

SUMMARY
Many cellular responses, such as autoimmunity and cytotoxicity, are controlled by receptors with cytoplasmic immunoreceptor tyrosine-based inhibition motifs (ITIMs). Here, we showed that binding of inhibitory natural killer (NK) cell receptors to human leukocyte antigen (HLA) class I on target cells induced tyrosine phosphorylation of the adaptor Crk, concomitant with dephosphorylation of the guanine exchange factor Vav1. Furthermore, Crk dissociated from the guanine exchange factor C3G and bound to the tyrosine kinase c-Abl during inhibition. Membrane targeting of a tyrosine-mutated form of Crk could overcome inhibition of NK cell cytotoxicity, providing functional evidence that Crk phosphorylation contributes to inhibition. The specific phosphorylation of Crk and its dissociation from a signaling complex, observed here with two types of inhibitory receptors, expands the signaling potential of the large ITIM-receptor family and reveals an unsuspected component of the inhibitory mechanism.
INTRODUCTION
Maintenance of a proper balance in cellular activation often requires control by inhibitory receptors. Many receptors provide negative regulation by recruiting the tyrosine phosphatases SHP-1 and SHP-2 to phosphorylated immunoreceptor tyrosine-based inhibition motifs (ITIMs) in the receptor cytoplasmic tail (Burshtyn et al., 1996; Long, 2008; Olcese et al., 1996). A few examples of the diverse functions under negative regulation by ITIM-containing receptors include T cell-mediated autoimmunity, controlled by programmed death-1 (PD-1) (Okazaki and Honjo, 2006); limitation of plasticity in the visual cortex by paired immunoglobin (Ig)-like receptor B (PIRB) (Syken et al., 2006); and red cell engulfment by macrophages, which is inhibited through binding of signal-regulatory protein α (SIRPα) to CD47 (Oldenborg et al., 2000). Natural killer (NK) cell activation and cytotoxicity are inhibited by human killer cell Ig-like receptors (KIR) and mouse Ly49 receptors, as well as the conserved CD94-NKG2A receptor, all of which bind to major histocompatibility complex (MHC) class I molecules on target cells (Lanier, 1998).
Given the importance of ITIM-containing receptors in controlling cellular responses, it is of obvious interest to elucidate the inhibitory signaling pathway. In the case of NK cell contact with target cells, cytotoxicity is induced by multiple activation pathways, which operate both in parallel and in synergy (Bryceson et al., 2005; Bryceson et al., 2006). This raises the question of how activation of NK cells through different signaling pathways can be blocked by a single inhibitory mechanism. The identification of Vav1 as a specific substrate of SHP-1 during inhibition of NK cells by engagement of HLA class I on target cells suggested that KIR blocks activation at a central common point in NK cell activation, rather than by dephosphorylation of several effector molecules involved in signaling by different receptors (Stebbins et al., 2003). Vav1 is a multifunctional protein, acting as an adaptor through SH2 and SH3 domains and as a guanine exchange factor (GEF) for the small GTPases Rac1 and Cdc42. A central role of Vav1 in T cell development and in formation of functional immune synapses is well established (Tybulewicz, 2005).
Besides their role in blocking NK cell cytotoxicity toward healthy cells, MHC class I-specific NK cell inhibitory receptors have been assigned another function. They adjust the responsiveness of each NK cell according to the strength of inhibitory signals received. For example, NK cells that express several inhibitory receptors are intrinsically more responsive, whereas NK cells devoid of inhibitory receptors for self MHC are hyporesponsive (Anfossi et al., 2006; Fernandez et al., 2005; Kim et al., 2005). Several models have been proposed to account for this property of ITIM-containing NK cell receptors, including a “licensing” model, which could involve an instructive signal (Yokoyama and Kim, 2006), and a “disarming” model, whereby lack of inhibitory signals results in unresponsiveness, akin to anergy (Gasser and Raulet, 2006). Although licensing of mouse NK cells by inhibitory Ly49 is ITIM dependent (Kim et al., 2005), the nature of the signal and the point at which it regulates NK cell activity are still unknown.
To better understand how inhibition is achieved, we examined changes in phosphorylated molecules associated with Vav1 during inhibition of NK cells by KIR. We sought to identify changes in early activation signals by using NK cells that were stimulated by direct contact with target cells. These target cells were either sensitive to lysis (activating conditions) or resistant to lysis because of inhibitory receptor binding to specific HLA class I ligand on the target. This experimental system, although technically challenging, avoids the artificial use of antibodies to ligate NK cell receptors and of solid supports to present ligands to NK cell receptors. The results revealed an unexpected signal induced by inhibition through KIR and CD94-NKG2A. During inhibition, we observed the loss of association of tyrosine-phosphorylated Vav1 with c-Cbl, and we observed the tyrosine phosphorylation of the small adaptor molecule Crk. We provide evidence that Crk phosphorylation induced specifically by engagement of ITIM-containing receptors contributes to inhibition of NK cells.
RESULTS
Vav1 Is a Target of Inhibition by KIR-SHP-1 in NK Cells
To explore the mechanism of inhibition by ITIM-containing NK cell receptors, we first wanted to substantiate the evidence that Vav1 is a selective substrate of SHP-1 during inhibition. The identification of Vav1 as a substrate of SHP-1 during inhibition of NK cells by target cells expressing a ligand of KIR had relied on the use of a “trapping” mutant of SHP-1 fused to KIR2DL1 in place of the cytoplasmic ITIMs (Stebbins et al., 2003). Because these results were obtained by expression of KIR-SHP-1 chimeras in the NK cell line YTS, it remained to be seen whether the selection of Vav1 as primary target for dephosphorylation during inhibition was representative of inhibition in other NK cells. To test this, we transfected the chimeric KIR2DL1-SHP-1 receptor, as well as the aspartic-acid-to-alanine substrate-trapping mutant SHP-1(DA), into the human NK cell line NK92 (Figure S1A available online). The IL-2-dependent cell line NK92 has a phenotype and properties similar to those of human IL-2-activated NK cells. Even a low expression of catalytically active KIR2DL1-SHP-1 was sufficient to inhibit lysis of 221 target cells expressing the HLA-Cw15 ligand of KIR2DL1, while retaining lysis of cells expressing HLA-Cw3, an allele not recognized by KIR2DL1 (Figure S1B). As expected, the catalytically inactive KIR2DL1-SHP-1(DA) did not inhibit lysis of 221-Cw15 cells (Figure S1B).
The substrate-trapping experiment, conducted in the physiological context of NK cell interaction with target cells (Stebbins et al., 2003), was carried out in parallel with YTS and NK92 cells expressing KIR2DL1-SHP-1(DA). Trapping of a tyrosine-phosphorylated protein in NK92-2DL1-SHP-1(DA), which comigrated with Vav1 and with Vav1 trapped in YTS cells, was evident (Figure S1C). Note that trapping of Vav1 by this approach implies that Vav1 is dephosphorylated during inhibition and, hence, that phosphorylated Vav1 should be absent during NK cell inhibition. The tyrosine-phosphorylated protein of ~125 kDa, detected after incubation with 221-Cw15 cells, is the chimeric KIR2DL1-SHP-1(DA) receptor itself (Stebbins et al., 2003). Therefore, trapping of a single dominant substrate, previously identified as Vav1 in YTS cells, occurred in both NK cell lines. We conclude that Vav1 dephosphorylation by SHP-1 during KIR-mediated inhibition is a general feature of the inhibitory signaling pathway.
Phosphorylated Vav1 Associates with c-Cbl during NK Cell Activation, but Not Inhibition
To investigate signaling events that occur during inhibition by an ITIM-containing receptor, further experiments were carried out with the unmanipulated (i.e., nonchimeric) inhibitory KIR2DL1 receptor expressed in YTS cells (Figure S2). YTS cells expressing KIR2DL1 (YTS-2DL1 cells) kill 221-Cw3 cells, but not 221-Cw15 cells (Figure S2). After incubation of YTS-2DL1 cells with target cells for 5 min at 37°C, immunoprecipitates (IPs) of Vav1 were probed with a phosphotyrosine antibody (Ab) to identify molecules associated with Vav1 during activation or inhibition of YTS-2DL1 cells (Figure 1A). Lysates of 221-Cw3, 221-Cw15, and YTS-2DL1 cells prior to mixing were also tested. Strong tyrosine phosphorylation of Vav1 was observed, and it was highest after activating conditions. In addition, a tyrosine-phosphorylated band at ~120 kDa (indicated by an asterisk in Figure 1A) was seen reproducibly in Vav1 IPs, specifically after stimulation of YTS-2DL1 cells with 221-Cw3 cells, but not with 221-Cw15 cells (Figure 1A). The phosphorylated protein of ~120 kDa was not seen in any of the cells prior to cell mixing (Figure 1A). Other tyrosine-phosphorylated bands in the 50 and 70 kDa range have not been seen reproducibly in several other experiments. Reprobing with Abs for several proteins in the range of molecular mass ~120 kDa indicated that the tyrosine-phosphorylated band comigrated with c-Cbl (data not shown). c-Cbl is known to bind the Vav1 SH2 domain through its phosphorylated YMTP motif at tyrosine 700 (Liu and Altman, 1998).
Figure 1. Tyrosine-Phosphorylated Proteins Associated with Vav1 and c-Cbl during Activation and Inhibition of YTS-2DL1 Cells.

YTS-2DL1 cells mixed with 221-Cw3 cells (2DL1+Cw3) and YTS-2DL1 cells mixed with 221-Cw15 cells (2DL1+Cw15) were incubated for 5 min at 37°C and lysed. Lysates of each cell before and after mixing were immunoprecipitated with Abs for Vav1 and c-Cbl, as indicated.
(A) Vav1 IPs were probed with anti-phosphotyrosine (p-Tyr) and reprobed with anti-Vav1.
(B) c-Cbl IPs were probed with anti-phosphotyrosine and reprobed with anti-c-Cbl.
(C) c-Cbl IPs were probed with anti-pY160-Vav1 and reprobed for c-Cbl. Data are representative of at least five independent experiments.
To test, conversely, whether phosphorylated Vav1 is associated with c-Cbl, we probed c-Cbl IPs for tyrosine-phosphorylated proteins by immunoblotting. Strong tyrosine phosphorylation of c-Cbl was observed, and it was highest after activating conditions. In addition, a tyrosine-phosphorylated band at ~95 kDa was seen specifically after stimulation of YTS-2DL1 cells with 221-Cw3 cells, but not with 221-Cw15 cells (indicated by an asterisk in Figure 1B). Reprobing with a Vav1 Ab revealed that the ~95 kDa band comigrated with Vav1 (data not shown). To confirm association of phospho-Vav1 with c-Cbl during activation, we probed c-Cbl IPs with an Ab specific for phosphorylated tyrosine 160 in Vav1 (pY160-Vav1). An increase in association of pY160-Vav1 with c-Cbl during activation was clearly detected (Figure 1C).
Formation of phosphorylated c-Cbl-Vav1 complexes during activation suggested that c-Cbl could be associated with Vav1 while Vav1 is dephosphorylated by SHP-1 during inhibition. Indeed, a faint but distinct tyrosine-phosphorylated protein of ~120 kDa, which comigrated with c-Cbl, has been seen reproducibly after mixing YTS-2DL1-SHP-1(DA) cells with 221-Cw15 cells, but not after mixing with 221-Cw3 cells (Figure S3). This phosphorylated protein of ~120 kDa was not pulled down with the null mutant 2DL1-SHP-1(RM), which does not bind phosphotyrosine (Figure S3). However, providing evidence of receptor engagement, phosphorylation of receptor 2DL1-SHP-1(RM) was clearly detected after mixing with 221-Cw15 cells, demonstrating that the lack of a tyrosine-phosphorylated ~120 kDa protein was not due to defective ligand engagement by 2DL1-SHP-1(RM) (Figure S3). These data suggest that phospho-Cbl is either a direct substrate of SHP-1 or associated with Vav1 during dephosphorylation by SHP-1.
Association of CrkII with c-Cbl, p130CAS, and C3G during Activation by Sensitive Target Cells
c-Cbl is a scaffold protein of the cytoskeleton; it promotes formation of a lamellipodium though its association with the SH2 domain of the small adaptor CrkII (Abassi and Vuori, 2002; Scaife and Langdon, 2000). CrkII, as well as the related CrkL, consists of an SH2 domain, which binds phosphorylated tyrosine 774 in the YDVP motif of c-Cbl, and two SH3 domains. Through its SH3 domain, CrkII recruits the GEF C3G, which activates the GTPase Rap1 (Chodniewicz and Klemke, 2004). Several YxxP motifs for binding of CrkII are also present in the scaffold protein p130CAS. Given the potential role of CrkII-C3G and of CrkII association with c-Cbl and p130CAS in activation of NK cell cytotoxicity, we examined such associations during activation and inhibition of YTS-2DL1 cells mixed with target cells. Clear association of CrkII with c-Cbl, p130CAS, and C3G was induced by activation, as detected by immunoblotting of CrkII in IPs (Figure 2A). CrkII association with c-Cbl, p130CAS, and C3G was either reduced or absent after incubation with 221-Cw15 cells. The basal amount of p130CAS-CrkII complex in unstimulated YTS-2DL1 cells was also dissociated during inhibition (Figure 2A). Therefore, c-Cbl-CrkII, p130CAS-CrkII, and CrkII-C3G signaling complexes are not present in YTS-2DL1 cells that are inhibited by HLA-Cw15 on target cells.
Figure 2. CrkII Is Associated with c-Cbl, p130CAS, and C3G during Activation but Phosphorylated during Inhibition.

YTS-2DL1 cells were mixed with target cells as described in Figure 1.
(A) Lysates were immunoprecipitated with Abs to c-Cbl, p130CAS, and C3G, as indicated, probed with anti-CrkII, and reprobed with Abs to the immunoprecipitated proteins.
(B) Lysates were immunoprecipitated with Abs to c-Abl, p-Tyr, and pY221-CrkII. Membranes were immunoblotted with anti-CrkII and then reprobed with Abs to the immunoprecipitated proteins.
(C) Whole-cell lysates of 221 cells, either fixed in 0.5% paraformaldehyde or not, were immunoblotted with CrkII Ab.
(D) Cell mixing was performed in parallel with unfixed and fixed 221 cells. Lysates were immunoprecipitated with 4G10 agarose and immunoblotted for CrkII. Data are representative of at least three independent experiments.
CrkII Phosphorylation and Association with c-Abl during KIR-Mediated Inhibition
Different mechanisms could account for the lack of c-Cbl-CrkII and CrkII-C3G complexes during inhibition. First, these associations may never form because of inhibition of a step required upstream of this signaling complex. Another potential mechanism is that inhibitory signals initiated by KIR2DL1 actively disassemble complexes of CrkII with c-Cbl and C3G, for instance, by de-phosphorylation of tyrosine 774 in c-Cbl. However, an alternative and well-established mechanism for dissociation of CrkII from c-Cbl is the phosphorylation of tyrosine 221 in CrkII by the tyrosine kinase c-Abl and the formation of a phospho-CrkII-Abl complex (Chodniewicz and Klemke, 2004). This mechanism is not dependent on dephosphorylation of pY774 in c-Cbl. CrkII phosphorylated at Y221 is autoinhibited by intramolecular binding of its own SH2 domain. Furthermore, C3G is displaced by the binding of the SH3 domain of c-Abl. We therefore examined whether CrkII phosphorylation and association with c-Abl occurred during inhibition of YTS-2DL1 cells.
Association of CrkII with c-Abl occurred during inhibition of YTS-2DL1 cells mixed with 221-Cw15 cells, as detected by immunoblotting of CrkII in c-Abl IPs (Figure 2B). Furthermore, tyrosine phosphorylation of CrkII was detected during inhibitory conditions, after mixing YTS-2DL1 cells with 221-Cw15 cells (Figure 2B). Thus, these results show that a specific phosphorylation event occurs upon interaction of an inhibitory KIR with an HLA class I ligand on target cells. It is likely that KIR-mediated inhibitory signals induce CrkII phosphorylation for the purpose of disassembling c-Cbl-CrkII-C3G and p130CAS-CrkII-C3G complexes.
The biochemical experiments performed here with lysates of NK cells mixed with target cells do not discriminate between proteins contributed by NK cells and those in target cells. It was therefore possible that engagement of HLA-Cw15 could somehow induce CrkII phosphorylation in 221 target cells. To address this possibility, phosphorylation of CrkII was examined after mixing YTS-2DL1 cells with 221 target cells that had been subjected to light fixation in paraformaldehyde. The amount of total CrkII released by lysis of 221 cells was much reduced after fixation (Figure 2C). Nevertheless, induction of CrkII tyrosine phosphorylation was clearly evident in lysates of YTS-2DL1 cells mixed with fixed 221-Cw15 cells (Figure 2D). The amount of phosphorylated CrkII was in fact greater after mixing with fixed target cells. These results support the conclusion that CrkII phosphorylation occurs in NK cells during inhibition by KIR2DL1.
CrkII Association with c-Abl during KIR-Mediated Inhibition of Primary NK Cells
An important question was whether dissociation of c-Cbl-CrkII and CrkII-C3G complexes and association of CrkII with c-Abl also occurred during inhibition of primary NK cells. Large numbers of primary NK cells were required to perform biochemical experiments after incubation with target cells. Furthermore, because each individual NK cell expresses a small repertoire of KIR and CD94-NKG2 receptors (Parham, 2005), it was necessary to isolate NK cells that express a defined inhibitory KIR. Another complication is that mAbs to KIR2DL1 (CD158a) and to KIR2DL2 and KIR2DL3 (CD158b) do not distinguish inhibitory KIRs from the activating isoforms KIR2DS1 and KIR2DS2, respectively. To identify inhibitory forms of KIR, we have generated a rabbit polyclonal antiserum, called cyt42/43, specific for the cytoplasmic tail of KIR2DL1 and KIR2DL2. Therefore, CD158a+ cyt42/43+ NK cells will be KIR2DL1+ and inhibited by HLA-Cw4 and HLA-Cw15 (Figure S4). By screening several individual donors, we identified one that expressed KIR2DL1 in the absence of KIR2DS1 on a high fraction of peripheral blood NK cells (Figure S4).
A large number of primary NK cells that uniformly express inhibitory KIR2DL1 were obtained by cell sorting (Figure S4). Phosphorylation of Vav1 at tyrosine 160 was detected in the IL-2-activated KIR2DL1+ NK cell population and increased after stimulation with sensitive 221 target cells (Figure 3A). After mixing with resistant 221-Cw15 cells, phosphorylation of Vav1 Y160 was even less than the basal amount in KIR2DL1+ cells (Figure 3A). In addition, CrkII association with c-Cbl and with C3G during activation, as well as association of CrkII with c-Abl during incubation with resistant 221-Cw15 cells, were clearly observed (Figure 3B). Some association of CrkII with c-Abl also occurred during activation of NK cells, a result consistent with the phosphorylation-dependent recruitment of CrkII (Abassi and Vuori, 2002; Chodniewicz and Klemke, 2004). We conclude that binding of inhibitory KIR2DL1 to an HLA class I ligand on target cells induces active association of CrkII with c-Abl in primary NK cells.
Figure 3. Formation of CrkII-c-Abl Complexes during Inhibition of Primary NK Cells.

KIR2DL1+ NK cells were mixed with 221 cells and 221-Cw15 cells for 5 min at 37°C. Cells lysates were immunoprecipitated with Abs to (A) Vav1 and (B) c-Cbl, C3G, and c-Abl. Blots were probed with Abs to (A) pY160-Vav1 and (B) CrkII and then reprobed for the indicated proteins. Data are representative of at least four independent experiments.
Inhibition by CD94-NKG2A Binding to HLA-E Induces Crk Phosphorylation and Association with c-Abl
Binding of the lectin-like receptor CD94-NKG2A to HLA-E inhibits target cell lysis and prevents proper NK cell immune-synapse formation (Masilamani et al., 2006). NKG2A and the Ig-like inhibitory KIR are not structurally related and have opposite transmembrane orientations, and yet they share ITIM sequences in their cytoplasmic tails. It was therefore of interest to test whether phospho-Vav1-c-Cbl, c-Cbl-Crk, and Crk-C3G associations were also inhibited through binding of CD94-NKG2A to HLA-E on target cells. The CD94-NKG2A+ NK cell line NKL was used to address this question. Whereas untransfected 221 cells were killed by NKL, expression of HLA-E on transfected 221 cells (221-E cells) provided protection from lysis by NKL cells (Figure S2).
After incubation of NKL cells with target cells for 5 min at 37°C, strong tyrosine phosphorylation of c-Cbl was apparent, more so with 221 cells than 221-E cells (Figure 4A). In addition, a phosphoprotein of ~95 kDa appeared during activating conditions only (indicated by an asterisk in Figure 4A). Association of phospho-Vav1 with c-Cbl during activation, but not inhibition, was demonstrated directly by probing the c-Cbl IP with the pY160 Vav1 Ab (Figure 4B). Therefore, association of phospho-Vav1 with c-Cbl is prevented or reversed during inhibition of NKL cells by CD94-NKG2A.
Figure 4. Tyrosine-Phosphorylated Proteins Associated with c-Cbl during Activation and Inhibition of NKL Cells.
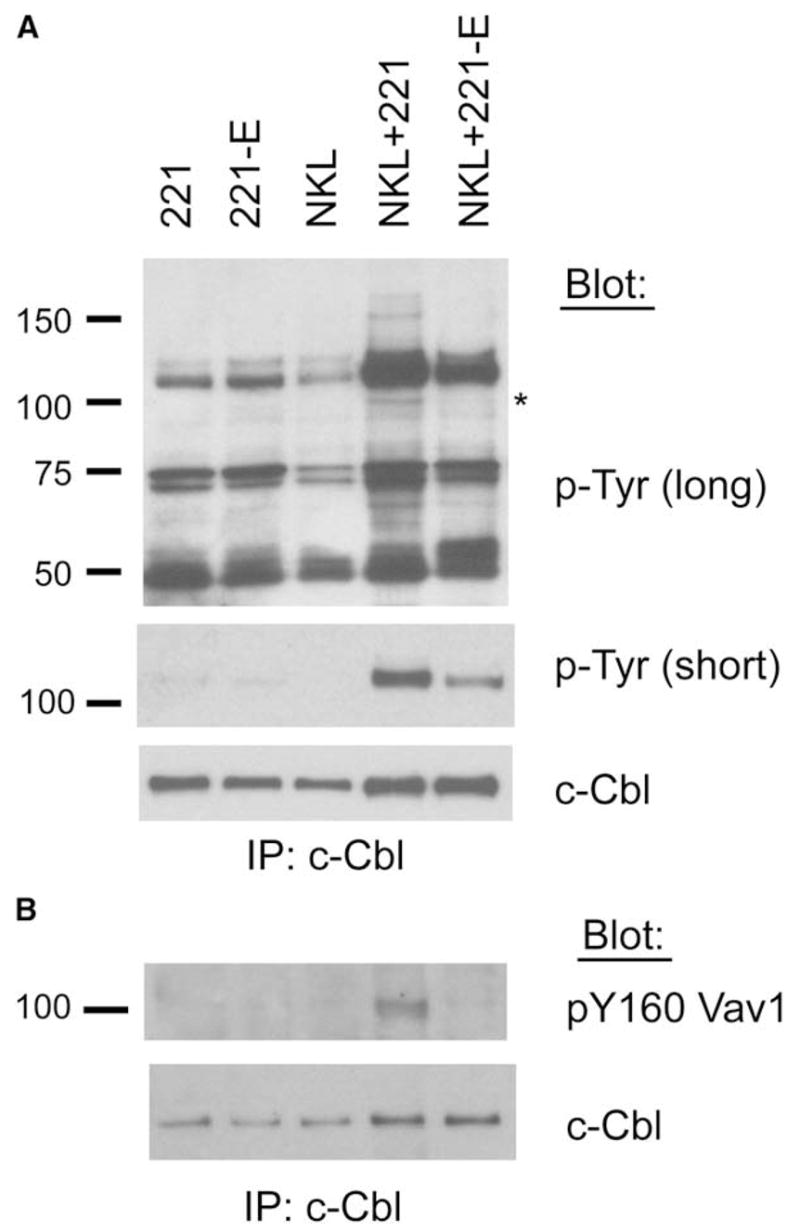
NKL cells were mixed with 221 or 221-E cells, incubated for 5 min at 37°C, and lysed. Lysates were immunoprecipitated with anti-c-Cbl.
(A) IPs were immunoblotted with anti-phosphotyrosine and reprobed with anti-c-Cbl. A long and a short exposure of the phosphotyrosine immunoblot are shown, as indicated.
(B) IPs were probed with anti-pY160-Vav1 and reprobed for c-Cbl. Data are representative of at least three independent experiments.
Association of Crk with c-Cbl, p130CAS, and C3G was then examined. The cell line NKL expresses mainly the lymphocyte-specific member of the Crk family, CrkL (data not shown). Formation of c-Cbl-CrkL and p130CAS-CrkL complexes was detected after incubation with 221 cells, but not 221-E cells (Figure 5A). Furthermore, association of CrkL with C3G was reduced when NKL cells were incubated with resistant 221-E target cells (Figure 5A). If CrkL forms ternary complexes by simultaneous binding to c-Cbl and C3G through its SH2 and SH3 domains, respectively, it may be possible to detect C3G in c-Cbl IPs. Indeed, C3G was detected in IPs of c-Cbl from lysates of NKL cells that had been incubated with 221 cells, but not 221-E cells (Figure 5B). These results are consistent with formation of a trimolecular complex c-Cbl-CrkL-C3G.
Figure 5. CrkL Phosphorylation and Association with c-Abl during Inhibition by CD94-NKG2A.
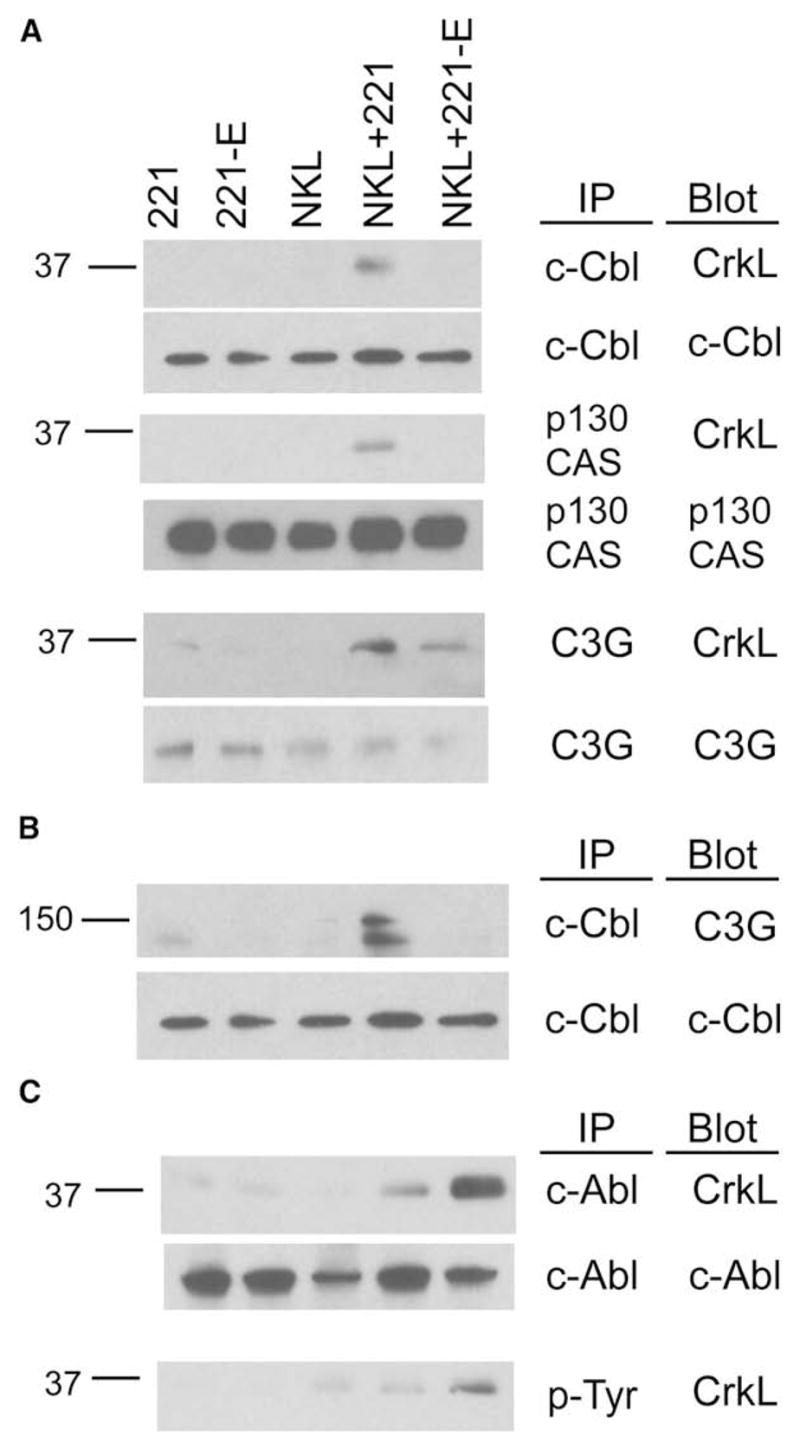
NKL cells were mixed with 221 or 221-E cells, incubated at 37°C for 5 min, and lysed.
(A) Lysates were immunoprecipitated with Abs to c-Cbl, p130CAS, and C3G. Membranes were immunoblotted with anti-CrkL and then reprobed with Abs to the immunoprecipitated proteins.
(B) Lysates were immunoprecipitated with anti-c-Cbl, and membranes were immunoblotted for C3G and reprobed for total c-Cbl.
(C) Lysates were immunoprecipitated with Abs to c-Abl and phosphotyrosine, and membranes were immunoblotted with anti-CrkL and then reprobed with Abs to c-Abl. Data are representative of at least three independent experiments.
Association of CrkL with c-Abl and tyrosine phosphorylation of CrkL were clearly detected after inhibition of NKL cells by 221-E cells (Figure 5C). The small amount of c-Abl-CrkL complex and of phosphorylated CrkL observed after activation by 221 cells is consistent with the phosphorylation-dependent membrane recruitment of Crk (Abassi and Vuori, 2002; Chodniewicz and Klemke, 2004). In conclusion, engagement of CD94-NKG2A by HLA-E has the same outcome as the inhibitory KIR2DL1-HLA-Cw15 interaction, in that they both prevent formation of Crk complexes with scaffold proteins c-Cbl and p130CAS and recruitment of C3G by Crk. Furthermore, they both induce specific tyrosine phosphorylation of the adapters CrkII and CrkL and their association with the tyrosine kinase c-Abl. We conclude that specific Crk tyrosine phosphorylation is a general feature of negative signaling by ITIM-containing receptors rather than a peculiar feature of inhibitory KIR.
Membrane Targeting of a Tyrosine Mutant of CrkII Overcomes Inhibition
If Crk phosphorylation and Crk dissociation from signaling complexes were to participate in the inhibitory mechanism of ITIM-containing receptors, it would follow that Crk has a positive role during activation of NK cells. To test this, we performed CrkII siRNA knockdown experiments with YTS-2DL1 cells. Out of six independent experiments, in which partial knockdown of CrkII was achieved, the cytotoxic activity of YTS-2DL1 cells toward 221 cells was reduced by 14% ± 5.6% (Figure S5). Loss-of-function experiments suffer from the possibility of indirect effects or nonspecific inhibition. Experiments in which function is restored provide more direct evidence for the role of a specific protein. We therefore investigated the possibility of restoring cytotoxic function in NK cells during inhibitory signaling by an ITIM-containing receptor.
The most important question here was whether phosphorylation of Crk and formation of Crk-Abl complexes contributes to inhibition of NK cell cytotoxicity. Simple experiments using the c-Abl inhibitor Gleevec (STI571) to prevent Crk phosphorylation by c-Abl were not an option because c-Abl inhibition compromised NK cell cytotoxicity toward sensitive target cells (data not shown). This was not a surprising result, given that c-Abl participates in signaling pathways, for example, by phosphorylation of ZAP70 downstream of the TCR (Zipfel et al., 2004), and participates in actin cytoskeleton remodeling through direct binding to F-actin (Woodring et al., 2003) and phosphorylation of WAVE2 (Stuart et al., 2006).
A tyrosine-mutated form of CrkII, which cannot be phosphorylated by c-Abl, has the potential to remain bound to phospho-Y774 in c-Cbl and to interfere with inhibition by KIR2DL1. However, such an approach was not feasible either because membrane recruitment of CrkII requires phosphorylation at Y221 (Abassi and Vuori, 2002). Instead, our chosen approach was to express in YTS-2DL1 cells a membrane-targeted mutant of CrkII, in which tyrosine 221 was mutated to a phenylalanine (Y221F). We designed this mutant to function independently of endogenous CrkII by endowing it with two properties: first, the ability to bypass the phosphorylation-dependent recruitment to the membrane (Abassi and Vuori, 2002), and second, resistance to phosphorylation by replacement of tyrosine 221 with phenylalanine. During inhibition, while endogenous CrkII would still be inactivated through phosphorylation and dissociation from c-Cbl, p130CAS, and C3G, the scaffold proteins and C3G should be free to associate with the membrane-targeted CrkII. To achieve membrane targeting of CrkII, we fused the 16 N-terminal amino acids of the kinase Lyn to the N terminus of CrkII (Lyn-Crk).
YTS-2DL1 cells were stably transfected with wild-type CrkII (Crk), CrkII-Y221F (Crk-YF), membrane-targeted CrkII (Lyn-Crk), and membrane-targeted Lyn-CrkII-Y221F (Lyn-Crk-YF). Stable clones expressing the different forms of CrkII were obtained readily, except for Lyn-Crk-YF, for which the few clones obtained expressed low amounts of Lyn-Crk-YF. YTS-2DL1 cells expressing CrkII and each of the three CrkII mutants were tested for their ability to receive inhibitory signals upon binding to HLA-Cw4 on target cells. Inhibition comparable to that obtained with YTS-2DL1 cells was observed with YTS-2DL1 cells expressing Lyn-Crk (Figure 6). In contrast, inhibition of YTS-2DL1 cells expressing membrane-targeted Lyn-Crk-YF was reduced (Figure 6). Several experiments were carried out with YTS-2DL1 cells expressing Crk and each of the three Crk mutants (Figure 6B). Inhibition was unimpaired in YTS-2DL1 cells expressing wild-type Crk, Crk-YF, and Lyn-Crk. In contrast, reduced inhibition was observed with YTS-2DL1 cells expressing Lyn-Crk-YF, and the loss of inhibition was more pronounced in those experiments in which inhibition of YTS-2DL1 cells was not complete (Figure 6B). The reversal of inhibition caused by membrane targeting of a nonphosphorylated CrkII implies that CrkII phosphorylation contributes to inhibition of NK cell cytotoxicity.
Figure 6. Membrane Targeting of CrkII-Y221F Overcomes Inhibition.

YTS-2DL1 cells, either untransfected (–) or transfected with wild-type CrkII (Crk), CrkII-Y221F (Crk-YF), Lyn-tagged wild-type CrkII (Lyn-Crk), and Lyn-tagged CrkII-Y221F (Lyn-Crk-YF), were tested for their ability to lyse 221-Cw3 and 221-Cw4 cells.
(A) Lysis of 221-Cw3 cells (black bars) and 221-Cw4 cells (shaded bars) at an effector-to-target ratio of 10.
(B) Inhibition of lysis, i.e., [(lysis of Cw3 − lysis of Cw4)/(lysis of Cw3)] × 100, observed in multiple experiments. Similar results were obtained in experiments with 221-Cw15 cells as resistant target cells. Each cluster of symbols represents a separate experiment. Left panel: Inhibition of YTS-2DL1 cells (triangles) is compared with inhibition of YTS-2DL1 cells transfected with Crk (open circles) or Crk-YF (filled circles). Right panel: Inhibition of YTS-2DL1 cells (triangles) is compared with inhibition of YTS-2DL1 cells transfected with membrane-targeted Lyn-Crk (open circles) or membrane-targeted Lyn-Crk-YF (filled circles).
DISCUSSION
Our study offers a new perspective on inhibitory signaling by ITIM-containing receptors. Using MHC class I-specific inhibitory receptors of NK cells as a model for ITIM signaling, we show that inhibition involves not only dephosphorylation of Vav1, but also phosphorylation of a regulatory tyrosine in the adaptor Crk. The fact that a specific tyrosine-phosphorylation event occurs in NK cells during inhibition by MHC class I-positive target cells begs a re-evaluation of the inhibitory mechanism and the signaling potential of ITIM-containing receptors.
Signals transmitted by inhibitory KIR2DL1 and by CD94-NKG2A were studied in the proper context of interactions between NK cells and target cells that express defined HLA ligands of these two inhibitory receptors. Signals were analyzed biochemically after engagement of activating and inhibitory receptors of NK cells by their natural ligands on target cells in the absence of Abs. Such an experimental approach entails technical challenges because signaling events occurring in a population of cells engaged in synapse formation with other cells are not synchronized and are weaker than after artificial crosslinking of receptors with Abs. The only difference between activating and inhibitory conditions in our experimental system was expression of HLA-C or HLA-E on target cells. Association of the adaptor Crk with the scaffold protein c-Cbl and the Rap1 GEF C3G, which were induced by contact of NK cells with sensitive target cells, did not occur during inhibitory NK-target cell contacts. Even more striking than the absence of specific signals were the tyrosine phosphorylation of Crk and the association of Crk with the tyrosine kinase c-Abl during inhibition only. Therefore, it is clear that tyrosine dephosphorylation by SHP-1 is not the only outcome of ITIM-mediated signaling and that ITIM-containing receptors can also induce specific tyrosine phosphorylation.
The specific tyrosine phosphorylation of Crk and its association with c-Abl were observed during inhibition by two unrelated receptors, the type I transmembrane, Ig-like KIR2DL1, and the type II transmembrane, lectin-like CD94-NKG2A. YTS cells expressing KIR2DL1 were tested with 221-Cw3 cells for activation and 221-Cw15 cells for inhibition, whereas 221 and 221-E target cells were used for a similar analysis of NKL cells, which express CD94-NKG2A. Therefore, the formal possibility that some unknown difference between 221-Cw3 and 221-Cw15 cells, besides expression of these two different HLA-C allotypes, would be responsible for changes in Crk phosphorylation in YTS-2DL1 cells is extremely unlikely, given that similar results were obtained with different target cells for the two NK cell lines. Crk phosphorylation and formation of Crk-Abl complexes appear to be a general feature of inhibition by different types of ITIM-containing receptors. The new mechanism described here is also relevant to inhibition of primary NK cells, in which formation of Crk-c-Abl complexes was detected during inhibition.
Because c-Cbl-Crk complexes contribute to actin reorganization and formation of lamellae (Abassi and Vuori, 2002; Bouton et al., 2001; Scaife and Langdon, 2000), dissociation of Crk could impair actin-dependent synapse formation between NK cells and target cells. Crk enhances lamellipodiae formation by recruiting C3G, a GEF for Rap1, or DOCK180, an activator of Rac1, to its SH3 domain (Buensuceso and O’Toole, 2000). Tyrosine phosphorylation of c-Cbl and p130CAS and formation of p130CAS-Crk complexes are induced by integrin signaling (Chodniewicz and Klemke, 2004; Liu and Altman, 1998). The role of GTPase Rap1 in promoting LFA-1-dependent adhesion through “inside-out” signaling (Katagiri et al., 2003) and the activation of Rap1 by C3G suggest that dissociation of Crk from c-Cbl and p130CAS may result in negative regulation of LFA-1 signals. Binding of inhibitory KIR to HLA-C on target cells does indeed inhibit LFA-1-mediated adhesion (Burshtyn et al., 2000).
A known mechanism for dissociation of Crk from c-Cbl and p130CAS is the phosphorylation of Y221 in Crk by the kinase c-Abl, rather than dephosphorylation of tyrosines in c-Cbl and p130CAS (Abassi and Vuori, 2002; Holcomb et al., 2006; Shishido et al., 2001). Crk phosphorylation by c-Abl involves a transient Crk-c-Abl complex, in which Crk is autoinhibited by its own SH2 domain bound to pY221 (Abassi and Vuori, 2002; Shishido et al., 2001). Consistent with the known functions of Crk and of Vav1, inhibition by ITIM-containing receptors blocks actin-dependent processes, such as actin cytoskeleton reorganization, phosphorylation of activation receptors NKG2D and 2B4 and their recruitment to cholesterol-enriched membrane domains, and lipid raft accumulation at the synapse (Dietrich et al., 2001; Endt et al., 2007; Fassett et al., 2001; Guerra et al., 2002; Masilamani et al., 2006; Watzl and Long, 2003). Therefore, inhibition of both Vav1 and Crk-mediated signals could serve to achieve more effective inhibition.
Besides its role in actin cytoskeleton remodeling, c-Cbl also serves as a negative regulator of immune responses through its E3 ubiquitin ligase activity (Liu, 2004; Thien and Langdon, 2005). c-Cbl-dependent ubiquitination of molecules, such as TCR ζ chain and Src-family kinases, targets these signaling components for degradation by proteasomes. c-Cbl-mediated ubiquitination also results in receptor internalization from the cell surface (Balagopalan et al., 2007; Naramura et al., 2002). Therefore, a contribution of c-Cbl to the inhibitory signal of KIR through ubiquitination of signaling components had to be considered. However, no ubiquitination of a number of proteins, including known substrates for ubiquitination and proteins involved in NK cell activation (c-Cbl, Vav1, CrkII, CrkL, SHP-1, Grb-2, c-Abl, KIR2DL1, Hck, and 2B4), could be detected in NK cells mixed with either sensitive or resistant target cells (data not shown).
There is general agreement that inhibition of NK cell cytotoxicity is achieved by dephosphorylation of signaling components at an early step in the activation pathway (Billadeau and Leibson, 2002; Long, 2008; Long et al., 2001). Activation signals that do not appear during inhibition either are not occurring because of a block at an earlier signaling step or are actively reversed by the inhibitory signaling pathway. Substrate-trapping experiments that identified Vav1 as substrate of SHP-1 in YTS cells (Stebbins et al., 2003) and in NK92 (this paper) imply that Vav1 is actively dephosphorylated during inhibition. In this study, we provide evidence for another active signaling process that occurs during inhibition and involves, surprisingly, tyrosine phosphorylation of Crk, despite recruitment and activation of the tyrosine phosphatase SHP-1. Inhibition via ITIM-containing receptors can no longer be viewed simply as a shutdown of tyrosine-phosphorylation events.
A functional experiment was designed to test whether Crk phosphorylation and dissociation from c-Cbl contributed to inhibition of NK cell cytotoxicity by KIR. The approach of choice was for us to engineer a membrane-targeted and tyrosine-mutated version of Crk (Lyn-Crk-YF) by fusion to the N-terminal sequence of the kinase Lyn, in order to bypass the phosphorylation-dependent Crk recruitment to the membrane. Despite very low expression of Lyn-Crk-YF in YTS-2DL1 cell clones, substantial lysis of target cells was restored during incubation with 221-Cw15 cells, which are normally protected through binding of KIR2DL1. Reversal of inhibition by expression of Lyn-Crk-YF was progressively greater as overall inhibition of YTS-2DL1 by 221-Cw15 cells was reduced, suggesting that two inhibitory mechanisms, such as Vav1 dephosphorylation and Crk phosphorylation, operate in parallel. It is worth noting that our experiment, in which cytotoxic function was restored, is a more stringent test of function than experiments in which function is lost through inhibition or loss of specific molecules.
Our results have revealed a second arm in the mechanism for inhibition of NK cell cytotoxicity (Figure 7). One relies on dephosphorylation of Vav1 by SHP-1, the other on the dissociation of Crk from c-Cbl and C3G. A new question raised by these findings is how ITIM-containing receptors orchestrate c-Abl recruitment, Crk phosphorylation, and disassembly of c-Cbl-Crk-C3G complexes. But regardless of the specific interactions in this pathway, it is striking that engagement of inhibitory KIR by HLA-C can result in a specific tyrosine-phosphorylation event.
Figure 7. Two Components of Inhibitory Signaling by ITIM-Containing Receptors.

SHP-1 recruited by tyrosine-phosphorylated inhibitory receptor (KIR) dephosphorylates tyrosines in Vav1 that regulate its GEF activity toward Rac1. Red indicates inhibitory signaling. Green indicates signaling for activation. Phosphorylated Vav1-c-Cbl complexes, Crk-c-Cbl and Crk-C3G complexes, form during activation. C3G has GEF activity toward Rap1. During inhibition, Crk is not associated with c-Cbl and C3G but is tyrosine phosphorylated and bound to the tyrosine kinase c-Abl. N indicates the N-terminal end of proteins. Only the C-terminal region of c-Cbl is shown. SH2 domain-phosphotyrosine (pY) interactions are shown. Circles represent SH3 domains.
The ability of an inhibitory KIR to promote tyrosine phosphorylation widens the scope of potential functions for the many receptors in the ITIM family, such as the proposed ITIM-dependent signal delivered by Ly49A to promote responsiveness of mouse NK cells (Kim et al., 2005). Our biochemical data indeed suggest that ITIM-dependent signaling could prevent NK cell unresponsiveness in the same manner that CD28 costimulation prevents T cell anergy. A hallmark of anergic T cells is the presence of constitutive c-Cbl-CrkL-C3G complexes and active Rap1 (Boussiotis et al., 1997). CD28 signaling suppresses TCR-dependent activation of Rap1 (Reedquist and Bos, 1998). ITIM-dependent inactivation of Rap1 by Crk phosphorylation may be used as a similar mechanism to promote NK cell responsiveness.
EXPERIMENTAL PROCEDURES
Cell Lines and Reagents
721.221 cells (referred to as 221 in this paper) and 221 cells transfected with HLA-Cw3, Cw4, Cw7, and Cw15 were obtained from J. Gumperz and P. Parham (Stanford University). 221 cells transfected with an HLA-E carrying the HLA-A2 signal peptide were obtained from D. Geraghty (Lee et al., 1998) (Fred Hutchinson Cancer Research Center, Seattle, WA). 221 transfectants were cultured in RPMI supplemented with glutamine, 10% fetal calf serum (FCS), and 50 μM 2-mercapto-ethanol. The human NK cell line YTS transfected with KIR2DL1 (YTS-2DL1, gift of G. Cohen, Massachusetts General Hospital, Boston, MA) (Cohen et al., 1999) was cultured in Iscove’s (IMDM) supplemented with glutamine, 12.5% FCS, and 50 μM 2-mercapto-ethanol. NKL cells (Robertson et al., 1996), obtained from M. Robertson (Indiana University Medical Center), were cultured in RPMI supplemented with glutamine, 10% FCS, 50 μM 2-mercapto-ethanol, and 100 U/ml rIL-2. Human NK cells were isolated from PBL with the MACS NK isolation kit (Miltenyi) and were grown in IMDM supplemented with 10% human AB serum (Valley Biomedicals), 10% human IL-2 (Hemagen), and 100 U/ml rIL-2 (Roche). KIR2DL1-positive NK cells were isolated from bulk NK cell populations with KIR2DL1 antibody and anti-mouse IgG Dynabeads (Dynal).
Antibodies
Antibodies were obtained from the following sources: Anti-KIR2DL1 (143211; R&D Systems); Anti-Vav1 (3130; Abcam); Anti-phosphotyrosine (4G10; Upstate); Anti-c-Cbl (A-9 and C-15), C3G (H-300), p130 CAS (N-20), c-myc (9E10), c-Abl (K12), and Vav1 (H211) (Santa Cruz); Anti-pY160 Vav-1 (Bio-source); Anti-c-Abl (8E9; BD Biosciences); Anti-c-Cbl (C9603; Sigma-Aldrich); and Anti-pY221 CrkII (3491; Cell Signaling). Mouse PE-conjugated and rabbit FITC-conjugated secondary antibodies for flow cytometry were from Jackson Immunoresearch. Mouse, rabbit, and goat antibodies conjugated to horseradish peroxidase (HRP) were from Santa Cruz. Streptavidin-HRP antibody was from GE Healthcare.
DNA Mutagenesis and Transfections
A myc-tagged rat CrkII cDNA (Cho and Klemke, 2000) (a gift of R. Klemke, University of California, San Diego) was subcloned into pCDNA3-hygro (Invitrogen) and subsequently mutagenized with Quick Change (Stratagene). Tyrosine 221 of CrkII was mutated to phenylalanine. An oligonucleotide encoding the 16 N-terminal amino acids of Lyn was inserted by “loop-in” mutagenesis, such that this membrane-targeting sequence of Lyn was inserted in place of the initiation methionine of CrkII and CrkII-Y221F. All final constructs were verified by sequencing. YTS-2DL1 cells were transfected by electroporation and placed under 400 μg/ml Hygromycin B selection. Bulk populations were subcloned and clones were screened for expression of CrkII by intracellular staining, immunoblotting, and RT-PCR.
Flow Cytometry
Cells were stained with directly conjugated antibodies or unlabeled antibodies followed by fluorescently labeled secondary antibodies (diluted 1 to 200) in phosphate-buffered saline (PBS) with 2.5% FCS and 0.1% NaN3. Samples were analyzed on a BD Facscan with FlowJO software (Treestar).
Cytotoxicity Assays
For Europium-based cytotoxicity assays, target cells were labeled with 40 μM BATDA (Perkin Elmer) for 30 min at 37°C. Target cells were washed in medium containing 1 mM Sulfinpyrazone (Sigma) and incubated with effector cells in the presence of Sulfinpyrazone for 2 hr at 37°C. Plates were mixed briefly and centrifuged at 1200 rpm for 2 min. 20 μl of supernatant was incubated with 200 μl of a 20% Europium solution (Perkin Elmer) in 0.3 M acetic acid for 5 min and analyzed with a Wallac (Perkin Elmer) plate reader.
NK-Target Cell Mixing Experiments
5 × 106 YTS-2DL1 and 5 × 106 transfected 221 cells were incubated separately on ice for 10 min. Cells were mixed and spun down, and media were aspirated. The cell pellet was then incubated on ice for 10 min. Cells were transferred to 37°C for 5 min, moved to ice, and lysed in 1 ml of lysis buffer (0.5% Triton X-100, 150 mM NaCl, 50 mM Tris-HCl [pH 7.4], 2 mM EGTA, 1 mM PMSF, and 1 mM NaVO3) for 20 min. Nuclei were pelleted, and supernatants were incubated with 1 μg antibody plus protein G Sepharose beads (GE Healthcare) for 1 hr. Beads were washed twice in lysis buffer and were resuspended in 20 μl of 1 × NuPage LDS sample buffer (Invitrogen) containing 50 mM DTT. Beads were incubated at 70°C for 10 min and then loaded on either NuPage MOPS or TA (Invitrogen) gels. Proteins were transferred to PVDF (Millipore) for 1 hr at 30 V in 1 × NuPage transfer buffer (Invitrogen). Membranes were blocked in PBS with 0.5% Tween 20 and 5% BSA for at least 30 min and then incubated with primary antibody plus NaN3 for at least 1 hr. Membranes were washed three times in PBS with 350 mM NaCl and 1% Tween 20 for 5 min. Incubations with 1 × 10−4 dilutions of secondary antibodies in a 1:1 mixture of blocking buffer and wash buffer were carried out for 30 min. Blots were washed three times in PBS with 1% Tween 20 for 5 min and developed with either Super Signal West Dura or Pico (Pierce). Membranes were exposed to Biomax MR film (Kodak) and developed. In some cases, target cells were fixed before mixing. Cells were washed twice in Hanks Balanced Salt Solution and incubated in 0.5% paraformaldehyde for 5 min at room temperature. Cells were washed three times in complete medium and then incubated at 37°C for 1 hr. Cells were washed once more before cell-mixing experiments.
Supplementary Material
Supplemental Data include Supplemental Experimental Procedures and five figures and can be found with this article online at http://www.immunity.com/cgi/content/full/29/4/578/DC1/.
Acknowledgments
We thank M. March and A. Shaw for useful discussions; G. Cohen, D. Geraghty, J. Gumperz, P. Parham, and M. Robertson for cell lines; and R. Klemke for a CrkII cDNA. This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases.
References
- Abassi YA, Vuori K. Tyrosine 221 in Crk regulates adhesion-dependent membrane localization of Crk and Rac and activation of Rac signaling. EMBO J. 2002;21:4571–4582. doi: 10.1093/emboj/cdf446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, Breso V, Frassati C, Reviron D, Middleton D, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25:331–342. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Balagopalan L, Barr VA, Sommers CL, Barda-Saad M, Goyal A, Isakowitz MS, Samelson LE. c-Cbl-mediated regulation of LAT-nucleated signaling complexes. Mol Cell Biol. 2007;27:8622–8636. doi: 10.1128/MCB.00467-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billadeau DD, Leibson PJ. ITAMs versus ITIMs: Striking a balance during cell regulation. J Clin Invest. 2002;109:161–168. doi: 10.1172/JCI14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussiotis VA, Freeman GJ, Berezovskaya A, Barber DL, Nadler LM. Maintenance of human T cell anergy: Blocking of IL-2 gene transcription by activated Rap1. Science. 1997;278:124–128. doi: 10.1126/science.278.5335.124. [DOI] [PubMed] [Google Scholar]
- Bouton AH, Riggins RB, Bruce-Staskal PJ. Functions of the adapter protein Cas: Signal convergence and the determination of cellular responses. Oncogene. 2001;20:6448–6458. doi: 10.1038/sj.onc.1204785. [DOI] [PubMed] [Google Scholar]
- Bryceson YT, March ME, Barber DF, Ljunggren HG, Long EO. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J Exp Med. 2005;202:1001–1012. doi: 10.1084/jem.20051143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryceson YT, March ME, Ljunggren HG, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev. 2006;214:73–91. doi: 10.1111/j.1600-065X.2006.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buensuceso CS, O’Toole TE. The association of CRKII with C3G can be regulated by integrins and defines a novel means to regulate the mitogen-activated protein kinases. J Biol Chem. 2000;275:13118–13125. doi: 10.1074/jbc.275.17.13118. [DOI] [PubMed] [Google Scholar]
- Burshtyn DN, Scharenberg AM, Wagtmann N, Rajagopalan S, Berrada K, Yi T, Kinet JP, Long EO. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitor receptor. Immunity. 1996;4:77–85. doi: 10.1016/s1074-7613(00)80300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burshtyn DN, Shin J, Stebbins C, Long EO. Adhesion to target cells is disrupted by the killer cell inhibitory receptor. Curr Biol. 2000;10:777–780. doi: 10.1016/s0960-9822(00)00568-6. [DOI] [PubMed] [Google Scholar]
- Cho SY, Klemke RL. Extracellular-regulated kinase activation and CAS/Crk coupling regulate cell migration and suppress apoptosis during invasion of the extracellular matrix. J Cell Biol. 2000;149:223–236. doi: 10.1083/jcb.149.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodniewicz D, Klemke RL. Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim Biophys Acta. 2004;1692:63–76. doi: 10.1016/j.bbamcr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity. 1999;10:661–671. doi: 10.1016/s1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- Dietrich J, Cella M, Colonna M. Ig-like transcript 2 (ILT2)/leukocyte Ig-like receptor 1 (LIR1) inhibits TCR signaling and actin cytoskeleton reorganization. J Immunol. 2001;166:2514–2521. doi: 10.4049/jimmunol.166.4.2514. [DOI] [PubMed] [Google Scholar]
- Endt J, McCann FE, Almeida CR, Urlaub D, Leung R, Pende D, Davis DM, Watzl C. Inhibitory receptor signals suppress ligation-induced recruitment of NKG2D to GM1-rich membrane domains at the human NK cell immune synapse. J Immunol. 2007;178:5606–5611. doi: 10.4049/jimmunol.178.9.5606. [DOI] [PubMed] [Google Scholar]
- Fassett MS, Davis DM, Valter MM, Cohen GB, Strominger JL. Signaling at the inhibitory natural killer cell immune synapse regulates lipid raft polarization but not class I MHC clustering. Proc Natl Acad Sci USA. 2001;98:14547–14552. doi: 10.1073/pnas.211563598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez NC, Treiner E, Vance RE, Jamieson AM, Lemieux S, Raulet DH. A subset of natural killer cells achieves self-tolerance without expressing inhibitory receptors specific for self-MHC molecules. Blood. 2005;105:4416–4423. doi: 10.1182/blood-2004-08-3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser S, Raulet DH. Activation and self-tolerance of natural killer cells. Immunol Rev. 2006;214:130–142. doi: 10.1111/j.1600-065X.2006.00460.x. [DOI] [PubMed] [Google Scholar]
- Guerra N, Michel F, Gati A, Gaudin C, Mishal Z, Escudier B, Acuto O, Chouaib S, Caignard A. Engagement of the inhibitory receptor CD158a interrupts TCR signaling, preventing dynamic membrane reorganization in CTL/tumor cell interaction. Blood. 2002;100:2874–2881. doi: 10.1182/blood-2002-02-0643. [DOI] [PubMed] [Google Scholar]
- Holcomb M, Rufini A, Barila D, Klemke RL. Deregulation of proteasome function induces Abl-mediated cell death by uncoupling p130CAS and c-CrkII. J Biol Chem. 2006;281:2430–2440. doi: 10.1074/jbc.M508454200. [DOI] [PubMed] [Google Scholar]
- Katagiri K, Maeda A, Shimonaka M, Kinashi T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat Immunol. 2003;4:741–748. doi: 10.1038/ni950. [DOI] [PubMed] [Google Scholar]
- Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song YJ, Yang LP, French AR, Sunwoo JB, Lemieux S, Hansen TH, Yokoyama WM. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature. 2005;436:709–713. doi: 10.1038/nature03847. [DOI] [PubMed] [Google Scholar]
- Lanier LL. NK cell receptors. Annu Rev Immunol. 1998;16:359–393. doi: 10.1146/annurev.immunol.16.1.359. [DOI] [PubMed] [Google Scholar]
- Lee N, Goodlett DR, Ishitani A, Marquardt H, Geraghty DE. HLA-E surface expression depends on binding of TAP-dependent peptides derived from certain HLA class I signal sequences. J Immunol. 1998;160:4951–4960. [PubMed] [Google Scholar]
- Liu YC. Ubiquitin ligases and the immune response. Annu Rev Immunol. 2004;22:81–127. doi: 10.1146/annurev.immunol.22.012703.104813. [DOI] [PubMed] [Google Scholar]
- Liu YC, Altman A. Cbl: Complex formation and functional implications. Cell Signal. 1998;10:377–385. doi: 10.1016/s0898-6568(97)00179-4. [DOI] [PubMed] [Google Scholar]
- Long EO. Negative signaling by inhibitory receptors: The NK cell paradigm. Immunol Rev. 2008;224:70–84. doi: 10.1111/j.1600-065X.2008.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long EO, Barber DF, Burshtyn DN, Faure M, Peterson M, Rajagopalan S, Renard V, Sandusky M, Stebbins CC, Wagtmann N, Watzl C. Inhibition of natural killer cell activation signals by killer cell immunoglobulin-like receptors (CD158) Immunol Rev. 2001;181:223–233. doi: 10.1034/j.1600-065x.2001.1810119.x. [DOI] [PubMed] [Google Scholar]
- Masilamani M, Nguyen C, Kabat J, Borrego F, Coligan JE. CD94/NKG2A inhibits NK cell activation by disrupting the actin network at the immunological synapse. J Immunol. 2006;177:3590–3596. doi: 10.4049/jimmunol.177.6.3590. [DOI] [PubMed] [Google Scholar]
- Naramura M, Jang IK, Kole H, Huang F, Haines D, Gu H. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat Immunol. 2002;3:1192–1199. doi: 10.1038/ni855. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006;27:195–201. doi: 10.1016/j.it.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Olcese L, Lang P, Vély F, Cambiaggi A, Marguet D, Bléry M, Hippen KL, Biassoni R, Moretta A, Moretta L, et al. Human and mouse killer-cell inhibitory receptors recruit PTP1C and PTPlD protein tyrosine phosphatases. J Immunol. 1996;156:4531–4534. [PubMed] [Google Scholar]
- Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- Parham P. MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol. 2005;5:201–214. doi: 10.1038/nri1570. [DOI] [PubMed] [Google Scholar]
- Reedquist KA, Bos JL. Costimulation through CD28 suppresses T cell receptor-dependent activation of the Ras-like small GTPase Rap1 in human T lymphocytes. J Biol Chem. 1998;273:4944–4949. doi: 10.1074/jbc.273.9.4944. [DOI] [PubMed] [Google Scholar]
- Robertson MJ, Cochran KJ, Cameron C, Le JM, Tantravahi R, Ritz J. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp Hematol. 1996;24:406–415. [PubMed] [Google Scholar]
- Scaife RM, Langdon WY. c-Cbl localizes to actin lamellae and regulates lamellipodia formation and cell morphology. J Cell Sci. 2000;113:215–226. doi: 10.1242/jcs.113.2.215. [DOI] [PubMed] [Google Scholar]
- Shishido T, Akagi T, Chalmers A, Maeda M, Terada T, Georgescu MM, Hanafusa H. Crk family adaptor proteins trans-activate c-Abl kinase. Genes Cells. 2001;6:431–440. doi: 10.1046/j.1365-2443.2001.00431.x. [DOI] [PubMed] [Google Scholar]
- Stebbins CC, Watzl C, Billadeau DD, Leibson PJ, Burshtyn DN, Long EO. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol Cell Biol. 2003;23:6291–6299. doi: 10.1128/MCB.23.17.6291-6299.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JR, Gonzalez FH, Kawai H, Yuan ZM. c-Abl interacts with the WAVE2 signaling complex to induce membrane ruffling and cell spreading. J Biol Chem. 2006;281:31290–31297. doi: 10.1074/jbc.M602389200. [DOI] [PubMed] [Google Scholar]
- Syken J, Grandpre T, Kanold PO, Shatz CJ. PirB restricts ocular-dominance plasticity in visual cortex. Science. 2006;313:1795–1800. doi: 10.1126/science.1128232. [DOI] [PubMed] [Google Scholar]
- Thien CB, Langdon WY. c-Cbl and Cbl-b ubiquitin ligases: Substrate diversity and the negative regulation of signalling responses. Biochem J. 2005;391:153–166. doi: 10.1042/BJ20050892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tybulewicz VL. Vav-family proteins in T-cell signalling. Curr Opin Immunol. 2005;17:267–274. doi: 10.1016/j.coi.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Watzl C, Long EO. Natural killer cell inhibitory receptors block actin cytoskeleton-dependent recruitment of 2B4 (CD244) to lipid rafts. J Exp Med. 2003;197:77–85. doi: 10.1084/jem.20020427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodring PJ, Hunter T, Wang JY. Regulation of F-actin-dependent processes by the Abl family of tyrosine kinases. J Cell Sci. 2003;116:2613–2626. doi: 10.1242/jcs.00622. [DOI] [PubMed] [Google Scholar]
- Yokoyama WM, Kim S. How do natural killer cells find self to achieve tolerance? Immunity. 2006;24:249–257. doi: 10.1016/j.immuni.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Zipfel PA, Zhang W, Quiroz M, Pendergast AM. Requirement for Abl kinases in T cell receptor signaling. Curr Biol. 2004;14:1222–1231. doi: 10.1016/j.cub.2004.07.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data include Supplemental Experimental Procedures and five figures and can be found with this article online at http://www.immunity.com/cgi/content/full/29/4/578/DC1/.