H, d, J = 8.8 Hz), 4.63 (2H, s), 4.11 (2H, t, J = 4.8 Hz), 4.03 (2H, m), 3.84 (2H, t, J = 4.8 Hz), 3.75 (3H, s), 3.01 (2H, t, J = 6.6 Hz); 13C NMR (100 MHz, CDCl3) δ: 179.0 (C=S), 153.6, 151.8, 151.3, 146.6, 141.0, 138.1, 128.8, 128.5, 127.7, 127.7, 117.7, 113.4, 112.8, 112.9, 111.6, 73.4, 68.9, 68.5, 55.6, 45.7, 30.0. Anal. Found: C, 56.07; H, 4.86; N, 7.37; S, 5.80%. C24H26BrN3O3S requires C, 55.82; H, 5.07; N, 8.14; S, 6.21.
4.5.9. N-(5-Bromo-2-pyridinyl)]-N′-{2-[5-methoxy-2-(2-propargyloxyethoxy)phenyl]ethyl}-thiourea (6i)
Using DMF/rt, (62%); mp 128–130 °C; IR (CHCl3) νmax 3693, 3416, 3307, 3165, 2961, 1506, 1475, 1223 cm−1; 1H NMR (300 MHz, CDCl3) δ: 11.19 (1H, br s, NH), 9.24 (1H, br s, NH), 8.07 (1H, d, J = 2.6 Hz), 7.66 (1H, dd, J = 2.6, 8.7 Hz), 6.81–6.77 (3H, m), 6.72 (1H, dd, J = 2.9, 9.2 Hz), 4.25 (2H, d, J = 2.4 Hz), 4.09 (2H, t, J = 4.8 Hz), 4.01 (2H, q, J = 6.6 Hz), 3.88 (2H, t, J = 4.8 Hz), 3.74 (3H, s), 2.99 (2H, t, J = 6.6 Hz), 2.45 (1H, t, J = 2.4 Hz); 13C NMR (75 MHz, CDCl3) δ: 178.9 (C=S), 153.6, 151.7, 151.1, 146.6, 141.0, 128.8, 117.6, 113.4, 112.9, 112.6, 111.5, 79.5, 74.7, 68.4, 68.2, 58.5, 55.5, 45.6, 29.9; HRMS (EI) m/z; found: 463.0578 (M+). C20H22BrN3O3S (M+) requires 463.0565. Anal. Found: C, 51.76; H, 4.72; N, 8.66; S, 6.86%. C20H22BrN3O3S requires C, 51.73; H, 4.78; N, 9.05; S, 6.91.
4.5.10. N-(5-Bromo-2-pyridinyl)-N′-{2-[5-methoxy-2-(2-(2-propargyloxyethoxy)ethoxy)phenyl]ethyl}-thiourea (6j)
Using DMF/rt, (60%); mp 91–92 °C; IR (CHCl3) νmax 3691, 3416, 3307, 3166, 2935, 1506, 1475, 1266 cm−1; 1H NMR (300 MHz, CDCl3) δ: 11.11 (1H, br s, NH), 8.67 (1H, br s, NH), 8.07 (1H, d, J = 2.4 Hz), 7.67 (1H, dd, J = 2.4, 8.7 Hz), 6.81–6.78 (2H, m), 6.73 (1H, dd, J = 3.0, 8.7 Hz), 6.67 (1H, d, J = 8.7 Hz), 4.21 (2H, d, J = 2.1 Hz), 4.07 (2H, t, J = 4.9 Hz), 4.01 (2H, q, J = 6.6 Hz), 3.85 (2H, t, J = 4.9 Hz), 3.73 (7H, m), 2.99 (2H, t, J = 6.6 Hz), 2.42 (1H, t, J = 2.1 Hz); 13C NMR (75 MHz, CDCl3) δ: 179.1 (C=S), 153.6, 151.6, 151.2, 146.8, 141.1, 128.8, 117.7, 113.2, 112.8, 112.6, 111.5, 79.6, 74.6, 70.6, 70.0, 69.2, 68.4, 58.5, 55.6, 45.7, 30.0; HRMS (EI) m/z; found: 507.0821 (M+). C22H26BrN3O4S (M+) requires 507.0827. Anal. Found: C, 52.02; H, 4.10; N, 7.87; S, 6.24%. C22H26BrN3O4S requires C, 51.97; H, 5.15; N, 8.26; S, 6.31.
4.5.11. N-(5-Bromo-2-pyridinyl)-N′-[2-(2-methoxycarbonylmethyloxy-5-methoxyphenyl)ethyl]-thiourea (6k)
Using THF/rt, (50%); mp 161–164 °C; IR (CHCl3) νmax 3681 (N–H), 3015, 1519, 1469 (C=S), 1212 (C–N) cm−1; 1H NMR (400 MHz, CDCl3) δ: 11.12 (1H, br s, NH), 8.32 (1H, br s, NH), 8.11 (1H, d, J = 2.4 Hz), 7.69 (1H, dd, J = 2.4, 9.1 Hz), 6.83 (1H, d, J = 2.4 Hz), 6.71 (2H, m), 6.61 (1H, d, J = 9.1 Hz), 4.62 (2H, s), 4.05 (2H, m), 3.78 (3H, s), 3.74 (3H, s), 3.06 (2H, t, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3 ppm) δ: 179.2 (C=S), 169.6, 154.2, 151.5, 150.3, 146.9, 141.2, 129.2, 117.8, 113.0, 112.7, 112,6, 111.6, 66.3, 55.5, 52.2, 45.7, 29.9; HRMS (ES) m/z; found: [M+H]+, 454.0437. Calcd. for C18H21BrN3O4S (M+H), 454.0436.
4.5.12. N-(5-Bromo-2-pyridinyl)-N′-[2-(2-cyanomethyloxy-5-methoxyphenyl)ethyl]-thiourea (6l)
Using THF/rt, (68%); mp 155–156 °C; IR (CHCl3) νmax 3681, 3413, 3022 (NH), 2239 (C≡N), 1505, 1469 (C=S), 1216 cm−1; 1H NMR (400 MHz, CDCl3) δ: 11.20 (1H, br s, NH), 8.72 (1H, br s, NH), 8.10 (1H, d, J = 2.4 Hz), 7.70 (1H, dd, J = 2.4 Hz, 8.8 Hz), 6.92 (1H, d, J = 8.8 Hz), 6.85 (1H, d, 3.2 Hz), 6.78 (1H, dd, J = 3.2, 8.8 Hz), 6.69 (1H, d, J = 8.8 Hz), 4.75 (2H, s), 3.99 (2H, q, J = 6.8 Hz), 3.76 (3H, s) 3.01 (2H, t, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ: 179.4 (C=S), 155.4, 151.6, 149.0, 146.7, 141.3, 129.8, 117.9, 115.4, 114.0, 113.3, 112.8, 111.9, 55.6, 54.9, 45.6, 29.6. Anal. Found: C, 48.38; H, 4.04; N, 12.38; S; 6.76%. C17H17BrN4O2S requires C, 48.46; H, 4.07; N, 13.30; S, 7.61.
4.5.13. N-(5-Bromo-2-pyridinyl)]-N′-{2-[2-(3-cyanopropyl-1-oxy)-5-methoxyphenyl]ethyl}-thiourea (6m)
Using THF/rt, (53%); mp 165–166 °C; IR (CHCl3) νmax, 3413 (NH), 3167, 2964 (C–H), 2246 (C≡N), 1505, 1465 (C=S), 1239 (C–N) cm−1; 1H NMR (400 MHz, CDCl3) δ: 11.26 (1 H, br s, NH), 9.34 (1H, br s, NH), 8.08 (1H, d, J = 2.4 Hz), 7.67 (1H, dd, J = 2.4 Hz, 8.8 Hz), 6.82 (1H, d, J = 3.2 Hz), 6.81 (1H, d, J = 8.8 Hz), 6.77 (1H, d, J = 8.8 Hz), 6.74 (1H, dd, J = 3.2 8.8 Hz), 3.99 (4H, m), 3.74 (3H, s), 2.98 (2H, t, J = 6.8 Hz), 2.62 (2H, t, J = 7.0 Hz), 2.14 (2H, m); 13C NMR (75 MHz, CDCl3) δ: 179.0 (C=S), 153.7, 151.7, 150.6, 146.5, 141.1, 128.4, 119.2, 117.7, 113.4, 112.7, 112.2, 111.5, 66.0, 55.5, 45.6, 29.9, 25.6, 14.4. Anal. Found: C, 50.61; H, 4.78; N, 12.37; S, 6.92%. C19H21BrN4O2S requires C, 50.78; H, 4.71; N, 12.47; S, 7.13.
4.5.14. N-(5-Bromo-2-pyridinyl)]-N′-{2-[2-(2-hydroxyethoxy)-5-methoxyphenyl]ethyl}-thiourea (6n)
Using THF/rt, (65%); mp 162–163 °C; IR (CHCl3) νmax 3623, 3413, 3181 (NH, OH), 2928, 1505 (C–H), 1472 (C=S) 1227 (C–N) cm−1; 1H NMR (400 MHz, CDCl3) δ: 11.16 (1H, br s, NH), 8.47 (1H, br s, NH), 8.12 (1H, d, J = 2.4 Hz), 7.70 (1H, dd, J = 2.4, 8.8 Hz), 6.81 (1H, d, J = 2.8 Hz), 6.77 (1H, d, J = 8.8 Hz), 6.73 (1H. dd, J = 2.8, 8.8 Hz), 6.66 (1H, d, J = 8.8 Hz), 3.99 (6H, m), 3.77 (3H, s), 3.03 (2H, t, J = 7.2 Hz), 2.66 (1H, br s); 13C NMR (75 MHz, CDCl3) δ: 179.4 (C=S), 153.7, 151.6, 151.3, 146.7, 141.3, 128.3, 117.4, 113.4, 112.8, 112.5, 111.7, 70.3, 61.5, 55.6, 45.7, 30.3. Anal. Found: C, 47.96; H, 4.61; N, 9.77; S, 7.01%. C17H20BrN3O3S requires C, 47.89; H, 4.75; N, 9.86; S, 7.52.
4.5.15. N-(5-Bromo-2-pyridinyl)]-N′-{2-[2-(3-hydroxypropyl-1-oxy)-5-methoxyphenyl]ethyl}-thiourea (6o)
Using THF/rt, (51%), mp 163–165 °C; IR (CHCl3) νmax 3681 (N–H), 3406 (O–H) 3014, 1509, 1472 (C=S), 1216 (C–N) cm−1; 1H NMR (400 MHz, CDCl3) δ: 11.15 (1H, br s, NH), 8.46 (1H, br s, NH), 8.10 (1H, d, J = 2.4 Hz), 7.68 (1H, dd, J = 2.4, 8.8 Hz), 6.81 (1H, d, J = 8.8 Hz), 6.81 (1H, d, J = 2.9 Hz), 6.74 (1H, dd, J = 2.9, 8.8 Hz), 6.62 (1H, d, J = 8.8 Hz), 4.05 (2H, t, J = 5.9 Hz), 4.00 (2H, m), 3.87 (2H, q, J = 5.9 Hz), 3.76 (3H, s), 2.98 (2H, t, J = 6.6 Hz), 2.05 (2H, quin, J = 5.9 Hz), 2.04 (1H, br s, OH); 13C NMR (101 MHz, CDCl3 ppm) δ: 179.1 (C=S), 153.5, 151.6, 151.3, 146.8, 141.2, 128.4, 117.7, 113.2, 112.7, 112.4, 111.6, 65.9, 60.1, 55.6, 45.8, 32.3, 30.0. Anal. Found: C, 49.37; H, 4.88; N, 9.08; S, 6.60%. C18H22BrN3O4S requires C, 49.09; H, 5.04; N, 9.54; S, 7.28.
4.6. Anti-HIV evaluation
The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: MT-2 cells and Nevirapine-Resistant HIV-1 (N119) from Dr. Douglas Richman and HTLV-IIIB/H9 from Dr. Robert Gallo.
Antiviral activity and cellular toxicity were determined using the MTT colorimetric method.19 MT-2 cells29 at a concentration of 1 × 105 cells per millilitre were infected with wild-type HIV-IIIB30 or Y181C mutant virus31 at a multiplicity of infection (MOI) of 0.1. Infected and mock-infected cells were incubated in growth medium (RPMI 1640, 10% dFBS, kanamycin) for 5 days with varying concentrations of each compound being tested in triplicate in a 96-well plate. MTT, a cell-permeable tetrazolium dye was then added to each well. After 5 h, acidified isopropanol was added to lyse the cells and stop the reaction. The plates were gently shaken overnight, and the absorbance measured at 595 nm on a plate reader. The average of these triplicate samples was then plotted versus inhibitor concentration to generate dose–response curves. The 50% effective concentration (EC50) and 50% cytotoxic concentration (CC50) of the compounds were defined as the concentrations required to inhibit viral replication and to reduce the number of viable cells by 50%, respectively.
4.7. Steady-state IC50 determination
6 nM RT (active sites based on pre-steady-state active site determination) was pre-incubated for at least 15 min with 1 μM 5′-radiolabeled primer/template prior to mixing with appropriate concentrations of inhibitor and allowed to incubate for a minimum of 15 additional minutes on ice. DMSO concentrations were kept constant at less than 2%. DMSO alone was added as a no inhibitor control for each set of experiments. Reactions were initiated with the addition of 5 μM dTTP and 10 mM MgCl2 and were quenched after 15 min at 37 °C with 0.3 MEDTA. All concentrations represent final concentrations after mixing. Reaction products were subjected to 20% denaturing polyacrylamide gel-electrophoresis and quantitated on a Bio-Rad Molecular Imager FX. Product formation was plotted as a function of inhibitor concentration and fitted to a hyperbola to generate IC50 curves. IC50 values are defined as the concentration of inhibitor that inhibits steady-state single nucleotide incorporation by 50%.
Supplemental Material
Acknowledgments
We thank the National Research Foundation of South Africa, the Third World Organization for Women in Science and the University of Cape Town for financial support. We also thank the National Institutes of Health (GM49551) for support to K.S.A.
Footnotes
Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2008.10.048.
References and notes
- 1.(a) Aquaro S, Svicher V, Schols D, Pollicita M, Antinori A, Balzarini J, Perno CF. J Leukoc Biol. 2006;80:1103. doi: 10.1189/jlb.0606376. [DOI] [PubMed] [Google Scholar]; (b) Meadows DC, Gervay-Hague J. ChemMedChem. 2006;1:16. doi: 10.1002/cmdc.200500026. [DOI] [PubMed] [Google Scholar]; (c) Stevens M, De Clercq E, Balzarini J. Med Res Rev. 2006;26:595. doi: 10.1002/med.20081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Pauwels R. Antiviral Res. 2006;71:77. doi: 10.1016/j.antiviral.2006.05.007. [DOI] [PubMed] [Google Scholar]; (b) De Clercq E. J Med Chem. 2005;48:1297. doi: 10.1021/jm040158k. [DOI] [PubMed] [Google Scholar]
- 3.(a) D'Cruz OJ, Uckun FM. J Enzyme Inhib Med Chem. 2006;21:329. doi: 10.1080/14756360600774413. [DOI] [PubMed] [Google Scholar]; (b) Waters L, John L, Nelson M. Int J Clin Prac. 2006;61:105. doi: 10.1111/j.1742-1241.2006.01146.x. [DOI] [PubMed] [Google Scholar]; (c) Zhou Z, Lin X, Madura JD. Infect Disord: Drug Targets. 2006;6:391. doi: 10.2174/187152606779025833. [DOI] [PubMed] [Google Scholar]; (d) Balzarini J. Curr Top Med Chem. 2004;4:921. doi: 10.2174/1568026043388420. [DOI] [PubMed] [Google Scholar]; (e) Pauwels R. Curr Opin Pharmacol. 2004;4:437. doi: 10.1016/j.coph.2004.07.005. [DOI] [PubMed] [Google Scholar]; (f) De Clercq E. Chem Biodiversity. 2004;1:44. doi: 10.1002/cbdv.200490012. [DOI] [PubMed] [Google Scholar]; (g) Campiani G, Ramunno A, Maga G, Nacci V, Fattorusso C, Catalanotti B, Morelli E, Novellino E. Curr Pharm Des. 2002;8:615. doi: 10.2174/1381612024607207. [DOI] [PubMed] [Google Scholar]
- 4.Muhanji CI, Hunter R. Curr Med Chem. 2007;14:1207. doi: 10.2174/092986707780597952. [DOI] [PubMed] [Google Scholar]
- 5.(a) Pedersen L, Jørgensen PT, Nielsen S, Hansen TH, Nielsen J, Pedersen EB. J Med Chem. 2005;48:1211. doi: 10.1021/jm040845b. [DOI] [PubMed] [Google Scholar]; (b) Velázquez S, Lobatón E, De Clercq E, Koontz DL, Mellors JW, Balzarini J, Camarasa MJ. J Med Chem. 2004;47:3418. doi: 10.1021/jm031045o. [DOI] [PubMed] [Google Scholar]; (c) Gavriliu D, Fossey C, Ciurea A, Delbederi Z, Sugeac E, Ladurée D, Schmidt S, Laumond G, Aubertin AM. Nucleosides Nucleotides Nucleic Acids. 2002;21:505. doi: 10.1081/NCN-120015066. [DOI] [PubMed] [Google Scholar]
- 6.(a) Pontikis R, Dollé V, Guillaumel J, Dechaux E, Note R, Nguyen CH, Legraverend M, Bisagni E, Aubertin AM, Grierson DS, Monneret C. J Med Chem. 2000;43:1927. doi: 10.1021/jm991125l. [DOI] [PubMed] [Google Scholar]; (b) Velázquez S, Tuñón V, Jimeno ML, Chamorro C, De Clercq E, Balzarini J, Camarasa MJ. J Med Chem. 1999;42:5188. doi: 10.1021/jm991092+. [DOI] [PubMed] [Google Scholar]; (c) Renoud-Grappin M, Fossey C, Fontaine G, Ladurée D, Aubertin AM, Kirn A. Antiviral Chem Chemother. 1998;9:205. doi: 10.1177/095632029800900302. [DOI] [PubMed] [Google Scholar]
- 7.Nanni RG, Ding J, Jacobo-Molina A, Hughes SH, Arnold E. Perspect Drug Discovery Des. 1993;1:129. [Google Scholar]
- 8.(a) Shuker S, Hajduk P, Meadow R, Fesik S. Science. 1996;274:1531. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]; (b) Ren J, Esnouf R, Garman E, Somers D, Ross C, Kirby I, Keeling J, Darby G, Jones Y, Stuart D, Stammers D. Nat Struct Mol Biol. 1995;2:293. doi: 10.1038/nsb0495-293. [DOI] [PubMed] [Google Scholar]; (c) Jacobo-Molina A, Arnold E. Biochemistry. 1991;30:6351. doi: 10.1021/bi00240a001. [DOI] [PubMed] [Google Scholar]; (d) Jacobo-Molina A, Ding J, Nanni RJ, Clark AD, Lu X, Jr, Tantillo C, Williams RL, Kamer G, Ferris AL, Clark P, Hizi A, Hughes SH, Arnold E. Proc Natl Acad Sci USA. 1993;90:6320. doi: 10.1073/pnas.90.13.6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruth JL, Cheng YC. J Biol Chem. 1982;257:10261. [PubMed] [Google Scholar]
- 10.Mao C, Sudbeck EA, Venkatachalam TK, Uckun FM. Bioorg Med Chem Lett. 1999;9:1593. doi: 10.1016/s0960-894x(99)00235-8. [DOI] [PubMed] [Google Scholar]
- 11.Hunter R, Muhanji CI, Hale I, Bailey CM, Basavapathruni A, Anderson KS. Bioorg Med Chem Lett. 2007;17:2614. doi: 10.1016/j.bmcl.2007.01.107. [DOI] [PubMed] [Google Scholar]
- 12.Sugeac E, Fossey C, Laduree D, Schmidt S, Laumond G, Aubertin AM. J Enzyme Inhib Med Chem. 2003;18:175. doi: 10.1080/1475636032000069846. [DOI] [PubMed] [Google Scholar]
- 13.Bell FW, Cantrell AS, Högberg M, Jaskunas SR, Johansson NG, Jordan CL, Kinnick MD, Lind P, Morin JM, Jr, Noréen R, Öberg B, Palkowitz JA, Parrish CA, Pranc P, Sahlberg C, Ternansky RJ, Vasileff RT, Vrang L, West SJ, Zhang H, Zhou XX. J Med Chem. 1995;38:4929. doi: 10.1021/jm00025a010. [DOI] [PubMed] [Google Scholar]
- 14.Vig R, Mao C, Venkatachalam TK, Tuel-Ahlgren L, Sudbeck EA, Uckun FM. Bioorg Med Chem. 1998;6:1789. doi: 10.1016/s0968-0896(98)00108-4. [DOI] [PubMed] [Google Scholar]
- 15.Mao C, Sudbeck EA, Venkatachalam TK, Uckun FM. Biochem Pharmacol. 2000;60:1251. doi: 10.1016/s0006-2952(00)00408-1. [DOI] [PubMed] [Google Scholar]
- 16.Agrofoglio LA, Gillaizeau I, Saito Y. Chem Rev. 2003;103:1875. doi: 10.1021/cr010374q. [DOI] [PubMed] [Google Scholar]
- 17.Glennon RA, Liebowitz SM, Leming-Doot D, Rosecrans JA. J Med Chem. 1980;23:990. doi: 10.1021/jm00183a006. [DOI] [PubMed] [Google Scholar]
- 18.Arnott G, Hunter R, Mbeki L, Mohamed E. Tetrahedron Lett. 2005;46:4023. [Google Scholar]
- 19.Pannecouque C, Daelemans D, De Clercq E. Nat Protoc. 2008;3:427. doi: 10.1038/nprot.2007.517. [DOI] [PubMed] [Google Scholar]
- 20.Barreiro G, Kim JT, Guimaraes CRW, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. J Med Chem. 2007;50:5324. doi: 10.1021/jm070683u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J Comput Chem. 1998;19:1639. [Google Scholar]
- 22.Ren J, Diprose J, Warren J, Esnouf RM, Bird LE, Ikemizu S, Slater M, Milton J, Balzarini J, Stuart DI, Stammers DK. J Biol Chem. 2000;275:5633. doi: 10.1074/jbc.275.8.5633. [DOI] [PubMed] [Google Scholar]
- 23.(a) Venkatachalam TK, Sudbeck E, Uckun FM. J Mol Struct. 2005;751:41. [Google Scholar]; (b) Sudbeck EA, Jennissen JD, Venkatachalam TK, Uckun FM. Acta Crystallogr Sect C: Cryst Struct Commun. 1999;C55:2122. doi: 10.1107/s0108270199012044. [DOI] [PubMed] [Google Scholar]
- 24.Udier-Blagović M, Tirado-Rives J, Jorgensen WL. J Am Chem Soc. 2003;125:6016. doi: 10.1021/ja034308c. [DOI] [PubMed] [Google Scholar]
- 25.Chen CL, Venkatachalam TK, Waurzyniak B, Chelstrom L, Uckun FM. Arzneimittel-Forschung. 2001;51:574. doi: 10.1055/s-0031-1300082. [DOI] [PubMed] [Google Scholar]
- 26.Dennington RD, Keith T, Millam J, Eppinnett K, Hovell WL, Gilliland R. GaussView, Version 3.09. Semichem, Inc.; Shawnee Mission, KS: 2003. [Google Scholar]
- 27.(a) Marsili M, Gasteiger J. Croat Chem Acta. 1981;53:601. [Google Scholar]; (b) Gasteiger J, Marsili M. Tetrahedron. 1980;36:3210. [Google Scholar]
- 28.Mehler EL, Solmajer T. Protein Eng. 1991;4:903. doi: 10.1093/protein/4.8.903. [DOI] [PubMed] [Google Scholar]
- 29.(a) Haertle T, Carrera CJ, Wasson DB, Sowers LC, Richmann DD, Carson DA. J Biol Chem. 1988;263:5870. [PubMed] [Google Scholar]; (b) Harada S, Koyanagi Y, Yamamoto N. Science. 1985;229:563. doi: 10.1126/science.2992081. [DOI] [PubMed] [Google Scholar]
- 30.(a) Popovic M, Read-Connole E, Gallo R. Lancet. 1984;ii:1472. doi: 10.1016/s0140-6736(84)91666-0. [DOI] [PubMed] [Google Scholar]; (b) Popovic M, Sarngadharan MG, Read E, Gallo RC. Science. 1984;224:497. doi: 10.1126/science.6200935. [DOI] [PubMed] [Google Scholar]; (c) Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, Josephs SF, Doran ER, Rafalski JA, Whitehorn EA, Baumeister K, Ivanoff L, Petteway SR, Jr, Pearson ML, Lautenberger JA, Papas TS, Ghrayab J, Chang NT, Gallo RC, Wong-Stall F. Nature. 1985;313:277. doi: 10.1038/313277a0. [DOI] [PubMed] [Google Scholar]
- 31.Richman D, Shih CK, Lowy I, Rose J, Prodanovic P, Goff S, Griffin J. Proc Natl Acad Sci USA. 1991;88:11241. doi: 10.1073/pnas.88.24.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.