

Abstract
Burgeoning research into early functional programming has opened a new perspective of developmental origins of disease and organ dysfunction. In examining the roots of this emerging paradigm, it appears that crucial observations were made already 25 years ago. Clear examples include the discoveries of foetal alcohol syndrome, cancer in daughters whose mothers had used diethylstilbestrol during pregnancy, and neurotoxicity of lead and methylmercury. However, this research was often considered controversial, scientific conclusions were cautiously hedged, and stakeholders demanded replication in excess. Recognition of the new paradigm was therefore delayed, preventive interventions even further so. In hindsight, and in light of the precautionary principle, the adverse effects incurred on public health and the environment would call for responsible judgment to balance reasonable scientific scepticism against the risks associated with inaction. The traditional scientific scrutiny whether confidence limits include the possibility of no effect must be modified to allow consideration of the likelihood of worst-case scenarios. Additional emphasis should be placed on the question: What could be known, given our study opportunities and methodologies? Our late insights into the early disease origins may then benefit science as well as prevention.
Introduction
The recognition of developmental origins of diseases and functional deficits constitutes a paradigm shift in toxicology and public health [1]. This new insight supersedes the past belief that development was generally a homogeneous and invariant sequence of developmental stages and instead stresses plasticity, continuity, and multicausality [2]. It will likely have tremendous impact on prevention and pollution abatement in the future, and this conference is testimony of the growing cross-disciplinary attention to this topic worldwide. However, when exploring the origins of the new concept, a paradox becomes obvious, viz., that very early research discoveries clearly foretold the new understanding, but their implications were largely ignored and therefore failed to cause a wider impact on research and prevention. One may appropriately ask why the paradigm of developmental programming surfaced so late. In order to consider this question, we need to ascertain the early origins of the early origins hypothesis. I will focus on the last 25 years, i.e. the time since I became professor and chair of environmental medicine in Odense, Denmark.
My own interest in environmental health was triggered by a TV news program from the first United Nations Environment Conference held in Stockholm in 1972. I saw victims from Minamata, Japan, who had travelled to Scandinavia to demonstrate the neurological consequences of developmental exposure to mercury pollution. Their suffering made a lasting impression on me, and I think on many viewers worldwide, that a mother could be fairly unharmed by the pollution, but would give birth to a child with serious poisoning, as reported by Japanese physicians [3].
The early origins of a paradigm
The serious consequences of methylmercury exposures during early development constituted a warning signal that adverse effects of toxicants depend on the developmental stage. This epidemic of a new neurological disease started in the 1950s in a Japanese fishing village; methylmercury accumulated in seafood was recognised as the cause of Minamata disease by 1960. However, the official approval of this conclusion by government took many more years to appear [3], even though developmental neurotoxicity due to methylmercury had been reported elsewhere years before [3,4]. While evidence of the vulnerability of the developing brain was growing, expert groups went no further than to conclude that the foetus may be more susceptible to methylmercury toxicity than the adult [5]. More recent studies, e.g. from the Faroes and New Zealand, suggested that methylmercury from seafood even far from point sources of pollution could also impact negatively on brain development [6]. Again, reviews by expert panels emphasised weaknesses in the research, and only in 2003 was it internationally accepted that a decreased methylmercury exposure limit was needed to protect against developmental toxicity.
Lead provides a parallel and even more compelling example. Lead poisoning was initially thought to be an acute disease that eventually cleared and left no sequelae. The pioneering study by Byers and Lord [7] showed that lasting brain damage occurred in lead-poisoned children, but had little immediate consequence. However, increased concerns about environmental lead exposure were raised when Patterson in 1965 [8] reported from geochemical studies that current lead exposures were 100-fold above ‘natural’ levels. This report inspired virtually hundreds of epidemiological studies to examine adverse health effects in children exposed to lead from petrol additives and other sources. Some studies were non-informative, mainly because of imprecise exposure assessment and deficient design. Highly convincing evidence was published in 1979 by Needleman and colleagues [9], who used the amount of lead retained in deciduous teeth as exposure marker to document that mental deficits in children were dose-related. Still, regulatory agencies and expert committees found reasons to raise doubt, and those concerns were of course highlighted by industry groups with an interest in promoting applications of lead.
A contentious atmosphere at scientific meetings stirred sharp debates, where alleged or real methodological flaws were used to raise doubt about scientific conclusions. In order to protect themselves against critique, scientists were particularly careful about mentioning relevant caveats and downplaying the significance of their findings. Linguists call this type of language ‘hedging’ [10]. The lay reader not familiar with this custom may erroneously believe that the results are more uncertain that they really are. When re-reading our own 1980s publications on lead neurotoxicity in children, I discover with dismay that we too frequently used double negations and words like may, maybe and perhaps. In light of current evidence on lead [11], many past reports were unnecessarily hedged and underestimated the impact of environmental lead exposure on brain development. As a result of the apparent uncertainties, the phase-out of lead additives in petrol, the most important global source of lead pollution, proceeded only slowly and has not yet been completed worldwide.
The cases of methylmercury and lead are not unique. Other impacts of neurotoxicity during development were discovered 25 or more years ago. The fetal alcohol syndrome was first described in case series in 1968 [12], and, during the subsequent years, dose-response relationships were observed for neurobehavioural delays associated with maternal alcohol intake [13]. Arsenic poisoning in infants from contaminated milk powder was found to cause permanent brain damage in adolescent survivors in the 1970s [14]. Although this experience was later forgotten, evidence is now emerging that developmental exposure to environmental arsenic may cause neurotoxicity. In regard to polychlorinated biphenyls (PCBs), the first report on cognitive deficits in children exposed from contaminated Great Lakes fish came in 1985 [15]. While PCBs were later banned, a recent report suggests that even lower-level current-day exposures may not be free of developmental neurotoxicity [16].
These studies focused on neurobehavioural effects in childhood or adolescence, but cumulated subclinical damage could potentially trigger or facilitate subsequent development of degenerative nervous system disease. Combined with the concept of reserve capacity [17, 18], developmental programming could involve a decreased resistance against the effects of aging, as suggested by animal models [19].
The experience from the studies of neurotoxicants is summed up in Fig. 1. The very first discoveries of clinical toxicity often occurred under occupational circumstances, later on followed by case reports involving children or pregnant women. As better studies were carried out, pollutant-induced toxicity was documented in children exposed to lower levels during early development. Deficits were thus observed at widespread and low exposures, thereby contributing to a silent epidemic of developmental neurotoxicity [20].
Fig 1. Delayed recognition of developmental effects in humans caused by environmental chemicals.
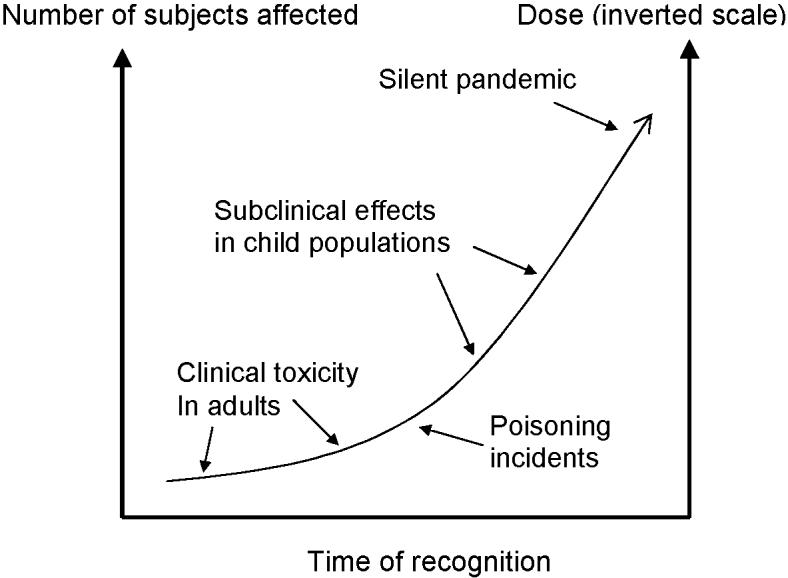
The scientific insight has developed over many years, starting with the first discoveries of occupational poisonings and other intoxications. Later studies of populations with environmental exposures have identified greater effects in subjects exposed during development, and at lower exposure levels, to which larger population groups are exposed. Lead, methylmercury, and several other substances have followed this sequence of events toward the upper right, but available information on most other environmental chemicals currently is limited to the bottom left of the figure. Redrawn from [20].
The new insights were not limited to brain development. One of the most remarkable findings of developmental origins of disease was the discovery in 1971 of clear-cell carcinoma in girls, whose mothers had used diethylstilbestrol during pregnancy [21]. Although a prescription drug, this substance was later used as a standard oestrogen for comparison with other industrial compounds. During the following years, long-term consequences of perinatal exposure to hormones began to be unravelled, e.g., by Bern and colleagues [22]. Skakkebæk reported in 1987 [23] that carcinoma in situ could be found in foetal testicular gonocytes, thus suggesting that this cancer could be of intrauterine origin. Ten years later, vom Saal and colleagues [24] happened upon adverse effects on reproductive system development in mice exposed to minute amounts on bisphenol A, a plastics component. Endocrine disruption due to industrial chemicals has now become a major concern in environmental risk assessment.
The roots of developmental vulnerability can be traced back even further. Measles infection during early pregnancy was first linked to congenital defects by an Australian ophthalmologist in 1941. After the advent of modern immunological techniques in the 1960s, the substantial risk of congenital defects from foetal measles infection was finally appreciated. The placenta had been thought to provide an excellent barrier against foreign compounds, including drugs, but an epidemic of congenital malformations occurred in the early 1960s and was found to be associated with the use of thalidomide against morning sickness in pregnancy. The idealised placental barrier - thought to be impenetrable by foreign compounds - was then realised to be a misconception [25].
Early insights in related fields of epidemiology also include Forsdahl's report in 1977 that infant mortality geographically was linked to subsequent adult mortality. Studies by Barker and colleagues during the following years showed that birth weight was a risk indicator for heart disease, obesity, and type 2 diabetes [26]. Important support for early origin of adult disease came from observations on a cohort born during the Dutch Hunger Winter 1944-1945 [27]. These experiences inspired further epidemiology research emphasising a life-course approach, where early events were linked to subsequent disease development [2]. Birth weight alone is no longer considered the key factor, as it poorly reflects intrauterine events and depends on several factors unrelated to developmental programming. As a recent addition, researchers have begun more systematically to address the toxic impacts on programming, e.g. due to maternal smoking during pregnancy [28].
The initial focus of this research was on the foetal origin of certain diseases in adulthood. However, the foetal period is completed at birth, which occurs at different stages in various species, and developmental processes may continue to be vulnerable to adverse effects also after birth. As suggested by Lucas [29], a preferred term would therefore be developmental programming, or developmental origin of human disease and dysfunction. The most recent years have provided a wealth of experimental data in support of this concept [1] and epigenetic mechanisms, which may be heritable, have been firmly supported as a possible mode of action [30].
Challenges to research
From this brief overview, it is clear that the current shift in paradigm has roots that extend back in time at least 25 years (Table 1). However, because of the dramatic implications of the developmental origins paradigm for both research and public health, the new concept challenged the balance in science between two conflicting obligations [31]. Good science is characterized by proper scepticism, although not to an extent so that no new ideas will be generated. Gullibility should not be so generous that it prevents the distinction between useful and worthless ideas. In regard to our research field, it seems that scientific scepticism in the past took precedence and that absence of evidence erroneously was thought to speak against the early origins hypothesis. Indeed, evidence has been only slowly emerging due to complexities of research. The first warning signs appeared from clinical and epidemiological research, but population studies are time-consuming, and animal models must be scrutinised in light of species differences in developmental stages.
Table 1.
Examples of early information on toxicants already available 20-25 years ago on the developmental origins of disease and organ dysfunction.
| Year | New insight | Reference |
|---|---|---|
| 1968 | Fetal alcohol syndrome first described in a case series | 12 |
| 1968 | Formal recognition that methylmercury caused Minamata disease | 3 |
| 1971 | Clear-cell carcinoma discovered in girls whose mothers used diethylstilbestrol during pregnancy | 21 |
| 1973 | Permanent damage found in survivors of infancy arsenic poisoning from milk powder | 14 |
| 1977 | Infant mortality and low birth weight first linked to adult mortality | 25 |
| 1979 | Dose-related mental deficits reported in children with past lead exposure at ‘background’ levels | 9 |
| 1985 | Cognitive deficits in children prenatally exposed to PCBs from maternal intake of Great Lakes fish | 15 |
| 1986 | Adverse effects observed in children exposed to “background” methylmercury from maternal fish intake during pregnancy | 6 |
| 1987 | Carcinoma in situ documented in fetal testicular tissue | 23 |
In addition to the long time interval between the causative exposure and the outcomes, epidemiologic research was severely hampered by common problems with sensitivity, specificity, precision, and statistical power. Further complicating this research, the exact time of exposure during development could affect the type of outcome. The lack of specificity meant that confounding could be present, and multifactorial causality would need to be considered. Due to all of these obstacles, studies undertaken in this field ran the risk of being both expensive and non-informative. As a further hurdle, such studies often take too long for academic achievement purposes, not to mention short-term grant support.
Epidemiology has sometimes been criticized for showing irrelevant associations (“black-box epidemiology”) [32], and almost unachievable goals for optimal epidemiology research have been promoted, with inspiration from vested industrial interests that could use such criteria to disqualify unwelcome research [33]. Because virtually all research is less than optimal in some way, hedging is a necessary rhetorical means of projecting due caution and modesty, when concluding from a study. In seeking the balance between the innovative but risky, and the correct but trivial, the researcher attempts to make the strongest claim for which there appears to be epistemic authority. In a contentious environment, the risks from making strong statements increase, thereby pushing inference towards the trivial with emphasis on caveats and needs for replication.
Epidemiology often seems an insensitive instrument to reveal links between hazardous environments and human disease. Evidence that, at first, appears to show no adverse effects of an environmental hazard, may later on turn out to be a false negative when larger and more decisive studies are carried out. Although the opposite may also occur, false positive assertions in environmental epidemiology appear to be comparatively rare. In general, epidemiology is inherently biased towards the null hypothesis due to such problems as low statistical power, overemphasis on a 5 % probability level, imprecise exposure data, and short-term or incomplete follow-up [34,35].
Further compounding this bias, scientific tradition demands less uncertainty by more research. When expert groups of scientists develop evaluations of environmental risks, they often recommend further research with emphasis on ideal practices of epidemiology, although unlikely to be feasible within a reasonable time span. Partially as a result of this desire for further information before reaching a firm conclusion, replication has been hailed as a mainstay in science, thereby augmenting the inertia. A review of published papers in environmental health journals will reveal that they primarily deal with a limited, rather stable list of pollutants. Lead is one of them, and PubMed now lists thousands of scientific publications on the human health effects of environmental lead exposure. Other important toxicants are poorly studied in comparison. Thus, both research attention and funding are directed away from new and emerging problems.
Late science lessons
Now that the notion of early origins is taking shape, it becomes easier to identify its roots and how the strengths and weaknesses of this field of research were misread, thereby delaying recognition of paradigm change. Scientific rigor has often been misunderstood as the unrealistic requirement for controlled studies that could furnish virtual statistical certainty. Likewise, inconclusive studies have been routinely labelled ‘negative’, instead of ‘non-informative’. Lead, mercury, and other environmental hazards did not become more toxic with time, but better science began to document effects at lower exposure levels; thresholds for toxicity may be very low, if they exist. After some latency, consensus was reached, and exposure limits were eventually revised downwards. Unfortunately, the delay in preventing hazardous exposures put large population groups at risk of developmental toxicity, and likely continues to do so today. This experience therefore challenges the traditional ideal of the sceptical ivory-tower scientist, who profusely hedges all his tentative conclusions.
The developmental origins paradigm suggests the need for a different emphasis in research, perhaps a new paradigm of science. A revised perspective could enjoy inspiration from the precautionary principle, which has triggered much new thinking on prevention in environmental health, but not yet influenced the practice and interpretation of environmental health science [34,36]. One consequence is that science needs to shed the traditional and monocular emphasis whether confidence limits include the possibility of no effect. This is far from being the only interest, although oftentimes this issue is considered the main success criterion. We need to scrutinise one or more worst-case scenarios: How serious could the adverse effects be, and how large an effect can be reasonably ruled out?
Another major consideration is of paramount importance. In interpreting research results, we must recognise that a phenomenon may well exist, even if we cannot see it. We must ask ourselves: What could be known, given our study opportunities and methodologies? Only then will we be able to properly judge new ideas and allow a promising paradigm to grow. As a related charge, we should critically consider the scientific tradition of replication and identify new ways to inspire innovative hypothesis testing.
Had these issues been considered 25 years ago, perhaps the paradigm shift that we are now witnessing would today have been a matter of history, from which prevention would already have benefited tremendously. Figure 2 illustrates how early research and precautionary action may be logically linked to subsequent risk assessment and evidence-based action. The experience from developmental programming indicates that a better and earlier connection between research and prevention is crucial, and that prudent intervention may be warranted in the absence of convincing evidence.
Fig. 2. Proposed linkage between environmental health research and prevention.
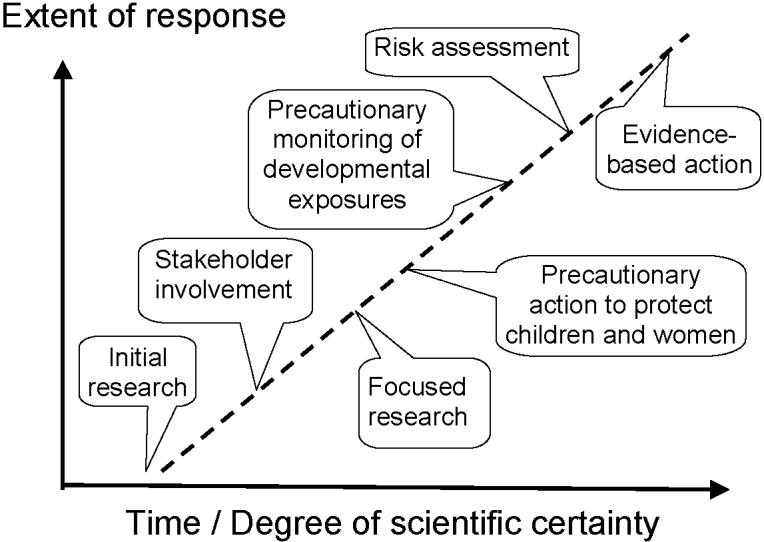
When a potential environmental toxicant issue is first suspected, initial research should aim at providing information on key issues that may lead it to a preliminary analysis, and this evidence should be combined with data from stakeholders, followed by appropriate precautionary actions as indicated, including subsequent monitoring and research. These efforts may eventually provide the basis for a formal risk assessment that will pave the way for evidence-based prevention that may strengthen or weaken steps already taken. Redrawn from [35].
Conclusions
With early foundations already laid at least 25 years ago, the paradigm of developmental origins of human disease and organ dysfunction has been slow to develop. The delayed recognition undoubtedly resulted in major costs to society due to the lack of intervention that could have been reasonably justified long ago. Now that this paradigm is gaining ground, its full prevention implications must be recognised. In addition, this experience suggests that undue scepticism and demands for endless replication should be countered by prudent and responsible inference that paves the way for more rapid recognition of early warning signs when human health and the environment are at risk.
Acknowledgments
The author's research is supported by the U.S. National Institute of Environmental Health Sciences (ES09797 and ES11687) and by the EU through its Sixth Framework Programme for RTD (contract no FOOD-CT-2006-016253, PHIME). The contents of this paper are solely the responsibility of the author and do not represent the official views of the funding agencies, which are not liable for any use that may be made of the information contained therein.
References
- 1.Heindel JJ. Role of exposure to environmental chemicals in the developmental basis of reproductive disease and dysfunction. Semin Reprod Med. 2006;24:168–77. doi: 10.1055/s-2006-944423. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Shlomo Y, Kuh D. A life course approach to chronic disease epidemiology: conceptual models, empirical challenges and interdisciplinary perspectives. Int J Epidemiol. 2002;31:285–93. [PubMed] [Google Scholar]
- 3.Harada M. Minamata disease: methylmercury poisoning in Japan caused by environmental pollution. Crit Rev Toxicol. 1995;25:1–24. doi: 10.3109/10408449509089885. [DOI] [PubMed] [Google Scholar]
- 4.Engleson G, Herner T. Alkyl mercury poisoning, Acta Paediatr. 1952;41:289–94. doi: 10.1111/j.1651-2227.1952.tb17033.x. [DOI] [PubMed] [Google Scholar]
- 5.Joint Expert Committee on Food Additives. World Health Organization . Evaluation of certain food additives and contaminants: twenty-second report of the Joint FAO/WHO JECFA. Geneva: 1978. [PubMed] [Google Scholar]
- 6.National Research Council . Toxicological Effects of Methylmercury. National Academy Press; Washington: 2000. [Google Scholar]
- 7.Byers RL, Lord EE. Late effects of lead poisoning on mental development. Am J Dis Child. 1943;66:471–83. [Google Scholar]
- 8.Patterson CC. Contaminated and natural lead environments of man. Arch Environ Health. 1965;11:344–60. doi: 10.1080/00039896.1965.10664229. [DOI] [PubMed] [Google Scholar]
- 9.Needleman HL, Gunnoe C, Leviton A, Reed R, Peresie H, Maher C, et al. Deficits in psychologic and classroom performance of children with elevated dentine lead levels. N Engl J Med. 1979;300:689–95. doi: 10.1056/NEJM197903293001301. [DOI] [PubMed] [Google Scholar]
- 10.Hyland K. Hedging in scientific research articles. John Benjamins; Amsterdam: 1998. [Google Scholar]
- 11.Lanphear BP, Hornung R, Khoury J, Yolton K, Baghurst P, Bellinger DC, et al. Low-level environmental lead exposure and children's intellectual function: an international pooled analysis. Environ Health Perspect. 2005;113:894–9. doi: 10.1289/ehp.7688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones KL, Smith DW. Recognition of the fetal alcohol syndrome in early infancy. Lancet. 1973;2:999–1001. doi: 10.1016/s0140-6736(73)91092-1. [DOI] [PubMed] [Google Scholar]
- 13.Streissguth AP, Sampson PD, Barr HM. Neurobehavioral dose-response effects of prenatal alcohol exposure in humans from infancy to adulthood. Ann N Y Acad Sci. 1989;562:145–58. doi: 10.1111/j.1749-6632.1989.tb21013.x. [DOI] [PubMed] [Google Scholar]
- 14.Dakeishi M, Murata K, Grandjean P. Lessons from arsenic poisoning of infants due to contaminated dried milk: a review. Environ Health. 2006;5:31. doi: 10.1186/1476-069X-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobson SW, Fein GG, Jacobson JL, Schwartz PM, Dowler JK. The effect of intrauterine PCB exposure on visual recognition memory. Child Dev. 1985;56:853–60. [PubMed] [Google Scholar]
- 16.Jacobson JL, Janisse J, Banerjee M, Jester J, Jacobson SW, Ager JW. A benchmark dose analysis of prenatal exposure to polychlorinated biphenyls. Environ Health Perspect. 2002;110:393–8. doi: 10.1289/ehp.02110393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reuhl KR. Delayed expression of neurotoxicity: the problem of silent damage. Neurotoxicology. 1991;12:341–6. [PubMed] [Google Scholar]
- 18.Grandjean P. Effects on reserve capacity, significance for exposure limits. Sci Total Environ. 1991;101:25–32. doi: 10.1016/0048-9697(91)90099-z. [DOI] [PubMed] [Google Scholar]
- 19.Barlow BK, Cory-Slechta DA, Richfield EK, Thiruchelvam M. The gestational environment and Parkinson's disease: evidence for neurodevelopmental origins of a neurodegenerative disorder. Reprod Toxicol. 2007;23:457–70. doi: 10.1016/j.reprotox.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 20.Grandjean P, Landrigan PJ. Developmental neurotoxicity of industrial chemicals. Lancet. 2006;368:2167–78. doi: 10.1016/S0140-6736(06)69665-7. [DOI] [PubMed] [Google Scholar]
- 21.Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med. 1971;284:878–81. doi: 10.1056/NEJM197104222841604. [DOI] [PubMed] [Google Scholar]
- 22.Mori T, Nagasawa H, Bern HA. Long-term effects of perinatal exposure to hormones on normal and neoplastic mammary growth in rodents: a review. J Environ Pathol Toxicol. 1979;3:191–205. [PubMed] [Google Scholar]
- 23.Skakkebaek NE, Berthelsen JG, Giwercman A, Muller J. Carcinoma-in-situ of the testis: possible origin from gonocytes and precursor of all types of germ cell tumours except spermatocytoma. Int J Androl. 1987;10:19–28. doi: 10.1111/j.1365-2605.1987.tb00161.x. [DOI] [PubMed] [Google Scholar]
- 24.vom Saal FS, Timms BG, Montano MM, Palanza P, Thayer KA, Nagel SC, et al. Prostate enlargement in mice due to fetal exposure to low doses of estradiol or diethylstilbestrol and opposite effects at high doses. Proc Natl Acad Sci USA. 1997;94:2056–61. doi: 10.1073/pnas.94.5.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dally A. Thalidomide: was the tragedy preventable? Lancet. 1998;351:1197–9. doi: 10.1016/S0140-6736(97)09038-7. [DOI] [PubMed] [Google Scholar]
- 26.Barker DJP. Fetal origins of cardiovascular and lung disease. Marcel Dekker; New York: 2001. [Google Scholar]
- 27.Susser M, Stein Z. Timing in prenatal nutrition: a reprise of the Dutch Famine Study. Nutr Rev. 1994;52:84–94. doi: 10.1111/j.1753-4887.1994.tb01395.x. [DOI] [PubMed] [Google Scholar]
- 28.Gillman MW, Barker D, Bier D, Cagampang F, Challis J, Fall C, et al. Meeting report on the 3rd International Congress on Developmental Origins of Health and Disease (DOHaD) Pediatr Res. 2007;61:625–9. doi: 10.1203/pdr.0b013e3180459fcd. [DOI] [PubMed] [Google Scholar]
- 29.Lucas A. Programming by early nutrition in man. Ciba Found Symp. 1991;56:38–50. [PubMed] [Google Scholar]
- 30.Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr. 2007;27:363–88. doi: 10.1146/annurev.nutr.27.061406.093705. [DOI] [PubMed] [Google Scholar]
- 31.Grandjean P. Seven deadly sins of environmental epidemiology and the virtues of precaution. Epidemiology. 2007 doi: 10.1097/EDE.0b013e31815be031. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taubes G. Epidemiology faces its limits. Science. 1995;269:164–9. doi: 10.1126/science.7618077. [DOI] [PubMed] [Google Scholar]
- 33.Ong EK, Glantz SA. Constructing “sound science” and “good epidemiology”: tobacco, lawyers, and public relations firms. Am J Publ Health. 2001;91:1749–57. doi: 10.2105/ajph.91.11.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grandjean P, Bailar JC, Gee D, Needleman HL, Ozonoff DM, Richter E, et al. Implications of the Precautionary Principle for research and policy-making. Am J Ind Med. 2004;45:382–5. doi: 10.1002/ajim.10361. [DOI] [PubMed] [Google Scholar]
- 35.Bailar JC. How to distort the scientific record without actually lying: truth, and the arts of science. Eur J Oncol. 2006;11:217–24. [Google Scholar]
- 36.Grandjean P. Implications of the precautionary principle for primary prevention and research. Annu Rev Publ Health. 2004;25:199–223. doi: 10.1146/annurev.publhealth.25.050503.153941. [DOI] [PubMed] [Google Scholar]