

Synopsis
Insulin resistance characterizes type 2 diabetes and the metabolic syndrome, disorders associated with an increased risk of death due to macrovascular disease. In the past few decades, research from both the basic science and clinical arenas has enabled evidenced-based use of therapeutic modalities such as statins and angiotensin converting enzyme inhibitors to reduce cardiovascular (CV) mortality in insulin resistant patients. Recently, promising drugs such as the thiazolidinediones have come under scrutiny for possible deleterious CV effects. Ongoing research has broadened our understanding of the pathophysiology of atherosclerosis, implicating detrimental effects of inflammation and the cellular stress response on the vasculature. In this review, we address current thinking that is shaping our molecular understanding of insulin resistance and atherosclerosis.
Keywords: insulin resistance, metabolic syndrome, atherosclerosis, cardiovascular disease, inflammation, cellular stress
Introduction
The term “insulin resistance” has existed in the medical vernacular nearly as long as the clinical use of insulin itself. In fact, the first report of a patient resistant to the effects of insulin was published in 1924, only two years after 14-year old Leonard Thompson became the first human to be successfully treated with insulin 1, 2. Although the early medical literature is replete with such examples, the etiology of this resistance was imprecise purification, varied absorption, and immune-mediated clearance of impure/canine insulin. In the modern era, insulin resistance most commonly denotes a condition in which there is an insufficient peripheral (e.g. muscle, liver, and adipose) tissue response to a given quantity of insulin. The progression of such a subnormal response is usually insidious, with affected individuals living subclinically for years with glucose levels nearly normal due to hypersecretion of insulin. In the subset of individuals with both chronic insulin resistance and pancreatic beta cell failure, glucose levels become sufficiently elevated to fulfill the diagnostic criteria for type 2 diabetes mellitus, the end-stage of the insulin resistance spectrum.
Although impaired glucose tolerance and hyperglycemia are classical manifestations of insulin resistance and type 2 diabetes, many patients present with several associated signs and laboratory abnormalities (i.e. abdominal obesity, elevated blood pressure, elevated triglycerides, and low HDL cholesterol). Insulin resistance is thought to play a role in the association of these metabolic phenotypes; indeed, such clustering of phenotypes has been termed the insulin resistance syndrome or the metabolic syndrome. The National Cholesterol Education Program’s Adult Treatment Panel III (ATPIII) and the World Health Organization (WHO) have rigorously defined the components of the metabolic syndrome in recently published/updated criteria 3–5.
One of the most devastating complications of long-standing insulin resistance and its associated metabolic derangements is progressive macrovascular pathology (namely atherosclerosis). Atherosclerosis remains the major cause of morbidity and mortality among diabetic patients. Diabetics have higher rates of coronary artery disease (CAD) and myocardial infarctions (MIs) than non-diabetic patients with similar risk factor profiles 6–8. Diabetics without a prior MI have a similar mortality risk to non-diabetics with a prior MI 6, 9. Revealing results from such large studies prompted the most recent ATPIII guidelines to designate diabetes as a coronary disease equivalent 3.
Although hyperglycemia is a hallmark of diabetes, it is not clear that elevated glucose levels alone contribute in a major way to cardiovascular disease (CVD) progression. Indeed, diabetic patients without the metabolic syndrome have less CVD than non-diabetics with the metabolic syndrome 10. It is becoming increasingly clear that metabolic derangements associated with insulin resistance are important contributors to atherogenesis (Figure 1). Regardless of which criteria are used to define this syndrome, afflicted patients are at approximately 2-to-4-fold increased risk for CVD mortality 11. Furthermore, there is a graded risk depending on the number of risk factors present 12.
Figure 1. The Central Role of Insulin Resistance in Vascular Disease.
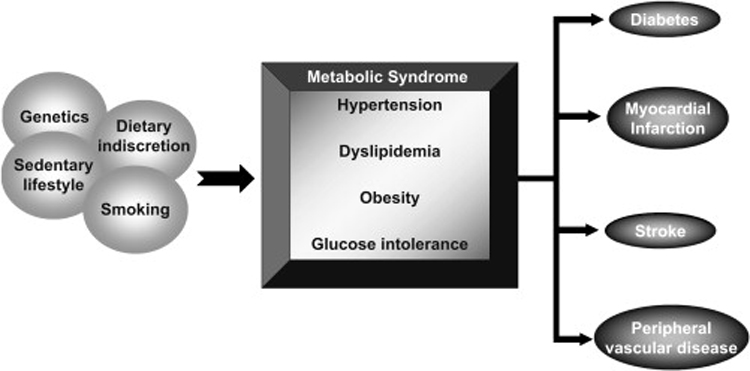
Multiple environmental and genetic factors contribute to the formation of the insulin resistance phenotype (aka the “metabolic syndrome”), marked by hypertension, dyslipidemia, obesity, and glucose intolerance. These risk factors in turn contribute to initiation and progression of type 2 diabetes and the macrovascular diseases (i.e. myocardial infarctions, strokes, and peripheral vascular disease).
The wealth of evidence in the past few decades points to a striking relation between insulin resistance and atherosclerosis. With this background, the goal of this review is three-fold: (1) to provide an overview of the currently utilized treatment strategies to reduce CVD mortality in insulin resistant patients, (2) to establish a framework for further defining the role of insulin resistance in vascular disease, and (3) to summarize preclinical data that may lead to future therapeutics.
Prevention of CVD in Insulin Resistance (Current Approaches)
Current primary prevention treatment modalities in patients with the metabolic syndrome and/or diabetes rely on risk factor modification (i.e. reduction of hyperlipidemia, hypertension, obesity, and lifestyle changes). Although beyond the focus of this review, a few treatment options are especially worthy of mention. Beginning in the 1990s, large clinical trials demonstrated the enormous clinical utility of two classes of drugs, statins and angiotensin-converting enzyme inhibitors (ACEi) /angiotensin receptor blockers (ARB), in diminishing the cardiovascular morbidity and mortality of insulin resistant/diabetic patients.
The first large secondary prevention (i.e. patients with a prior MI) trial to demonstrate the CV benefits of statins (in this case simvastatin) was the 4S trial 13. This was followed by WOSCOPS, the first large primary prevention trial to demonstrate statin benefits in patients with no known CVD but including many with metabolic profiles resembling incipient insulin resistance 14. These findings have been corroborated by numerous other trials showing the utility of statins as a class in different patient subgroups. One of the most surprising statin trials was the Heart Protection Study, a comparison of simvastatin versus placebo in a cohort of approximately 20,000 patients with known CVD or DM 15. In addition to showing similar CV mortality benefits as prior studies, this trial also clearly demonstrated that patients with the lowest category of LDL cholesterol levels (<116 mg/dl) derived benefits from statin therapy that was comparable to benefits in statin-treated hypercholesterolemic patients. In this regard, there is accumulating evidence that statins are more than simply cholesterol-lowering agents, having beneficial effects on endothelial dysfunction, inflammation, and the coagulation cascade as well as platelet function (reviewed in 16).
It has been known for some time that inhibition of the renin-angiotensin system is critical for reducing CV mortality and improving cardiac function in patients with left ventricular dysfunction/ischemic cardiomyopathy 17, 18. What remained unclear was whether ACEi and ARBs would be effective in patients with high risk features (including known vascular disease and diabetes) without overt heart failure. The HOPE and LIFE trials were two large randomized studies designed to address the effectiveness of the ACEi ramipril and the ARB losartan, respectively, in such patients 19, 20. The significant decrease in CV mortality observed with treatment in these trials has elevated this class of drugs to first-line agents in the treatment of hypertension in diabetics, even in the absence of heart failure.
Is Modulation of Insulin Resistance Beneficial for CVD Prevention?
As described above, the classical approach to the treatment of insulin resistance and diabetes is risk-factor modification (i.e. directed treatment of the most skewed parameters of the disorders). Despite clear successes with this approach, it represents a reaction to long-standing metabolic derangements rather than treatment for a potentially unifying process, such as insulin resistance. Thiazolidinediones are a class of drugs known to improve insulin resistance that were found to act by binding to Peroxisome-Proliferator-Activated Receptor gamma (PPARγ), a discovery providing a new mechanistic approach to diabetes treatment 21. PPARs are ligand-activated nuclear receptors of which PPARγ is one subtype. As PPARγ is highly expressed in adipose tissue where it is involved in lipid homeostasis (especially fatty acid uptake/storage), it stood to reason its activation might enhance insulin-sensitivity by directing toxic free fatty away from other insulin-dependent organs and into adipose tissue. This hypothesis was supported by some studies in humans focusing on glucose metabolism. Although trogilitazone, the first thiazolidinedione to be approved for clinical use, was withdrawn due to hepatotoxicity, the continued success of other members of this class of drugs, rosiglitazone and pioglitazone, was heralded by some as a revolution in diabetes and insulin resistance management 22.
Although not supported by disease-related outcome data, the use of thiazolidinediones in diabetic subjects was expected to not only improve surrogate end-points such as glycemic control but also end-points such as macrovascular events. The utility of these agents came under scrutiny when a meta-analysis of rosiglitazone trials showed increased CV mortality 23. Although pioglitazone faired better in the 5000+-patient PROACTIVE trial (non-significant reduction in CV events) 24 and a followup meta-analysis (a statistically significant decrease in death, MI, and strokes) 25, concerns over the lack of prospective outcomes data with thiazolidinediones and the known increased risk of volume overload/heart failure with these drugs 26 have raised doubts about this class of therapeutic agents.
Despite the current controversy about thiazolidinediones, an understanding of the molecular underpinnings driving insulin resistance and their relation to atherosclerosis is pivotal in the development of rational drug design. There are several aspects of insulin resistance that are actively being pursued in hopes of better understanding atherosclerotic events.
Insulin Resistance and Proatherogenic Lipids
Despite the traditional focus on LDL and CVD risk, a portion of the connection between blunted insulin signaling, abnormal lipid metabolism, and atherosclerosis appears to mediated by aberrations in triglyceride/VLDL and HDL levels instead of LDL 27. Derangements in adipocyte and hepatocyte function play a central role in these abnormalities 28, 29.
Insulin resistance reduces the ability of adipose tissue to clear/store circulating lipids, in part because of reduced lipoprotein lipase enzyme activity. Abnormal adipocyte insulin signaling also results in inappropriate lipolysis even during times of nutrient excess. The result is paradoxical elevations in serum triglycerides as well as circulating free fatty acids (FFA). In conjunction with elevations in apolipoprotein B (apoB) (thought to be due to posttranslational stabilization of the protein) and enhanced lipogenesis by the liver, increases in apoB-containing/ triglyceride-rich lipids (primarily present as VLDL particles) are a hallmark of patients with insulin resistance 30, 31.
Several trials have demonstrated the CV risks associated with the hypertriglyceridemia of insulin resistance. Hypertriglyceridemia has the strongest correlation with CVD among the five components of the metabolic syndrome 32. Although the significance of fasting triglycerides (as an independent factor) with respect to CV events has been a matter of debate 27, elevated nonfasting triglycerides, a state classically associated with insulin resistance as described above, significantly elevates CV mortality risk 33, 34.
Insulin resistance can also have adverse consequences on LDL and HDL metabolism. Although elevations in LDL cholesterol levels are not a hallmark of insulin resistance/diabetes, the composition and possibly proatherogenic function of LDL is affected. The hypertriglyceridemia/VLDL excess brought on by insulin resistance expedites cholesteryl ester transfer protein (CETP)-mediated exchange of LDL cholesteryl ester for VLDL triglyceride. In turn, the newly acquired LDL triglyceride undergoes lipolysis by hepatic and lipoprotein lipase, resulting in LDL particles that are smaller in size, more dense, and depleted of their usual cholesteryl ester content 35, 36. A similar mechanism involving CETP-mediated exchange of lipids also occurs between VLDL and HDL; however, in addition to the eventual creation of small/dense particles, HDL clearance is also enhanced with the end result being low levels of dysfunctional HDL particles 37, 38.
The Role of Insulin Resistance in Inflammatory Signaling and Atherosclerosis
An obvious connection between insulin resistance and atherosclerosis is derived from observations that obesity and insulin resistance often occur in concert with significant increases in inflammatory mediators 39, 40. Atherosclerosis acts in many ways like an inflammatory condition with prominent cellular infiltration and robust cytokine expression 41. One of the first links between obesity, insulin resistance, and inflammation was the demonstration that mouse adipose tissue can produce TNFα, its production is proportional to the degree of obesity, and neutralization of the TNF-receptor can significantly decreased obesity-induced insulin resistance 42. Numerous subsequent studies have debunked the initial view of the adipocyte as a stagnant lipid storage entity; adipose tissue is now known to be an active secretory organ, dynamically producing a variety of pro-inflammatory mediators such as TNFα, IL-1, IL-6, MCP-1, and PAI-1, factors traditionally associated with inflammatory cells 43. Similar to TNFα the production of these “adipokines” is a function of the health of the tissue and under conditions of nutrient excess, obesity, and progressive insulin resistance, there is substantial proinflammatory adipokine production 44–48. A substantial body of work has shown that these inflammatory mediators perpetuate the insulin resistance phenotype and result in deleterious effects on the vasculature (reviewed in 43). A detailed review of the effects of proatherogenic signaling including the role of protein kinase C (PKC) has recently appeared 49, but two of the major pathways likely to be clinically relevant to vascular disease in insulin resistance involve the Nuclear Factor kappa B (NF-κB) and c-Jun N-terminal kinase (JNK) cascades.
NF-κB Signaling and Atherogenesis
A central mediator of inflammatory signaling in the vasculature of an insulin resistance individual is the NF-κB family of nuclear transcription factors (Figure 2). The classical and most prominent member of this family is a heterodimer of the p65/RelA and p50 proteins. In the cytoplasm, this dimer is bound to IκB proteins in an inactive state 50. The arrival of extracellular signals (including several of the cytokines described above including TNFα and IL-1), stimulates the membrane-associated IκB kinase (IKK) to serine-phosphorylate IκB, thus facilitating its proteosomal degradation and liberating NF-κB to undergo nuclear translocation and set into motion a potent feed-forward production of pro-inflammatory transcripts 51, 52. A global survey of such NF-κB targets, as has been performed by both individual target gene assessment and more broad expression profiling approaches, reveals striking proatherogenic features. Broad categories include mononuclear cell chemoattractants, vascular adhesion molecules, mediators of chemotaxis, inducers of the monocyte to macrophage differentiation program, stimulators of smooth muscle cell proliferation, angiogenic factors, and amplification of proteases involved in the breakdown of the intercellular matrix, spanning the entire gamut of mediators required for plaque formation, progression, and destabilization 53.
Figure 2. Critical Proatherogenic Signaling Events at the Vasculature in the Setting of Insulin Resistance.
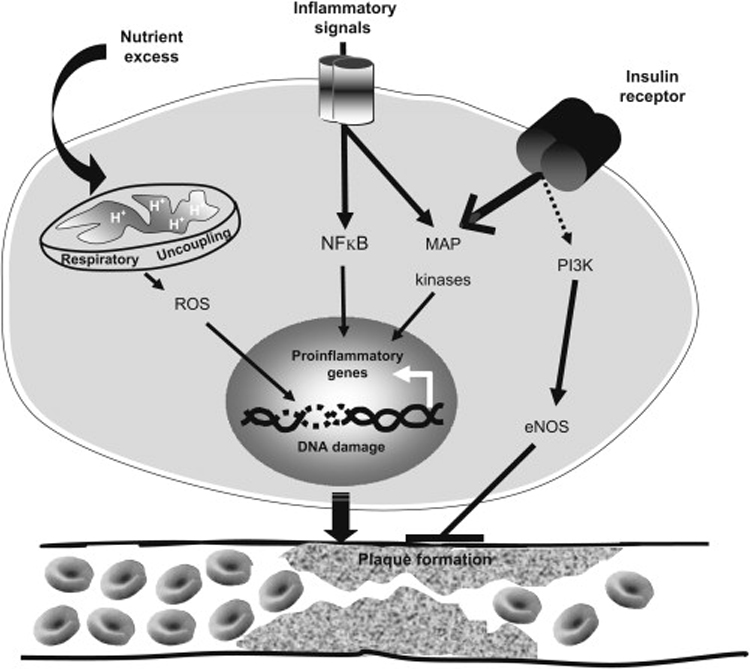
The secretion of proinflammatory mediators initiated by systemic insulin resistance stimulate several intracellular signaling cascades, of which NFκB and JNK (a member of the MAP kinase family) are prominent examples; these potent inducers of proinflammatory genes create a feedforward cycle of signaling that provokes several aspects of atherogenesis. In addition, insulin resistance also selectively interferes with PI-3-kinase-mediated insulin receptor signaling, in turn suppressing eNOS activity in favor of the proproliferative/proatherogenic MAP kinases. Finally, states of nutrient excess dysregulate mitochondrial energy balance thus promoting respiratory uncoupling and the generation of reactive oxygen species (ROS); this can wreak havoc on the integrity of the genome and blunt vascular defenses against atherogenesis.
Although the potential for cytokine-mediated vascular damage is evident, attempts at studying the NF-κB pathway with mouse models have produced contradictory results. For example, genetic targeting of p55, one of two TNFα receptors and the predominant mediator of inflammation actually protects mice against atherosclerosis 54. Furthermore, a partial macrophage-specific deletion of IKK2 leading to ~50% reduction in NF-κB activation increases the extent and the complexity of atherosclerotic lesions in mice 55 while bone marrow reconstitution experiments with NF-κB1-null macrophages in mice fed a high fat diet leads to decreased atheromatous plaque formation but an increased cellular infiltration 56. These confounding results point to the tremendous complexity of the inflammatory response and our current inability to tease out such nuances with available technologies.
JNK signaling and Atherosclerosis
JNK is a member of the mitogen-activated protein (MAP) kinase family (which also includes extracellular signal-related kinase (ERK1/2), big MAP kinase (BMK1), and p38 MAP kinase) (reviewed in 57). Although these serine/threonine kinases are responsive to a variety of stimuli, JNK signaling is potently activated by mediators of inflammatory and stress responses including cytokines and environmental stresses. Upon activation, JNK can phosphorylate a host of transcription factors, including the one for which it is named (c-Jun component of the AP-1 transcription factor) thereby triggering robust gene expression. Similar to NF-κB signaling, cytokine-induced JNK signaling fuels a feed-forward cycle of proinflammatory/proatherogenic factor production including TNFα, IL-2, matrix metalloproteinases, and adhesion molecules 58–60.
Initial data suggesting a role for JNK in atherosclerosis were based on observations that the macrophages and smooth muscle cells of human and animal model atheromatous plaques had prominently activated JNK signaling 61, 62. Further evidence was provided by Ricci et al. by creating mice with macrophage-specific ablation of two of the JNK family members, JNK1 and JNK2, in an ApoE-null background 63. Interestingly, only the JNK2(−/−) mice had significantly reduced atherosclerotic burden; this was due to defective foam cell formation proposed to occur via reduced uptake of modified LDL.
Direct Effects of Insulin Resistance on the Vasculature and Atherogenesis
One of the early events in the pathophysiology of atherosclerosis appears to be endothelial dysfunction, a state of blunted vasodilatory capacity and reduced ability to protect against platelet aggregation, blood cell adhesion, and smooth muscle proliferation. A pivotal factor involved in vascular health is nitric oxide (NO), which is dynamically controlled by signaling processes 64, 65. As patients with either the metabolic syndrome or diabetes have impaired NO-mediated vasodilation 66–68, insulin resistance may have direct pathogenic effects on the vasculature. Insulin is a major stimulus for NO-mediated vasodilation 69.
In many tissues including the vasculature, insulin effects can be generally divided into two pathways, involving either PI-3-kinase/Akt or MAP kinase signaling. Upon ligand binding, the insulin receptor activates a signaling cascade involving insulin receptor substrate (IRS) adapter molecules, PI-3-kinase, and Akt, subsequently leading in the endothelium to the phosphorylation/activation of eNOS, a process independent of classical calcium-based eNOS signaling 70, 71. In contradistinction, the growth factor functions of insulin occur via its MAP kinase-dependent arm and are more involved with cellular migration, vascular smooth muscle cell proliferation, and prothrombotic states 72. During insulin resistant states, there is a selective disruption of vascular PI-3-kinase/Akt-mediated signaling; interestingly, insulin-mediated MAP kinase signaling continues unabated 73, 74 and can even further blunt PI-3-kinase signaling by serine-phosphorylation of IRS-1 75. Thus, the development of insulin resistance and compensatory hyperinsulinemia can progressively shift the balance of insulin signaling toward a mitogenic state that may contribute to atherosclerosis (Figure 2).
Are the Direct Effects of Insulin Resistance Always Pro-Atherogenic or Can Insulin Resistance Play a Paradoxical Protective Role?
As discussed above, the direct effects of insulin on the vasculature implicate insulin resistance in vascular/endothelial dysfunction. However, it should noted that there are limited data extending this connection to atherosclerosis; regardless of the experimental system, most published studies use NO-mediated vascular reactivity/signaling as endpoints rather than more direct ones such as plaque formation/atherosclerotic burden. When one highlights studies assessing the role of insulin signaling in atherogenesis, several interesting observations arise.
ApoE-null mice with selective inactivation of the insulin receptor (IR) in the myeloid lineage have smaller atherosclerotic lesions 76. In a different approach, bone marrow transplantation of LDLR-null mice with insulin receptor-deficient cells did not affect the number of lesions but created more complex/necrotic lesions 77. Although whole-animal IRS-2 null mice on an ApoE-null background have increased plaque burden, repopulation of the hematopoietic system of lethally-irradiated ApoE-null mice with IRS-2(−/−) cells mitigates the atherosclerotic phenotype 76. Deletion of the PI-3-kinase p110γ gene in mice, which in contrast to the PI-3-kinase p110α and p110β genes is predominantly expressed in hematopoietic and muscle cells, leads to diminished atherosclerosis 78. These observations highlight the complexity of insulin signaling at the level of the vasculature and demonstrate the potential disparity between systemic insulin resistance and its direct effects on the forming atherosclerotic lesion.
Emerging Themes: The Concept of “Protective Macrophages” in Insulin Resistance and Atherosclerosis
The discovery of adipose tissue as a secretory organ, capable of producing inflammatory markers especially in states of nutrient excess/obesity was an important step in understanding the initiation of insulin resistance. In the past few years, this concept has been advanced and refined by implicating specific cellular events in the obesity-insulin resistance link. Immunohistochemical analysis of obesity-induced pathological changes reveals a progressive accumulation of bone-marrow derived macrophages in the adipose tissue matrix over time 79, 80. These macrophages are the predominant source of adipose tissue TNFα production and a significant contributor to the production of IL-6 as well several other inflammatory markers 80. Additionally, this macrophage infiltration is observed before overt manifestations of insulin resistance as determined by compensatory increases in insulin levels 79.
It has been known for some time that macrophages are present in numerous tissues even under normal noninflammatory conditions 81. These resident macrophages are phenotypically different from the more familiar “classically activated”/M1-macrophages (i.e. those elicited during acute inflammatory responses) and thus are termed “alternatively activated” or M2-macrophages. Whereas M1 macrophages are activated under inflammatory settings and express numerous proinflammatory markers, M2 macrophages are activated by different stimuli (e.g. IL-4 and IL-13) and actually produce anti-inflammatory mediators (e.g. IL-10 and IL-1 Receptor Antagonist) that are involved in tissue healing/remodeling and seem to oppose the effects of their M1 counterparts 82, 83. Interestingly, these observations were recently highlighted in adipose tissue. On a normal diet, the adipose tissue of lean mice is populated with “alternatively activated” macrophages, whereas diet-induced obesity produces a shift toward the M1 phenotype 84. Curiously, this shift is abrogated in obese C-C motif chemokine receptor-2 (CCR2)-null mice, suggesting that loss of this monocyte chemoattractant receptor presumably disables the robust recruitment of proinflammatory macrophages 84. This finding extends previous data showing the protective effect of MCP-1 and CCR-2 KO mice in diet-induced insulin resistance 85, 86.
Recently, Odegaard, et al. provided mechanistic insight into this phenotypic switch by demonstrating that PPARγ is required for the presence and maturation of alternatively activated macrophages in adipose tissue 87. Furthermore, their macrophage-specific PPARγ-KO mice were more prone to diet-induced obesity and insulin resistance 87. Using a similar gene targeting strategy, Hevener et al. provided a different perspective on macrophage-specific PPARγ signaling, showing impaired hepatic and skeletal muscle insulin sensitivity, signaling, and lipid accumulation 88. Interestingly, the insulin-sensitizing effects of TZDs were only partially effective in these mice, indicating that macrophage PPARγ signaling is at least partially required to achieve the full effects of TZDs on insulin sensitivity 88
There is also evidence that alternatively activated macrophages may be involved in atherosclerosis 89. Both M1 and M2 markers are present in human atherosclerotic plaques and are produced by distinct pools of mononuclear cells in these plaques. Also, PPARγ activators such as TZDs are able to drive the differentiation of monocytes into an M2 phenotype, although they are neither able to increase plaque M2 markers nor able to cause a switch of M1 cells to an M2 phenotype 89. Given the ability of macrophages to take on diametrically opposite fates, the prospect of harnessing such an ability to mitigate proatherogenic states is exciting. Another direct link between macrophages, adipocytes, and inflammation has recently emerged in the form of the adipocyte binding protein aP2 (also known as FABP4). Chemically inhibiting the function of aP2, a protein expressed in macrophages in addition to adipocytes, can suppress inflammatory pathways, decrease atherosclerosis, and treat type 2 diabetes in mice 90.
Insulin Resistance, Oxidative/Mitochondrial Stress, and Atherosclerosis
Mitochondria are the major source of ATP production by harboring both the enzymes of the tricyclic acid cycle and oxidative phosphorylation. The continuous flow of electrons from complex I through IV of the electron transport chain and pumping of protons though the inner mitochondrial membrane is contingent on steady-state nutrient delivery. In cases of nutrient excess, the surplus of effluxed protons leads to slowed electron transport chain kinetics and augmentation of alternative electron accepting mechanisms 91. Enhanced production of reactive oxygen species (ROS) such as superoxide is a direct by-product of this process and a contributing factor to vascular dysfunction in insulin resistance (Figure 2) 92.
As the predominant site of ROS production, the mitochondrion is a susceptible target for oxidative damage and several lines of evidence point to mitochondrial dysfunction in promoting atherogenesis. Studies in cell culture and rodent models show that increased free fatty acid oxidation in aortic endothelium induces enhanced mitochondrial superoxide generation and inactivation of anti-atherogenic factors (e.g. eNOS and prostacyclin synthase); direct inhibition of carnitine palmitoyl transferase I (the rate-limiting mitochondrial enzyme in fatty acid oxidation) reverses this effect 93. Atherosclerotic lesions from patients undergoing vascular surgery and those from ApoE-null mice show increased mitochondrial DNA damage; this process is observed even in young mice before the overt development of atherosclerotic plaques 94. Additionally, superoxide dismutase heterozygous-null mice (SOD2+/−) in the ApoE(−/−) background have accelerated atherosclerosis 94. In this regard, the fact that mitochondria lack a robust mismatch repair system might make mitochondrial DNA especially susceptible to ROS damage and mitochondrial dysfunction 95.
Another feature of mitochondrial bioenergetics relevant to atherosclerosis is the observed heterogeneity of metabolism in the vasculature (i.e. different areas of an arterial wall have varied ATP-generating efficiency probably due to variable uncoupling of respiration and oxidative phosphorylation) 96. Differences in adequate oxygen delivery to medium and large-size arteries have been proposed to underlie this heterogeneity 96, 97. Intriguingly, although respiratory uncoupling seems to occur in all blood vessels, it is increased in the aorta of atheroma-prone pigeons 98. Also, a dearth of essential fatty acids, a marker of atherosclerotic lesions, heightens respiratory uncoupling 99, 100.
The uncoupling proteins are a family of transporters found in the inner mitochondrial membrane which dissipate the mitochondrial proton-motive force by allowing protons back into the mitochondrial matrix 101. In a direct test of the relationship between respiratory uncoupling and vascular dysfunction, Bernal-Mizrachi et al. created smooth-muscle transgenic mice overexpressing UCP1 102; the choice of smooth muscle cells as the site of uncoupling was based on smooth muscle being a major site for ROS production 91. These “uncoupled” mice showed significant signs of vascular dysfunction with overt hypertension and when in an ApoE-null background, increased atherosclerosis 102. Furthermore, superoxide production was elevated and NO availability was decreased.
Taken together, adiposity and insulin resistance create a state of nutrient excess, skewing normally functioning mitochondrial bioenergetics into excess ROS production and by respiratory uncoupling mechanisms, inefficient generation of ATP; the result is vascular dysfunction and a proatherogenic state. In this regard, it is interesting to note that caloric restriction can induce mitochondrial biogenesis, decrease ROS, and lead to more efficient ATP production 103, 104.
Insulin Resistance, Genomic Stress, and Atherosclerosis
Damage to mitochondrial DNA is not the only consequence of excessive production of ROS; the nuclear genome is also susceptible to damage and alterations of relevant genes involved in DNA repair and stress response contribute to insulin resistance, vascular dysfunction, and atherosclerosis (reviewed in 105). Patients with established CAD and diabetes have increased markers of genomic instability and oxidative DNA damage in peripheral blood mononuclear cells 106, 107. FISH analysis of human carotid endarterectomy (CEA) plaques shows chromosomal instability with polyploidy and deletions 108, 109. Markers of oxidative DNA damage such as 8-oxo-deoxyguanosine (8-oxo-dG) are elevated in CEA specimens and this is accompanied by overexpression of several DNA repair proteins 110. Similarly, diet-induced atherosclerotic plaques in rabbits show increased 8-oxo-dG, DNA strand breaks, and DNA repair enzymes; these markers of DNA damage are significantly but not entirely reversed with normalization of the diet 111.
These data suggesting a causal link between DNA damage and atherosclerosis are corroborated by the phenotypes of several accelerated aging and/or DNA damage disorders 112. Werner syndrome occurs due to mutations in the WRN gene, encoding a DNA helicase with several functions in DNA replication and repair 113. In addition to premature aging, Werner patients develop insulin resistance, atherosclerosis, and valvular heart disease 112, 114, a phenotype that is partially recapitulated in a mouse model of the disease 115. Patients with Hutchinson-Gilford Progeria syndrome (HGPS), the prototypical accelerated aging syndrome, also develop insulin resistance and premature atherosclerosis but at even earlier time points than Werner patients and often succumb to MI or strokes by their early teenage years 116, 117. HGPS is caused by mutations in the Lamin A gene, encoding proteins essential for the integrity of the nuclear lamina/nuclear membrane 117. The disruption of nuclear lamina is not solely structural and is now known to affect transcriptional regulation, genomic stability, and DNA repair. Recent data show that HGPS patients accumulate a farnesylated precursor of Lamin A that likely imparts genomic instability by impairing formation of DNA repair foci 118, 119. A farnesyltransferase inhibitor can reverse many of the structural and phenotypic abnormalities of a mouse model of HGPS 120. As statins are also inhibitors of the pathway important for farnesylation, this raises the intriguing possibility that part of the non-lipid mediated effects of statins are directed at enhancing genomic stability. Another relevant human disease is Ataxia-Telangiectasia (AT), caused by mutations in ATM, a protein kinase with central roles in the response to DNA damage 121. In addition to progressive ataxia and significant predisposition to cancers, many AT patients also develop insulin resistance 122, 123. Although AT patients die early from a variety of cancers (median age of death 20) 124, carriers of ATM mutations (estimated to comprise 1.4–2% of the population) have higher CV mortality 125. In support of a direct pathogenic role for ATM in metabolic and vascular regulation, ATM-null mice in an ApoE-null background indeed develop insulin resistance and accelerated atherosclerosis 126. Chloroquine, used to treat malaria but also a known activator of ATM, was able to protect against atherosclerosis and many of the metabolic effects of diet-induced insulin resistance in mouse models 126.
Summary
The metabolic syndrome and diabetes are in large part varied manifestations of an underlying process known as insulin resistance. Normally insulin sensitive metabolic organs develop a progressive inability to respond to this signal with resultant metabolic derangements. Cardiovascular disease is associated with insulin resistant states, although the presence of a myriad of insulin signaling pathways potentially affecting vascular function precludes a simple explanation for this association. Treatment of insulin resistant patients with drugs such as statins, ACEi, and ARBs can yield profound improvements in CV mortality. The development of insulin sensitizers such as the thiazolidinediones has been conceptually exciting, but recent data showing lack of/modest benefit or possibly even an increase in CV events combined with a propensity to exacerbate heart failure have dampened enthusiasm for this class of drugs.
Insulin resistant people tend to have increased adiposity, so novel strategies that exploit the relationship between adipocytes and the inflammatory process in the vasculature to treat atherosclerosis are attractive. Interfering with cellular stress pathways, including those that involve damage to mitochondria and the nuclear genome, may also prove to be useful in the quest for developing new approaches to treat atherosclerosis in people with insulin resistance.
Acknowledgments
This work was supported by Grants HL083762 and DK076729 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Falta WA. Uber Einen Insulinrefraktaren fall von Diabetes Mellitus. Klin Wchnschr. 1924;3:1315–1317. [Google Scholar]
- 2.Levinson PD. Eighty years of insulin therapy: 1922–2002. Med Health R I. 2003 Apr;86(4):101–106. [PubMed] [Google Scholar]
- 3.Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III) JAMA. 2001 May 16;285(19):2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 4.Alberti KG, Zimmet P, Shaw J. Metabolic syndrome--a new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet Med. 2006 May;23(5):469–480. doi: 10.1111/j.1464-5491.2006.01858.x. [DOI] [PubMed] [Google Scholar]
- 5.Grundy SM, Cleeman JI, Daniels SR, et al. Circulation. 17. Vol. 112. 2005. Oct 25, Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement; pp. 2735–2752. [DOI] [PubMed] [Google Scholar]
- 6.Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998 Jul 23;339(4):229–234. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 7.Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care. 1993 Feb;16(2):434–444. doi: 10.2337/diacare.16.2.434. [DOI] [PubMed] [Google Scholar]
- 8.Kannel WB, McGee DL. Diabetes and cardiovascular risk factors: the Framingham study. Circulation. 1979 Jan;59:8–13. doi: 10.1161/01.cir.59.1.8. [DOI] [PubMed] [Google Scholar]
- 9.Vaccaro O, Eberly LE, Neaton JD, Yang L, Riccardi G, Stamler J. Impact of diabetes and previous myocardial infarction on long-term survival: 25-year mortality follow-up of primary screenees of the Multiple Risk Factor Intervention Trial. Arch Intern Med. 2004 Jul 12;164(13):1438–1443. doi: 10.1001/archinte.164.13.1438. [DOI] [PubMed] [Google Scholar]
- 10.Alexander CM, Landsman PB, Teutsch SM, Haffner SM. NCEP-defined metabolic syndrome, diabetes, and prevalence of coronary heart disease among NHANES III participants age 50 years and older. Diabetes. 2003 May;52(5):1210–1214. doi: 10.2337/diabetes.52.5.1210. [DOI] [PubMed] [Google Scholar]
- 11.Hunt KJ, Resendez RG, Williams K, Haffner SM, Stern MP. National Cholesterol Education Program versus World Health Organization metabolic syndrome in relation to all-cause and cardiovascular mortality in the San Antonio Heart Study. Circulation. 2004 Sep 7;110(10):1251–1257. doi: 10.1161/01.CIR.0000140762.04598.F9. [DOI] [PubMed] [Google Scholar]
- 12.Sattar N, Gaw A, Scherbakova O, et al. Circulation. 4. Vol. 108. 2003. Jul 29, Metabolic syndrome with and without C-reactive protein as a predictor of coronary heart disease and diabetes in the West of Scotland Coronary Prevention Study; pp. 414–419. [DOI] [PubMed] [Google Scholar]
- 13.Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994 Nov 19;344(8934):1383–1389. [PubMed] [Google Scholar]
- 14.Shepherd J, Cobbe SM, Ford I, et al. N Engl J Med. 20. Vol. 333. 1995. Nov 16, Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group; pp. 1301–1307. [DOI] [PubMed] [Google Scholar]
- 15.Collins R, Armitage J, Parish S, Sleight P, Peto R. Effects of cholesterol-lowering with simvastatin on stroke and other major vascular events in 20536 people with cerebrovascular disease or other high-risk conditions. Lancet. 2004 Mar 6;363(9411):757–767. doi: 10.1016/S0140-6736(04)15690-0. [DOI] [PubMed] [Google Scholar]
- 16.Ray KK, Cannon CP. The potential relevance of the multiple lipid-independent (pleiotropic) effects of statins in the management of acute coronary syndromes. J Am Coll Cardiol. 2005 Oct 18;46(8):1425–1433. doi: 10.1016/j.jacc.2005.05.086. [DOI] [PubMed] [Google Scholar]
- 17.Yusuf S, Pepine CJ, Garces C, et al. Effect of enalapril on myocardial infarction and unstable angina in patients with low ejection fractions. Lancet. 1992 Nov 14;340(8829):1173–1178. doi: 10.1016/0140-6736(92)92889-n. [DOI] [PubMed] [Google Scholar]
- 18.Pfeffer MA, Braunwald E, Moye LA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N Engl J Med. 1992 Sep 3;327(10):669–677. doi: 10.1056/NEJM199209033271001. [DOI] [PubMed] [Google Scholar]
- 19.Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000 Jan 20;342(3):145–153. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- 20.Lindholm LH, Ibsen H, Dahlof B, et al. Cardiovascular morbidity and mortality in patients with diabetes in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002 Mar 23;359(9311):1004–1010. doi: 10.1016/S0140-6736(02)08090-X. [DOI] [PubMed] [Google Scholar]
- 21.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995 Jun 2;270(22):12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 22.Yki-Jarvinen H. Thiazolidinediones. N Engl J Med. 2004 Sep 9;351(11):1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- 23.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007 Jun 14;356(24):2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 24.Dormandy JA, Charbonnel B, Eckland DJ, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005 Oct 8;366(9493):1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- 25.Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA. 2007 Sep 12;298(10):1180–1188. doi: 10.1001/jama.298.10.1180. [DOI] [PubMed] [Google Scholar]
- 26.Lago RM, Singh PP, Nesto RW. Congestive heart failure and cardiovascular death in patients with prediabetes and type 2 diabetes given thiazolidinediones: a meta-analysis of randomised clinical trials. Lancet. 2007 Sep 29;370(9593):1129–1136. doi: 10.1016/S0140-6736(07)61514-1. [DOI] [PubMed] [Google Scholar]
- 27.Szapary PO, Rader DJ. The triglyceride-high-density lipoprotein axis: an important target of therapy? Am Heart J. 2004 Aug;148(2):211–221. doi: 10.1016/j.ahj.2004.03.037. [DOI] [PubMed] [Google Scholar]
- 28.McGarry JD. What if Minkowski had been ageusic? An alternative angle on diabetes. Science. 1992 Oct 30;258(5083):766–770. doi: 10.1126/science.1439783. [DOI] [PubMed] [Google Scholar]
- 29.Ginsberg HN. REVIEW: Efficacy and mechanisms of action of statins in the treatment of diabetic dyslipidemia. J Clin Endocrinol Metab. 2006 Feb;91(2):383–392. doi: 10.1210/jc.2005-2084. [DOI] [PubMed] [Google Scholar]
- 30.Adiels M, Boren J, Caslake MJ, et al. Overproduction of VLDL1 driven by hyperglycemia is a dominant feature of diabetic dyslipidemia. Arterioscler Thromb Vasc Biol. 2005 Aug;25(8):1697–1703. doi: 10.1161/01.ATV.0000172689.53992.25. [DOI] [PubMed] [Google Scholar]
- 31.Sparks JD, Sparks CE. Insulin regulation of tricylglycerol-rich lipoprotein synthesis and secretion. Biochim Biophys Acta. 1994 Nov 17;1215(1–2):9–32. doi: 10.1016/0005-2760(94)90088-4. [DOI] [PubMed] [Google Scholar]
- 32.Ninomiya JK, L'Italien G, Criqui MH, Whyte JL, Gamst A, Chen RS. Association of the metabolic syndrome with history of myocardial infarction and stroke in the Third National Health and Nutrition Examination Survey. Circulation. 2004 Jan 6;109(1):42–46. doi: 10.1161/01.CIR.0000108926.04022.0C. [DOI] [PubMed] [Google Scholar]
- 33.Bansal S, Buring JE, Rifai N, Mora S, Sacks FM, Ridker PM. Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA. 2007 Jul 18;298(3):309–316. doi: 10.1001/jama.298.3.309. [DOI] [PubMed] [Google Scholar]
- 34.Nordestgaard BG, Been M, Sehnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, iscemic heart disease, and death in men and women. JAMA. 2007 Jul 18;298(3):299–308. doi: 10.1001/jama.298.3.299. [DOI] [PubMed] [Google Scholar]
- 35.Sattar N, Williams K, Sniderman AD, D'Agostino R, Jr, Haffner SM. Comparison of the associations of apolipoprotein B and non-high-density lipoprotein cholesterol with other cardiovascular risk factors in patients with the metabolic syndrome in the Insulin Resistance Atherosclerosis Study. Circulation. 2004 Oct 26;110(17):2687–2693. doi: 10.1161/01.CIR.0000145660.60487.94. [DOI] [PubMed] [Google Scholar]
- 36.Austin MA, King MC, Vranizan KM, Krauss RM. Atherogenic lipoprotein phenotype. A proposed genetic marker for coronary heart disease risk. Circulation. 1990 Aug;82(2):495–506. doi: 10.1161/01.cir.82.2.495. [DOI] [PubMed] [Google Scholar]
- 37.Rashid S, Watanabe T, Sakaue T, Lewis GF. Mechanisms of HDL lowering in insulin resistant, hypertriglyceridemic states: the combined effect of HDL triglyceride enrichment and elevated hepatic lipase activity. Clin Biochem. 2003 Sep;36(6):421–429. doi: 10.1016/s0009-9120(03)00078-x. [DOI] [PubMed] [Google Scholar]
- 38.Golay A, Zech L, Shi MZ, Chiou YA, Reaven GM, Chen YD. High density lipoprotein (HDL) metabolism in noninsulin-dependent diabetes mellitus: measurement of HDL turnover using tritiated HDL. J Clin Endocrinol Metab. 1987 Sep;65(3):512–518. doi: 10.1210/jcem-65-3-512. [DOI] [PubMed] [Google Scholar]
- 39.Festa A, D'Agostino R, Jr, Howard G, Mykkanen L, Tracy RP, Haffner SM. Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS) Circulation. 2000 Jul 4;102(1):42–47. doi: 10.1161/01.cir.102.1.42. [DOI] [PubMed] [Google Scholar]
- 40.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006 Jul;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999 Jan 14;340(2):115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 42.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993 Jan 1;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 43.Lau DC, Dhillon B, Yan H, Szmitko PE, Verma S. Adipokines: molecular links between obesity and atheroslcerosis. Am J Physiol Heart Circ Physiol. 2005 May;288(5):H2031–H2041. doi: 10.1152/ajpheart.01058.2004. [DOI] [PubMed] [Google Scholar]
- 44.Roytblat L, Rachinsky M, Fisher A, et al. Raised interleukin-6 levels in obese patients. Obes Res. 2000 Dec;8(9):673–675. doi: 10.1038/oby.2000.86. [DOI] [PubMed] [Google Scholar]
- 45.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995 May;95(5):2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Esposito K, Pontillo A, Di Palo C, et al. Effect of weight loss and lifestyle changes on vascular inflammatory markers in obese women: a randomized trial. JAMA. 2003 Apr 9;289(14):1799–1804. doi: 10.1001/jama.289.14.1799. [DOI] [PubMed] [Google Scholar]
- 47.Festa A, D'Agostino R, Jr, Tracy RP, Haffner SM. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes. 2002 Apr;51(4):1131–1137. doi: 10.2337/diabetes.51.4.1131. [DOI] [PubMed] [Google Scholar]
- 48.Christiansen T, Richelsen B, Bruun JM. Monocyte chemoattractant protein-1 is produced in isolated adipocytes, associated with adiposity and reduced after weight loss in morbid obese subjects. Int J Obes (Lond) 2005 Jan;29(1):146–150. doi: 10.1038/sj.ijo.0802839. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz EA, Reaven PD. Molecular and signaling mechanisms of atherosclerosis in insulin resistance. Endocrinol Metab Clin North Am. 2006 Sep;35(3):525–549. doi: 10.1016/j.ecl.2006.06.005. viii. [DOI] [PubMed] [Google Scholar]
- 50.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 51.Karin M. The beginning of the end: IkappaB kinase (IKK) and NF-kappaB activation. J Biol Chem. 1999 Sep 24;274(39):27339–27342. doi: 10.1074/jbc.274.39.27339. [DOI] [PubMed] [Google Scholar]
- 52.Thurberg BL, Collins T. The nuclear factor-kappa B/inhibitor of kappa B autoregulatory system and atherosclerosis. Curr Opin Lipidol. 1998 Oct;9(5):387–396. doi: 10.1097/00041433-199810000-00002. [DOI] [PubMed] [Google Scholar]
- 53.Kempe S, Kestler H, Lasar A, Wirth T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005;33(16):5308–5319. doi: 10.1093/nar/gki836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schreyer SA, Peschon JJ, LeBoeuf RC. Accelerated atherosclerosis in mice lacking tumor necrosis factor receptor p55. J Biol Chem. 1996 Oct 18;271(42):26174–26178. doi: 10.1074/jbc.271.42.26174. [DOI] [PubMed] [Google Scholar]
- 55.Kanters E, Pasparakis M, Gijbels MJ, et al. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2003 Oct;112(8):1176–1185. doi: 10.1172/JCI18580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanters E, Gijbels MJ, van der Made I, et al. Hematopoietic NF-kappaB1 deficiency results in small atherosclerotic lesions with an inflammatory phenotype. Blood. 2004 Feb 1;103(3):934–940. doi: 10.1182/blood-2003-05-1450. [DOI] [PubMed] [Google Scholar]
- 57.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001 Apr;81(2):807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 58.Sumara G, Belwal M, Ricci R. "Jnking" atherosclerosis. Cell Mol Life Sci. 2005 Nov;62(21):2487–2494. doi: 10.1007/s00018-005-5253-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ventura JJ, Kennedy NJ, Lamb JA, Flavell RA, Davis RJ. c-Jun NH(2)-terminal kinase is essential for the regulation of AP-1 by tumor necrosis factor. Mol Cell Biol. 2003 Apr;23(8):2871–2882. doi: 10.1128/MCB.23.8.2871-2882.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov. 2003 Jul;2(7):554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- 61.Nishio H, Matsui K, Tsuji H, Tamura A, Suzuki K. Immunohistochemical study of the phosphorylated and activated form of c-Jun NH2-terminal kinase in human aorta. Histochem J. 2001 Mar;33(3):167–171. doi: 10.1023/a:1017952310800. [DOI] [PubMed] [Google Scholar]
- 62.Metzler B, Hu Y, Dietrich H, Xu Q. Increased expression and activation of stress-activated protein kinases/c-Jun NH(2)-terminal protein kinases in atherosclerotic lesions coincide with p53. Am J Pathol. 2000 Jun;156(6):1875–1886. doi: 10.1016/S0002-9440(10)65061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ricci R, Sumara G, Sumara I, et al. Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science. 2004 Nov 26;306(5701):1558–1561. doi: 10.1126/science.1101909. [DOI] [PubMed] [Google Scholar]
- 64.Hartge MM, Kintscher U, Unger T. Endothelial dysfunction and its role in diabetic vascular disease. Endocrinol Metab Clin North Am. 2006 Sep;35(3):551–560. doi: 10.1016/j.ecl.2006.06.006. viii–ix. [DOI] [PubMed] [Google Scholar]
- 65.Hsueh WA, Quinones MJ. Role of endothelial dysfunction in insulin resistance. Am J Cardiol. 2003 Aug 18;92(4A):10J–17J. doi: 10.1016/s0002-9149(03)00611-8. [DOI] [PubMed] [Google Scholar]
- 66.Williams SB, Cusco JA, Roddy MA, Johnstone MT, Creager MA. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J Am Coll Cardiol. 1996 Mar 1;27(3):567–574. doi: 10.1016/0735-1097(95)00522-6. [DOI] [PubMed] [Google Scholar]
- 67.Steinberg HO, Tarshoby M, Monestel R, et al. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. J Clin Invest. 1997 Sep 1;100(5):1230–1239. doi: 10.1172/JCI119636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perticone F, Ceravolo R, Candigliota M, et al. Obesity and body fat distribution induce endothelial dysfunction by oxidative stress: protective effect of vitamin C. Diabetes. 2001 Jan;50(1):159–165. doi: 10.2337/diabetes.50.1.159. [DOI] [PubMed] [Google Scholar]
- 69.Scherrer U, Randin D, Vollenweider P, Vollenweider L, Nicod P. Nitric oxide release accounts for insulin's vascular effects in humans. J Clin Invest. 1994 Dec;94(6):2511–2515. doi: 10.1172/JCI117621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Montagnani M, Ravichandran LV, Chen H, Esposito DL, Quon MJ. Insulin receptor substrate-1 and phosphoinositide-dependent kinase-1 are required for insulin-stimulated production of nitric oxide in endothelial cells. Mol Endocrinol. 2002 Aug;16(8):1931–1942. doi: 10.1210/me.2002-0074. [DOI] [PubMed] [Google Scholar]
- 71.Zeng G, Nystrom FH, Ravichandran LV, et al. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation. 2000 Apr 4;101(13):1539–1545. doi: 10.1161/01.cir.101.13.1539. [DOI] [PubMed] [Google Scholar]
- 72.Takeda K, Ichiki T, Tokunou T, et al. Critical role of Rho-kinase and MEK/ERK pathways for angiotensin II-induced plasminogen activator inhibitor type-1 gene expression. Arterioscler Thromb Vasc Biol. 2001 May;21(5):868–873. doi: 10.1161/01.atv.21.5.868. [DOI] [PubMed] [Google Scholar]
- 73.Jiang ZY, Lin YW, Clemont A, et al. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J Clin Invest. 1999 Aug;104(4):447–457. doi: 10.1172/JCI5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Montagnani M, Golovchenko I, Kim I, et al. Inhibition of phosphatidylinositol 3-kinase enhances mitogenic actions of insulin in endothelial cells. J Biol Chem. 2002 Jan 18;277(3):1794–1799. doi: 10.1074/jbc.M103728200. [DOI] [PubMed] [Google Scholar]
- 75.Gual P, Gremeaux T, Gonzalez T, Le Marchand-Brustel Y, Tanti JF. MAP kinases and mTOR mediate insulin-induced phosphorylation of insulin receptor substrate-1 on serine residues 307, 612 and 632. Diabetologia. 2003 Nov;46(11):1532–1542. doi: 10.1007/s00125-003-1223-4. [DOI] [PubMed] [Google Scholar]
- 76.Baumgartl J, Baudler S, Scherner M, et al. Myeloid lineage cell-restricted insulin resistance protects apolipoproteinE-deficient mice against atherosclerosis. Cell Metab. 2006 Apr;3(4):247–256. doi: 10.1016/j.cmet.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Han S, Liang CP, DeVries-Seimon T, et al. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 2006 Apr;3(4):257–266. doi: 10.1016/j.cmet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 78.Chang JD, Sukhova GK, Libby P, et al. Deletion of the phosphoinositide 3-kinase p110gamma gene attenuates murine atherosclerosis. Proc Natl Acad Sci U S A. 2007 May 8;104(19):8077–8082. doi: 10.1073/pnas.0702663104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003 Dec;112(12):1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003 Dec;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003 Jul;19(1):71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 82.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003 Jan;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 83.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005 Dec;5(12):953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 84.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007 Jan;117(1):175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006 Jun;116(6):1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weisberg SP, Hunter D, Huber R, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006 Jan;116(1):115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007 Jun 28;447(7148):1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hevener AL, Olefsky JM, Reichart D, et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest. 2007 Jun;117(6):1658–1669. doi: 10.1172/JCI31561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bouhlel MA, Derudas B, Rigamonti E, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007 Aug;6(2):137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 90.Furuhashi M, Tuncman G, Gorgun CZ, et al. Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature. 2007 Jun 21;447(7147):959–965. doi: 10.1038/nature05844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002 Jan;82(1):47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 92.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005 Jun;54(6):1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 93.Du X, Edelstein D, Obici S, Higham N, Zou MH, Brownlee M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J Clin Invest. 2006 Apr;116(4):1071–1080. doi: 10.1172/JCI23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ballinger SW, Patterson C, Knight-Lozano CA, et al. Mitochondrial integrity and function in atherogenesis. Circulation. 2002 Jul 30;106(5):544–549. doi: 10.1161/01.cir.0000023921.93743.89. [DOI] [PubMed] [Google Scholar]
- 95.Mason PA, Matheson EC, Hall AG, Lightowlers RN. Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res. 2003 Feb 1;31(3):1052–1058. doi: 10.1093/nar/gkg167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Levin M, Leppanen O, Evaldsson M, Wiklund O, Bondjers G, Bjornheden T. Mapping of ATP, glucose, glycogen, and lactate concentrations within the arterial wall. Arterioscler Thromb Vasc Biol. 2003 Oct 1;23(10):1801–1807. doi: 10.1161/01.ATV.0000092872.54026.8D. [DOI] [PubMed] [Google Scholar]
- 97.Bjornheden T, Levin M, Evaldsson M, Wiklund O. Evidence of hypoxic areas within the arterial wall in vivo. Arterioscler Thromb Vasc Biol. 1999 Apr;19(4):870–876. doi: 10.1161/01.atv.19.4.870. [DOI] [PubMed] [Google Scholar]
- 98.Santerre RF, Nicolosi RJ, Smith SC. Respiratory control in preatherosclerotic susceptible and resistant pigeon aortas. Exp Mol Pathol. 1974 Jun;20(3):397–406. doi: 10.1016/0014-4800(74)90069-0. [DOI] [PubMed] [Google Scholar]
- 99.Klein PD, Johnson RM. Phosphorus metabolism in unsaturated fatty acid-deficient rats. J Biol Chem. 1954 Nov;211(1):103–110. [PubMed] [Google Scholar]
- 100.Cornwell DG, Panganamala RV. Atherosclerosis: an intracellular deficiency in essential fatty acids. Prog Lipid Res. 1981;20:365–376. doi: 10.1016/0163-7827(81)90069-2. [DOI] [PubMed] [Google Scholar]
- 101.Echtay KS. Mitochondrial uncoupling proteins-What is their physiological role? Free Radic Biol Med. 2007 Nov 15;43(10):1351–1371. doi: 10.1016/j.freeradbiomed.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 102.Bernal-Mizrachi C, Gates AC, Weng S, et al. Vascular respiratory uncoupling increases blood pressure and atherosclerosis. Nature. 2005 May 26;435(7041):502–506. doi: 10.1038/nature03527. [DOI] [PubMed] [Google Scholar]
- 103.Lopez-Lluch G, Hunt N, Jones B, et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A. 2006 Feb 7;103(6):1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nisoli E, Tonello C, Cardile A, et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005 Oct 14;310(5746):314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 105.Mahmoudi M, Mercer J, Bennett M. DNA damage and repair in atherosclerosis. Cardiovasc Res. 2006 Jul 15;71(2):259–268. doi: 10.1016/j.cardiores.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 106.Botto N, Rizza A, Colombo MG, et al. Evidence for DNA damage in patients with coronary artery disease. Mutat Res. 2001 Jun 27;493(1–2):23–30. doi: 10.1016/s1383-5718(01)00162-0. [DOI] [PubMed] [Google Scholar]
- 107.Botto N, Masetti S, Petrozzi L, et al. Elevated levels of oxidative DNA damage in patients with coronary artery disease. Coron Artery Dis. 2002 Aug;13(5):269–274. doi: 10.1097/00019501-200208000-00004. [DOI] [PubMed] [Google Scholar]
- 108.Matturri L, Cazzullo A, Turconi P, et al. Chromosomal alterations in atherosclerotic plaques. Atherosclerosis. 2001 Feb 15;154(3):755–761. doi: 10.1016/s0021-9150(00)00488-3. [DOI] [PubMed] [Google Scholar]
- 109.Casalone R, Granata P, Minelli E, et al. Cytogenetic analysis reveals clonal proliferation of smooth muscle cells in atherosclerotic plaques. Hum Genet. 1991 Jun;87(2):139–143. doi: 10.1007/BF00204169. [DOI] [PubMed] [Google Scholar]
- 110.Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation. 2002 Aug 20;106(8):927–932. doi: 10.1161/01.cir.0000026393.47805.21. [DOI] [PubMed] [Google Scholar]
- 111.Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. Oxidative DNA damage and repair in experimental atherosclerosis are reversed by dietary lipid lowering. Circ Res. 2001 Apr 13;88(7):733–739. doi: 10.1161/hh0701.088684. [DOI] [PubMed] [Google Scholar]
- 112.Capell BC, Collins FS, Nabel EG. Mechanisms of cardiovascular disease in accelerated aging syndromes. Circ Res. 2007 Jul 6;101(1):13–26. doi: 10.1161/CIRCRESAHA.107.153692. [DOI] [PubMed] [Google Scholar]
- 113.Gray MD, Shen JC, Kamath-Loeb AS, et al. The Werner syndrome protein is a DNA helicase. Nat Genet. 1997 Sep;17(1):100–103. doi: 10.1038/ng0997-100. [DOI] [PubMed] [Google Scholar]
- 114.Yamada K, Ikegami H, Yoneda H, Miki T, Ogihara T. All patients with Werner's syndrome are insulin resistant, but only those who also have impaired insulin secretion develop overt diabetes. Diabetes Care. 1999 Dec;22(12):2094–2095. doi: 10.2337/diacare.22.12.2094. [DOI] [PubMed] [Google Scholar]
- 115.Massip L, Garand C, Turaga RV, Deschenes F, Thorin E, Lebel M. Increased insulin, triglycerides, reactive oxygen species, and cardiac fibrosis in mice with a mutation in the helicase domain of the Werner syndrome gene homologue. Exp Gerontol. 2006 Feb;41(2):157–168. doi: 10.1016/j.exger.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 116.DeBusk FL. The Hutchinson-Gilford progeria syndrome. Report of 4 cases and review of the literature. J Pediatr. 1972 Apr;80(4):697–724. doi: 10.1016/s0022-3476(72)80229-4. [DOI] [PubMed] [Google Scholar]
- 117.Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003 May 15;423(6937):293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu Y, Rusinol A, Sinensky M, Wang Y, Zou Y. DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J Cell Sci. 2006 Nov 15;119(Pt 22):4644–4649. doi: 10.1242/jcs.03263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Manju K, Muralikrishna B, Parnaik VK. Expression of disease-causing lamin A mutants impairs the formation of DNA repair foci. J Cell Sci. 2006 Jul 1;119(Pt 13):2704–2714. doi: 10.1242/jcs.03009. [DOI] [PubMed] [Google Scholar]
- 120.Fong LG, Frost D, Meta M, et al. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006 Mar 17;311(5767):1621–1623. doi: 10.1126/science.1124875. [DOI] [PubMed] [Google Scholar]
- 121.Savitsky K, Bar-Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995 Jun 23;268(5218):1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 122.Schalch DS, McFarlin DE, Barlow MH. An unusual form of diabetes mellitus in ataxia telangiectasia. N Engl J Med. 1970 Jun 18;282(25):1396–1402. doi: 10.1056/NEJM197006182822503. [DOI] [PubMed] [Google Scholar]
- 123.Bar RS, Levis WR, Rechler MM, et al. Extreme insulin resistance in ataxia telangiectasia: defect in affinity of insulin receptors. N Engl J Med. 1978 May 25;298(21):1164–1171. doi: 10.1056/NEJM197805252982103. [DOI] [PubMed] [Google Scholar]
- 124.Morrell D, Cromartie E, Swift M. Mortality and cancer incidence in 263 patients with ataxia-telangiectasia. J Natl Cancer Inst. 1986 Jul;77(1):89–92. [PubMed] [Google Scholar]
- 125.Su Y, Swift M. Mortality rates among carriers of ataxia-telangiectasia mutant alleles. Ann Intern Med. 2000 Nov 21;133(10):770–778. doi: 10.7326/0003-4819-133-10-200011210-00009. [DOI] [PubMed] [Google Scholar]
- 126.Schneider JG, Finck BN, Ren J, et al. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006 Nov;4(5):377–389. doi: 10.1016/j.cmet.2006.10.002. [DOI] [PubMed] [Google Scholar]