

Abstract
Protection of mitochondrial proteins from glycation by endogenous dicarbonyl compounds, methylglyoxal and glyoxal, was found recently to prevent increased formation of reactive oxygen species and oxidative and nitrosative damage to the proteome during aging and produce life extension in the nematode Caenorhabditis elegans. This suggests that dicarbonyl glycation damage to the mitochondrial proteome may be a preceding event to mitochondrial dysfunction leading to oxidative stress. Future research will address the functional charges in mitochondrial proteins that are the targets for dicarbonyl glycation.
Keywords: advanced glycation end-product (AGE), dicarbonyl, glycation, mitochondrion, oxidative stress
Dicarbonyl glycation of mitochondrial proteins as a cause of mitochondrial dysfunction and oxidative stress
In our recent research with collaborators, we found evidence of life extension of the nematode Caenorhabditis elegans with overexpression of the gene for glyoxalase 1 (Glo1) [1]. Silencing of Glo1 decreased lifespan. Glo1 is a further example of a ‘vitagene’, a gene which with manipulated change in expression produces life extension [2]. Glo1 is a glutathione-dependent enzyme that catalyses the metabolism of reactive dicarbonyl-glycating agents, MG (methylglyoxal) and glyoxal [3]. It thereby prevents the glycation of proteins by these dicarbonyl compounds. Proteins of mitochondria were found to be major targets of dicarbonyl glycation: increased glycation of mitochondrial proteins was associated with increased formation of ROS (reactive oxygen species) and increased proteome damage by oxidative and nitrosative processes. There was a decline of Glo1 expression in C. elegans and increased formation of mitochondrial ROS in normal aging. Overexpression of Glo1 in C. elegans decreased dicarbonyl glycation of mitochondrial proteins, decreased the formation of ROS and proteome markers of dicarbonyl glycation and also markers of oxidative and nitrosative damage (methionine sulfoxide and 3-nitrotyrosine residues respectively) with concomitant life extension. This indicated, for the first time, that dicarbonyl glycation may be the critical damage to the mitochondrial proteome that triggers increased ROS and oxidative damage in aging. It may therefore be important to prevent increased dicarbonyl glycation to achieve a decrease in formation of ROS by mitochondrial dysfunction. This has important implications for current understanding of ROS and oxidative damage in an attempt to achieve healthy aging and prevent vascular disease, particularly in diabetes and renal failure. An important future consideration is what are the likely targets of dicarbonyl glycation in mitochondria?
Protein glycation
Glycation is a major cause of spontaneous damage to cellular and extracellular proteins in physiological systems, affecting 0.1–0.2% of lysine and arginine residues [4,5]. For some proteins with limited protein turnover, such as lens fibre cells, the extent of protein glycation may be up to 10-fold higher [6]. Most mitochondrial proteins are short-lived, however, with turnovers from 10–30 min to 3–5 days [7]. Quality control of the mitochondrial proteome involves mitochondrial proteases, leading to release of peptides from mitochondria [8], ubiquitination and proteolysis of outer membrane, mitochondrial fission and fusion, and finally autophagy of damaged mitochondria (mitophagy) [9].
Glycation adducts are formed in non-enzymatic spontaneous reactions of monosaccharides with proteins. Glucose can enter mitochondria via GLUT1 (glucose transporter 1), which also transports vitamin C into mitochondria [10]. Probably the most important glycating agents to consider for glycation damage to the mitochondrial proteome are reactive dicarbonyls such as glyoxal, MG and 3DG (3-deoxyglucosone) (Figure 1). Dicarbonyls are formed endogenously by lipid peroxidation and the degradation of glycolytic intermediates and glycated proteins. Dicarbonyl compounds are potent glycating agents, 200–50 000-fold more reactive than glucose. The physiological concentrations of dicarbonyls are typically 10 000–50 000-fold lower than glucose, yet nevertheless dicarbonyl compounds remain important precursors of AGEs (advanced glycation end-products) in physiological systems. MG was suggested to have low permeability of mitochondrial membranes as it inhibited d-β-hydroxybutyrate dehydrogenase activity in inverted inner mitochondrial membrane vesicles better than in intact mitochondria [11], but this may have been be due to decreased inhibitory activity with intact mitochondria owing to binding to GSH of MG.
Figure 1. Major pathways for the formation of AGEs in physiological systems and precursor dicarbonyl metabolites.
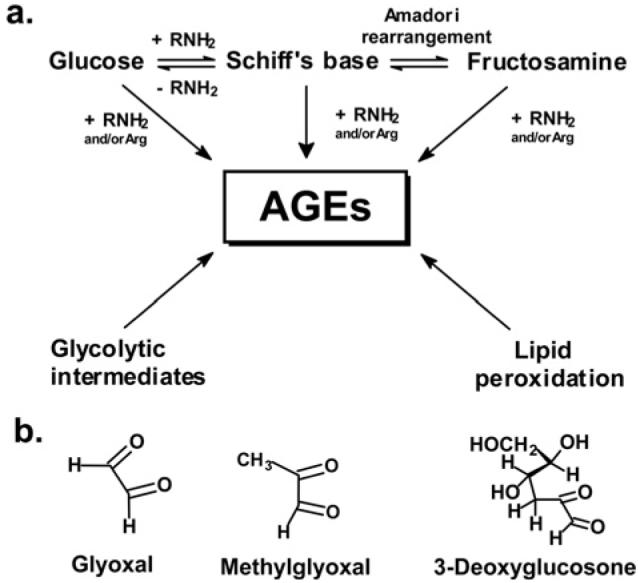
(a) Formation of early and advanced glycation adducts from glucose and glycolytic intermediates and products of lipid peroxidation. (b) Physiological reactive dicarbonyl-glycating agents.
Glycation by glucose and other monosaccharides is directed to amino groups of lysine and N-terminal amino acid residues of proteins, whereas glycation by dicarbonyl compounds is mainly, but not exclusively, directed to arginine residues [12].
Early-stage glycation adducts and AGEs
The initial glycation adducts formed by glycation by glucose and other monosaccharides are called early-stage glycation adducts. The predominant early glycation adduct is FL (Nε-fructosyl-lysine). Schiff base and FL adducts degrade slowly in further advanced reactions to form many different glycation adducts. These adducts are known collectively as AGEs. Dicarbonyls react with proteins directly to also form AGEs. Important AGEs quantitatively are hydroimidazolones derived from arginine residues modified by glyoxal, MG and 3DG: G-H1 [Nδ-(5-hydro-4-imidazolon-2-yl)ornithine], MG-H1 [Nδ-(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine] and 3DG-H {Nδ-[5-hydro-5-(2,3,4-trihydroxybutyl)-4-imidazolon-2-yl]ornithine and related structural isomers}. Other important and widely-studied AGEs are CML (Nε-carboxymethyl-lysine) and CEL (Nε-carboxyethyl-lysine), and the protein cross-links, pentosidine and glucosepane [5,13–16]. Further AGEs and related derivatives of emerging importance are CMC (Nε-carboxymethylcysteine) [17], CMA (Nε-carboxymethylarginine) [18] and ornithine [19], the latter formed as a degradation product of hydroimidazolones (Figure 2).
Figure 2. Protein glycation adduct residues.

(a) Early glycation adducts. (b) AGEs. For the corresponding free adducts at physiological pH, the N-terminal amino group is protonated −NH3+ and the C-terminal carbonyl is a carboxylate −CO2− moiety. DOLD, 3-deoxyglucosone-derived lysine dimer; GOLD, glyoxal-derived lysine dimer; MOLD, methylglyoxal-derived lysine dimer.
Enzymatic defence against glycation
Protein glycation in physiological systems is prevented and repaired by components of the enzymatic defence against glycation [20]. It involves enzymatic activities that suppress the formation of glycation adducts and repair sites of early glycation: Glo1 and AKRds (aldo–keto reductases) and aldehyde dehydrogenases detoxify reactive dicarbonyl-glycating agents [21,22], and amadoriase and fructosamine 3-phosphokinase that catalyse the removal of FL residues and free adducts [23-25] (Figure 3). The enzymatic defence against glycation suppresses damage to biological macromolecules, but it is an imperfect defence; glycation adducts of proteins, nucleotides and basic phospholipids are formed in normal physiological states. The enzymatic defence against glycation is overwhelmed in some disease states, and glycation adduct concentrations increase. The decline of expression of enzymes of the enzymatic defence against glycation may be key to increased protein glycation in the aging phenotype.
Figure 3. The enzymatic defence against glycation.
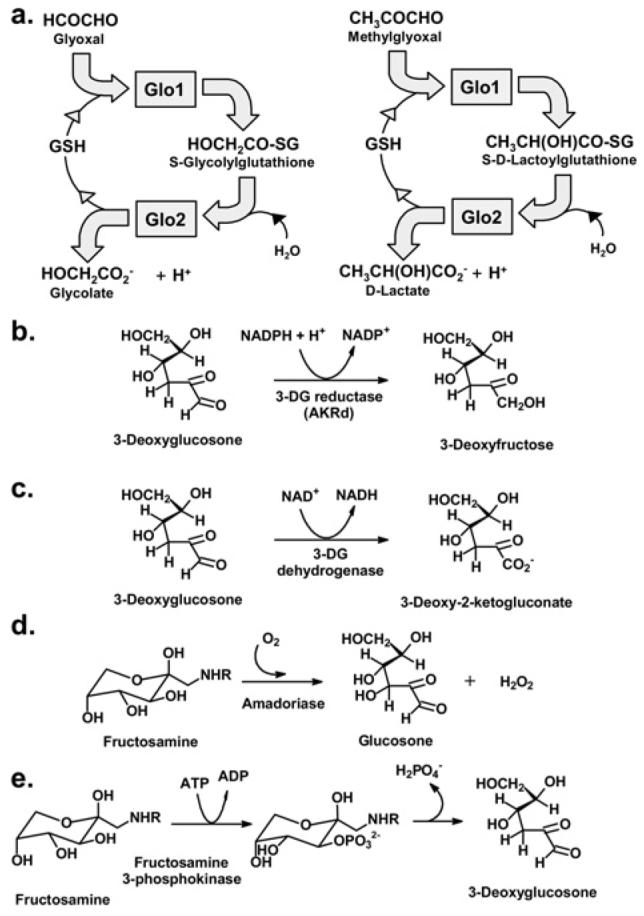
(a) Metabolism of glyoxal and MG by the glyoxalase system. (b) Metabolism of 3DG by 3-deoxyglucosone reductase, an AKRd. (c) Metabolism of 3DG by 3-deoxyglucosone dehydrogenase. (d, e) Repair of early glycated proteins by Amadoriase and fructosamine-3-phosphokinase.
Glycation of mitochondrial proteins
Proteins modified by AGE residues in mitochondria have been detected by immunoassay and by GC–MS. Immunoassay of AGEs suffers problems of incomplete characterization of antibody epitope recognition. For example, the 6D12 monoclonal antibody used to quantify the CML residues in rat liver mitochondrial protein [26,27] detects both CML and CEL residues with different affinities (the later with higher affinity than the former [28]) and cannot be used to quantify AGE residue content of proteins. Solutions of dried milk protein powder, a rich source of CML and other AGE residues [29], was also used to suppress non-specific antibody binding and hence probably produced interferences in these studies. Immunohistochemistry of tissues with 6D12 and anti-AGE antibodies 1F6, and 2A2 all showed mitochondrial immunoreactivity in human tissues [30].
Estimates of AGE residues of mitochondrial protein of improved analytical security came form the studies of Pamplona et al. [31] using stable-isotopic-dilution analysis/GC–MS. They found 1–2 mmol of CML/mol of lysine in rat heart mitochondrial protein [31]; CML residue content determined by 6D12 immunoassay was approx. 10-fold over estimates [26]. CEL residue content of mitochondrial protein was approx. 0.5 mmol/mol of lysine. Both CML and CEL residue content of mitochondrial protein decreased with caloric restriction [32]. Quantitative immunoblotting of AGE residue content of mitochondrial proteins has been performed with one of the best characterized monoclonal antibodies against AGE residues: monoclonal antibody 1H7G5 which recognizes MG-derived hydroimidazolone MG-H1 [33]. Quantitative immunoblotting with this antibody showed MG-H1 residues of mitochondrial protein in wild-type C. elegans and decreased MG-H1 residues of mitochondrial protein in transgenic C. elegans with overexpression of Glo1 [1]. Similar immunoblotting showed MG-H1 residue content of protein of mitochondria isolated from rat renal cortex. MG-H1 residue content of mitochondrial proteins was increased in diabetic rats [34].
The proteins modified by AGE residues have, as yet, only been detected by immunoreactivity with anti-AGE antibodies. This has not been corroborated by MS analysis of glycated peptides after limited proteolysis. It has been suggested that glutamate dehydrogenase is a target for AGE modification by immunoblotting with antibody 6D12 [35]. Several other mitochondrial proteins were found to be susceptible to modification by MG [34] (Table 1).
Table 1.
Mitochondrial proteins susceptible to modification by advanced glycation and thiol oxidation
| Mitochondrial proteins modified by AGEs | AGE | Mitochondrial proteins susceptible to thiol oxidation |
|---|---|---|
| Enoyl-CoA hydratase, mitochondrial | MG-H1 [34] | Enoyl-CoA hydratase, short chain 1, mitochondrial [39] |
| Complex III, core protein I | Complex III, core protein I | |
| NADH-ubiquinone oxidoreductase 30 kDa subunit, complex I | Acyl-CoA dehydrogenase, very long chain | |
| F1-ATPase, chain G | Carnitine acetyltransferase | |
| Electron flavoprotein β-subunit | Mitochondrial acyl-CoA thioesterase 2 | |
| Cytochrome c1, complex III | Mitochondrial trifunctional protein, α-subunit | |
| Glutamate dehydrogenase | CML/CEL [35] | Propionyl-CoA carboxylase, α-chain |
| Pyruvate dehydrogenase kinase, isoenzyme 2 |
Effect of physiological dicarbonyl compounds on mitochondrial function
Incubation of rat renal mitochondria with MG (10–200 μM) for only 5 min at 24 °C produced a concentration-dependent decrease in state 3 respiration, an increase and then a decrease in state 4 respiration and a decrease in respiratory control quotient [36]. With such a short incubation time, it is likely that these effects were due to cysteinyl thiol modifications [37]. Reactive surface thiols are present in proteins of complexes I, II and IV [38], and proteins susceptible to thiol oxidation have been identified [39] (Table 1). The cellular concentrations of MG is usually approx. 2–4 μM [40]. Approx. 25% of MG is expected to be bound reversibly to GSH and 50% to protein thiols (assuming cellular concentrations of GSH and protein thiols of 3 and 6 mM respectively [41] and a thiol/MG dissociation constant of 3 mM [3]). It is therefore unclear whether physiological concentrations of MG have an acute effect on mitochondrial respiration; any effect is likely to be a modest decrease of effect on state 3 respiration. Similarly, the acute effects of MG and glyoxal (200 μM–5 mM) on the mitochondrial transition pore protein are of unlikely physiological relevance [42]. The steady-state levels of MG-H1 residues of mitochondrial proteins, as found in C. elegans and rat renal cortical mitochondria [1,34], as MG-H1 is the most important AGE residue quantitatively in physiological systems [43] and is associated with functional impairment of modified proteins [40,44].
MG modification of the mitochondrial proteome
There are estimated to be approx. 1500 proteins in the mitochondrial proteome. Only 13 proteins involved in oxidative phosphorylation are encoded by the mitochondrial genome; the others are nuclear-encoded, synthesized in the cytosol and targeted to mitochondria [45]. Seven proteins have, to date, been suggested as susceptible targets of advanced glycation and AGE residue formation (Table 1). We do not yet understand the full consequences of increased modification of mitochondrial proteins by MG. Enoyl-CoA hydratase is involved in fatty acid β-oxidation. Core protein I of complex III was susceptible to S-carboxymethylation and modification inhibited electron-transport activity [46]. Other targets for dicarbonyl glycation are subunits of complex I and F1-ATPase. Mitochondrial electron flavoprotein β-subunit was a further subject for dicarbonyl glycation. Modification of a single arginine residue is known to impede electron transfer [47]. Cytochrome c1 is part of the cytochrome bc1 complex of complex III, a 30 kDa membrane-bound c-type cytochrome protein of mitochondria that functions as an electron donor to cytochrome c [48].
Summary
Dicarbonyl glycation of proteins and the glyoxalase system has been overlooked as an influential factor on oxidative stress. Life extension and decreased mitochondrial ROS with overexpression of Glo1 in C. elegans has suggested that non-oxidative dicarbonyl glycation may be a critical upstream event of mitochondrial dysfunction and oxidative stress. This has implications for current hypotheses of the cause of mitochondrial dysfunction and oxidative stress in vascular cells during hyperglycaemia in diabetes, linked to chronic vascular complications, and to mitochondrial dysfunction and oxidative stress in aging. Modification of mitochondrial proteins with associated functional impairment may have a critical role in the formation of mitochondrial ROS and also in the activation of the mitochondrial pathway of apoptosis in some circumstances. It is now important to characterize proteins that are susceptible to dicarbonyl glycation and functional impairment associated with the ‘dicarbonyl proteome’ of mitochondria.
Acknowledgments
We thank the BBSRC (Biotechnology and Biological Sciences Research Council) (U.K.), Wellcome Trust, British Heart Foundation, Diabetes UK, Cancer Research UK, British Council and European Community 6th Framework Programme for support for our dicarbonyl glycation-related research.
Abbreviations used
- AGE
advanced glycation end-product
- AKRd
aldo–keto reductase
- CEL
Nε-carboxyethyl-lysine
- CML
Nε-carboxymethyl-lysine
- 3DG
3-deoxyglucosone
- FL
Nε-fructosyl-lysine
- Glo1
glyoxalase 1
- MG
methylglyoxal
- MG-H1
MG-derived hydroimidazolone, Nδ-(5-hydro-5-methyl-4-imidazolon-2-yl)ornithine
- ROS
reactive oxygen species
References
- 1.Morcos M, Du X, Pfisterer F, Hutter H, Sayed AAR, Thornalley P, Ahmed N, Baynes J, Thorpe S, Kukudov G, et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell. 2008;7:260–269. doi: 10.1111/j.1474-9726.2008.00371.x. [DOI] [PubMed] [Google Scholar]
- 2.Calabrese V, Guagliano E, Sapienza M, Panebianco M, Calafato S, Puleo E, Pennisi G, Mancuso C, Butterfield DA, Stella AG. Redox regulation of cellular stress response in aging and neurodegenerative disorders: role of vitagenes. Neurochem. Res. 2007;32:757–773. doi: 10.1007/s11064-006-9203-y. [DOI] [PubMed] [Google Scholar]
- 3.Thornalley PJ. The glyoxalase system in health and disease. Mol. Aspects Med. 1993;14:287–371. doi: 10.1016/0098-2997(93)90002-u. [DOI] [PubMed] [Google Scholar]
- 4.Thornalley PJ. The clinical significance of glycation. Clin. Lab. 1999;45:263–273. [Google Scholar]
- 5.Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R, Dawnay A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 2003;375:581–592. doi: 10.1042/BJ20030763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmed N, Thornalley PJ, Dawczynski J, Franke S, Strobel J, Stein G, Haik GM. Methylglyoxal-derived hydroimidazolone advanced glycation end-products of human lens proteins. Invest. Ophthalmol. Visual Sci. 2003;44:5287–5292. doi: 10.1167/iovs.03-0573. [DOI] [PubMed] [Google Scholar]
- 7.Bota DA, Davies KJA. Protein degradation in mitochondria: implications for oxidative stress, aging and disease: a novel etiological classification of mitochondrial proteolytic disorders. Mitochondrion. 2001;1:33–49. doi: 10.1016/s1567-7249(01)00005-8. [DOI] [PubMed] [Google Scholar]
- 8.Augustin S, Nolden M, Muller S, Hardt O, Arnold I, Langer T. Characterization of peptides released from mitochondria: evidence for constant proteolysis and peptide efflux. J. Biol. Chem. 2005;280:2691–2699. doi: 10.1074/jbc.M410609200. [DOI] [PubMed] [Google Scholar]
- 9.Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306–314. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.KC S, Cárcamo JM, Golde DW. Quality control of mitochondria: protection against neurodegeneration and ageing. FASEB J. 2005;19:1657–1667. doi: 10.1096/fj.05-4107com. [DOI] [PubMed] [Google Scholar]
- 11.Latruffe N, Elkebbaj MS, Moussard C, Gaudemer Y. Permeability of inner mitochondrial membrane to arginine reagents. FEBS Lett. 1982;144:273–278. doi: 10.1016/0014-5793(82)80653-4. [DOI] [PubMed] [Google Scholar]
- 12.Thornalley PJ. Dicarbonyl intermediates in the maillard reaction. Ann. N.Y. Acad. Sci. 2005;1043:111–117. doi: 10.1196/annals.1333.014. [DOI] [PubMed] [Google Scholar]
- 13.Ahmed N, Argirov OK, Minhas HS, Cordeiro CA, Thornalley PJ. Assay of advanced glycation endproducts (AGEs): surveying AGEs by chromatographic assay with derivatization by 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate and application to Nε-carboxymethyllysine- and Nε-(1-carboxyethyl)lysine-modified albumin. Biochem. J. 2002;364:1–14. doi: 10.1042/bj3640001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thorpe SR, Baynes JW. CML: a brief history. Int. Congr. Ser. 2002;1245:91–99. [Google Scholar]
- 15.Sell DR, Monnier VM. Structure elucidation of a senescence cross-link from human extracellular matrix: implication of pentoses in the aging process. J. Biol. Chem. 1989;264:21597–21602. [PubMed] [Google Scholar]
- 16.Biemel KM, Friedl DA, Lederer MO. Identification and quantification of major Maillard cross-links in human serum albumin and lens protein: evidence for glucosepane as the dominant compound. J. Biol. Chem. 2002;277:24907–24915. doi: 10.1074/jbc.M202681200. [DOI] [PubMed] [Google Scholar]
- 17.Zeng J, Davies MJ. Evidence for the formation of adducts and S-(carboxymethyl)cysteine on reaction of α-dicarbonyl compounds with thiol groups on amino acids, peptides, and proteins. Chem. Res. Toxicol. 2005;18:1232–1241. doi: 10.1021/tx050074u. [DOI] [PubMed] [Google Scholar]
- 18.Odani H, Iijima K, Nakata M, Miyata S, Kusunoki H, Yasuda Y, Hiki Y, Irie S, Maeda K, Fujimoto D. Identification of Nω -carboxymethylarginine, a new advanced glycation endproduct in serum proteins of diabetic patients: possibility of a new marker of aging and diabetes. Biochem. Biophys. Res. Commun. 2001;285:1232–1236. doi: 10.1006/bbrc.2001.5322. [DOI] [PubMed] [Google Scholar]
- 19.Sell DR, Monnier VM. Conversion of arginine into ornithine by advanced glycation in senescent human collagen and lens crystallins. J. Biol. Chem. 2004;279:54173–54184. doi: 10.1074/jbc.M408946200. [DOI] [PubMed] [Google Scholar]
- 20.Thornalley PJ. Glyoxalase I – structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003;31:1343–1348. doi: 10.1042/bst0311343. [DOI] [PubMed] [Google Scholar]
- 21.Shinohara M, Thornalley PJ, Giardino I, Beisswenger PJ, Thorpe SR, Onorato J, Brownlee M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Invest. 1998;101:1142–1147. doi: 10.1172/JCI119885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki K, Koh YH, Mizuno H, Hamaoko R, Taniguchi N. Overexpression of aldehyde reductase protects PC12 cells from the cytotoxicity of methylglyoxal or 3-deoxyglucosone. J. Biochem. (Tokyo) 1998;123:353–357. doi: 10.1093/oxfordjournals.jbchem.a021944. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi M, Pischetsrieder M, Monnier VM. Isolation, purification, and characterization of amadoriase isoenzymes (fructosyl amine-oxygen oxidoreductase EC 1.5.3) from Aspergillus sp. J. Biol. Chem. 1997;272:3437–3443. doi: 10.1074/jbc.272.6.3437. [DOI] [PubMed] [Google Scholar]
- 24.Delpierre G, Rider MH, Collard F, Stroobant V, Vanstapel F, Santos H, Van Schaftingen E. Identification, cloning, and heterologous expression of a mammalian fructosamine-3-kinase. Diabetes. 2000;49:1627–1634. doi: 10.2337/diabetes.49.10.1627. [DOI] [PubMed] [Google Scholar]
- 25.Szwergold BS, Howell S, Beisswenger PJ. Human fructosamine-3-kinase: purification, sequencing, substrate specificity, and evidence of activity in vivo. Diabetes. 2001;50:2139–2147. doi: 10.2337/diabetes.50.9.2139. [DOI] [PubMed] [Google Scholar]
- 26.Bakala H, Delaval E, Hamelin M, Bismuth J, Borot-Laloi C, Corman B, Friguet B. Changes in rat liver mitochondria with aging: Lon protease-like reactivity and Nε-carboxymethyllysine accumulation in the matrix. Eur. J. Biochem. 2003;270:2295–2302. doi: 10.1046/j.1432-1033.2003.03598.x. [DOI] [PubMed] [Google Scholar]
- 27.Alikhani ZB, Alikhani M, Boyd CM, Nagao K, Trackman PC, Graves DT. Advanced glycation end products enhance expression of pro-apoptotic genes and stimulate fibroblast apoptosis through cytoplasmic and mitochondrial pathways. J. Biol. Chem. 2005;280:12087–12095. doi: 10.1074/jbc.M406313200. [DOI] [PubMed] [Google Scholar]
- 28.Koito W, Araki T, Horiuchi S, Nagai R. Conventional antibody against Nε-(carboxymethyl)lysine (CML) shows cross-reaction to Nε-(carboxyethyl)lysine (CEL): immunochemical quantification of CML with a specific antibody. J. Biochem. (Tokyo) 2004;136:831–837. doi: 10.1093/jb/mvh193. [DOI] [PubMed] [Google Scholar]
- 29.Drusch S, Faist V, Erbersdobler H. Determination of Nε-carboxymethyllysine in milk products by a modified reversed-phase HPLC method. Food Chem. 1999;65:547–553. [Google Scholar]
- 30.Ling X, Sakashita N, Takeya M, Nagai R, Horiuchi S, Takahashi K. Immunohistochemical distribution and subcellular localization of three distinct specific molecular structures of advanced glycation end products in human tissues. Lab. Invest. 1998;78:1591–1606. [PubMed] [Google Scholar]
- 31.Pamplona R, Portero-Otin M, Bellmunt MJ, Gredilla R, Barja G. Aging increases Nε-(carboxymethyl)lysine and caloric restriction decreases Nε-(carboxyethyl)lysine and Nε-(malondialdehyde)lysine in rat heart mitochondrial proteins. Free Radical Res. 2002;36:47–54. doi: 10.1080/10715760210165. [DOI] [PubMed] [Google Scholar]
- 32.Pamplona R, Portero-Otin M, Requena J, Gredilla R, Barja G. Oxidative, glycoxidative and lipoxidative damage to rat heart mitochondrial proteins is lower after 4 months of caloric restriction than in age-matched controls. Mech. Ageing Dev. 2002;123:1437–1446. doi: 10.1016/s0047-6374(02)00076-3. [DOI] [PubMed] [Google Scholar]
- 33.Giardino I, Thornalley PJ, Edelstein D, Brownlee M. Generation and characterisation of an antibody against AGEs that induce endothelial dysfunction. Diabetes. 1998;47:A123. [Google Scholar]
- 34.Rosca MG, Mustata TG, Kinter MT, Ozdemir AM, Kern TS, Szweda LI, Brownlee M, Monnier VM, Weiss MF. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am. J. Physiol. Renal Physiol. 2005;289:F420–F430. doi: 10.1152/ajprenal.00415.2004. [DOI] [PubMed] [Google Scholar]
- 35.Hamelin M, Mary J, Vostry M, Friguet B, Bakala H. Glycation damage targets glutamate dehydrogenase in the rat liver mitochondrial matrix during aging. Eur. J. Biochem. 2007;274:5949–5961. doi: 10.1111/j.1742-4658.2007.06118.x. [DOI] [PubMed] [Google Scholar]
- 36.Rosca MG, Monnier VM, Szweda LI, Weiss MF. Alterations in renal mitochondrial respiration in response to the reactive oxoaldehyde methylglyoxal. Am. J. Physiol. Renal Physiol. 2002;283:F52–F59. doi: 10.1152/ajprenal.00302.2001. [DOI] [PubMed] [Google Scholar]
- 37.Lo TWC, Westwood ME, McLellan AC, Selwood T, Thornalley PJ. Binding and modification of proteins by methylglyoxal under physiological conditions: a kinetic and mechanistic study with Nα-acetylarginine, Nα-acetylcysteine, and Nα-acetyllysine, and bovine serum albumin. J. Biol. Chem. 1994;269:32299–32305. [PubMed] [Google Scholar]
- 38.Lin TK, Hughes G, Muratovska A, Blaikie FH, Brookes PS, Darley-Usmar V, Smith RAJ, Murphy MP. Specific modification of mitochondrial protein thiols in response to oxidative stress: a proteomics approach. J. Biol. Chem. 2002;277:17048–17056. doi: 10.1074/jbc.M110797200. [DOI] [PubMed] [Google Scholar]
- 39.Hurd TR, Prime TA, Harbour ME, Lilley KS, Murphy MP. Detection of reactive oxygen species-sensitive thiol proteins by redox difference gel electrophoresis: implications for mitochondrial redox signaling. J. Biol. Chem. 2007;282:22040–22051. doi: 10.1074/jbc.M703591200. [DOI] [PubMed] [Google Scholar]
- 40.Dobler D, Ahmed N, Song LJ, Eboigbodin KE, Thornalley PJ. Increased dicarbonyl metabolism in endothelial cells in hyperglycemia induces anoikis and impairs angiogenesis by RGD and GFOGER motif modification. Diabetes. 2006;55:1961–1969. doi: 10.2337/db05-1634. [DOI] [PubMed] [Google Scholar]
- 41.Mistry P, Merazga Y, Spargo DJ, Riley PA, McBrien DCH. The effects of cisplatin on the concentration of protein thiols and glutathione in the rat kidney. Cancer Chemother. Pharmacol. 1991;28:277–282. doi: 10.1007/BF00685535. [DOI] [PubMed] [Google Scholar]
- 42.Speer O, Morkunaite-Haimi S, Liobikas J, Franck M, Hensbo L, Linder MD, Kinnunen PKJ, Wallimann T, Eriksson O. Rapid suppression of mitochondrial permeability transition by methylglyoxal: role of reversible arginine modification. J. Biol. Chem. 2003;278:34757–34763. doi: 10.1074/jbc.M301990200. [DOI] [PubMed] [Google Scholar]
- 43.Thornalley PJ. Quantitative screening of protein glycation, oxidation, and nitration adducts by LC-MS/MS: protein damage in diabetes, uremia, cirrhosis, and Alzheimer's disease. In: Dalle-Donne I, Scaloni A, Butterfield DA, editors. Redox Proteomics. Wiley; Hoboken: 2006. pp. 681–728. [Google Scholar]
- 44.Ahmed N, Dobler D, Dean M, Thornalley PJ. Peptide mapping identifies hotspot site of modification in human serum albumin by methylglyoxal involved in ligand binding and esterase activity. J. Biol. Chem. 2005;280:5724–5732. doi: 10.1074/jbc.M410973200. [DOI] [PubMed] [Google Scholar]
- 45.Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, Wiley S, Murphy AN, Gaucher SP, Capaldi RA, Gibson BW, Ghosh SS. Characterization of the human heart mitochondrial proteome. Nat. Biotech. 2003;21:281–286. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- 46.Gellerfors P, Lunden M, Nelson BD. Evidence for a function of core protein in complex III from beef-heart mitochondria. Eur. J. Biochem. 1976;67:463–468. doi: 10.1111/j.1432-1033.1976.tb10711.x. [DOI] [PubMed] [Google Scholar]
- 47.Parker AR. A single arginine residue is required for the interaction of the electron transferring flavoprotein (ETF) with three of its dehydrogenase partners. Mol. Cell. Biochem. 2003;254:91–100. doi: 10.1023/a:1027349303797. [DOI] [PubMed] [Google Scholar]
- 48.Xia D, Yu CA, Kim H, Xian JZ, Kachurin AM, Zhang L, Yu L, Deisenhofer J. Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria. Science. 1997;277:60–66. doi: 10.1126/science.277.5322.60. [DOI] [PMC free article] [PubMed] [Google Scholar]