

Abstract
17β-Estradiol (E2) has been implicated to be neuroprotective in a variety of neurodegenerative disorders, although the mechanism remains poorly understood. The current study sheds light on this issue by demonstrating that low physiological levels of E2 protects the hippocampus CA1 against global cerebral ischemia by preventing elevation of dickkopf-1 (Dkk1), an antagonist of the Wnt/β-catenin signaling pathway, which is a principal mediator of neurodegeneration in cerebral ischemia and Alzheimer's disease. E2 inhibition of Dkk1 elevation correlated with a reduction of phospho-β-catenin and elevation of nuclear β-catenin levels, as well as enhancement of Wnt-3, suggesting E2 activation of the Wnt/β-catenin signaling pathway. In agreement, the β-catenin downstream prosurvival factor, survivin, was induced by E2 at 24 and 48 h after cerebral ischemia, an effect observed only in surviving neurons because degenerating neurons lacked survivin expression. E2 suppression of Dkk1 elevation was found to be caused by attenuation of upstream c-Jun N-terminal protein kinase (JNK)/c-Jun signaling, as E2 attenuation of JNK/c-Jun activation and a JNK inhibitor significantly blocked Dkk1 induction. Tau hyperphosphorylation has been implicated to have a prodeath role in Alzheimer's disease and cerebral ischemia, and E2 attenuates tau hyperphosphorylation. Our study demonstrates that tau hyperphosphorylation is strongly induced after global cerebral ischemia, and that E2 inhibits tau hyperphosphorylation by suppressing activation of the JNK/c-Jun/Dkk1 signaling pathway. Finally, exogenous Dkk1 replacement via intracerebroventricular administration completely reversed E2-induced neuroprotection, nuclear β-catenin induction, and phospho-tau attenuation, further suggesting that E2 inhibition of Dkk1 is a critical mechanism underlying its neuroprotective and phospho-tau regulatory effects after cerebral ischemia.
Keywords: 17β-estradiol, cerebral ischemia, hippocampus, survivin, Alzheimer's disease, neuronal cell death
Introduction
The steroid hormone, 17β-estradiol (E2), has been implicated to be neuroprotective in a variety of neurodegenerative disorders such as cerebral ischemia, Alzheimer's disease, and Parkinson's disease (Brann et al., 2007). With respect to cerebral ischemia, it is well known that women are “protected” against stroke relative to men, at least until menopause (Roquer et al., 2003; Murphy et al., 2004; Niewada et al., 2005), and that after menopause, women reportedly have a worse stroke outcome compared with males (Di Carlo et al., 2003; Niewada et al., 2005). Furthermore, E2 has been shown to be neuroprotective in rodent models of both global and focal cerebral ischemia (Simpkins et al., 1997; Miller et al., 2005; Brann et al., 2007). E2 has also been suggested to delay onset of Alzheimer's disease and to be neuroprotective in Parkinson's disease (Henderson, 2006; Brann et al., 2007).
The mechanism of E2 neuroprotection in cerebral ischemia is unclear, but a role for mediation by both estrogen receptor-α (ER-α) and ER-β has been implicated (Miller et al., 2005), as well as ER-induced genomic and nongenomic signaling (Alkayed et al., 2001; Jover-Mengual et al., 2007) and induction of antiapoptotic proteins such as Bcl (Jover et al., 2002; Brann et al., 2007). In Alzheimer's disease, E2 also suppresses hyperphosphorylation of tau, a principal component in neurofibrillary tangle formation (Wen et al., 2004; Alvarez-de-la-Rosa et al., 2005; Simpkins et al., 2005). Hyperphosphorylation of tau also occurs in cerebral ischemia (Wen et al., 2004), which may explain a 4- to 12-fold higher prevalence of dementia that is observed in stroke patients (Tatemichi et al., 1992).
Just how E2 could be neuroprotective against a variety of neurodegenerative diseases is unclear. It is possible that it modulates a common factor that induces neurodegeneration in the various disorders. Along these lines, recent studies have provided evidence that neurodegeneration in stroke, Alzheimer's disease, and epilepsy, is mediated by induction of the neurodegenerative factor dickkopf-1 (Dkk1) (Caricasole et al., 2004; Cappuccio et al., 2005; Scali et al., 2006; Busceti et al., 2007). Dkk1 acts as an antagonist of the canonical Wnt pathway by binding to the Wnt receptor, thus preventing Wnt activation of Wnt/β-catenin signaling. Recent studies showed that Dkk1 is strongly induced in the hippocampal CA1 after global cerebral ischemia, and administration of Dkk1 antisense oligonucleotides protects the CA1 against cerebral ischemia-induced neuronal death (Cappuccio et al., 2005). Dkk1 has also been shown to induce tau hyperphosphorylation, and Dkk1 is expressed in degenerating neurons in the brain from Alzheimer's disease patients (Caricasole et al., 2004; Scali et al., 2006).
Based on these findings, we hypothesized that E2 may be neuroprotective in neurodegenerative disorders and regulate tau phosphorylation through regulation of Dkk1. Our study shows that E2 prevents cerebral ischemia induction of Dkk1 in the CA1 region, activates β-catenin signaling, and induces the prosurvival factor, survivin. Our study further demonstrates that E2 prevention of Dkk1 elevation is caused by inhibition of upstream JNK/c-Jun signaling, and that E2 attenuates tau hyperphosphorylation via attenuation of the JNK-c-Jun/Dkk1 pathway.
Materials and Methods
Global cerebral ischemia.
Adult Sprague Dawley female rats were bilaterally ovariectomized 1 week before performance of global cerebral ischemia. At the time of ovariectomy, placebo or 17β-estradiol time-release pellets (0.025 mg; 21 d release pellet) (Innovative Research of America) were implanted subcutaneously in the upper midback region under the skin. For cerebral ischemia, all animals (excepting sham control) underwent four-vessel occlusion global cerebral ischemia, performed as described previously (Pulsinelli and Brierley, 1979; Pulsinelli and Buchan, 1988; Zhang et al., 2006a). Briefly, animals were anesthetized using chloral hydrate (350 mg/kg, i.p.), and both common carotid arteries (CCAs) of the rat were separated. A SILASTIC ligature was placed loosely around each artery without interrupting the blood flow, and the incision was then closed. Immediately after this procedure, both vertebral arteries at the level of the alar foramina were permanently occluded using electrocautery. After 24 h recovery, the bilateral CCAs were exposed under light anesthesia with 3% isoflurane and occluded with aneurysm clips to induce 10 min forebrain ischemia. Animals that lost their righting reflex within 30 s and whose pupils were dilated and unresponsive to light during cerebral ischemia were selected for the experiments, because this indicates successful global ischemia. The clips were then removed and the blood flow through the arteries was confirmed before the wound was sutured. Rectal temperature was maintained at 36.5–37.5°C throughout the experiment with a thermal blanket. The animals of the sham group underwent identical procedures except that the CCAs were simply exposed, but there were no occlusion.
Histochemical analysis of neuroprotection.
Histological examination of the ischemic brain was performed by neuronal-specific nuclear protein (NeuN) and Fluoro-Jade B as described previously by our laboratory (Zhang et al., 2006b; Wakade et al., 2008). The animals were anesthetized with chloral hydrate and perfused with 0.9% saline containing 10 U/ml of heparin, followed by fixation with cold 4% paraformaldehyde in 0.1 m phosphate buffer (PB). Brains were postfixed in the same fixative overnight at 4°C and cryoprotected with 30% sucrose in 0.1 m PB, pH 7.4, for 24–36 h. Coronal sections (20 μm) were collected through the entire dorsal hippocampus (∼2.5–4.5 mm posterior from bregma, ∼100 sections per brain) from animals killed at 7 d after ischemia or sham operation, and every fifth section was collected and stained. Briefly, sections were washed for 10 min in PBS followed by 0.1% PBS-Triton X-100 for additional 10 min. After incubation with blocking solutions containing 10% donkey for 1 h at room temperature in PBS containing 0.1% Triton X-100, sections were exposed overnight at 4°C to mouse anti-NeuN monoclonal antibody (1:500; Millipore). Sections were washed for 4 × 10 min, followed by incubation with Alexa Fluor 594 donkey anti-mouse antibody (1:500; Invitrogen) for 1 h at room temperature. Sections were then washed in PBS two times for 5 min each and then incubated for ∼20 min with fluorescent Fluro-Jade B (AG310; Millipore) stain diluted in PBS as recommended by the manufacturer. Staining solution was removed and sections washed with PBS-Triton X-100, then PBS, and finally with water, and mounted using water-based mounting medium containing antifading agents. Images were captured on an LSM510 Meta confocal laser microscope (Carl Zeiss) as described previously by our laboratory (Wakade et al., 2008).
The number of NeuN-positive CA1 neurons per 1 mm length of the medial CA1 pyramidal cell layer was counted bilaterally in five sections per animal. Cell counts from the right and left hippocampus on each of the five sections were averaged to provide the mean value. A mean ± SE was calculated from the data in each group and statistical analysis performed as described below.
DAB staining.
For DAB staining, sections were incubated with 10% normal goat/horse serum in PBS containing 0.1% Triton X-100 and 0.3% H2O2 for 1 h at room temperature to block nonspecific surfaces. Sections were then incubated with the primary antibodies overnight at 4°C in PBS containing 0.1% Triton X-100. The antibodies used were as follows: polyclonal rabbit anti-p-β-catenin (1:100; Cell Signaling Technology), anti-survivin (1:100; Cell Signaling Technology), anti-Wnt3 (1:50; Santa Cruz Biotechnology), and anti-phospho-tau (1:2000; PHF1; gift from Dr. Peter Davies, Albert Einstein College of Medicine, Bronx, NY). Afterward, sections were washed with the same buffer followed by incubation with secondary biotinylated goat anti-rabbit or horse anti-mouse antibodies (Vector Laboratories) at a dilution of 1:200 in PBS containing 0.1% Triton X-100 for 1 h at room temperature. Sections were then washed, followed by incubation with ABC reagents for 1 h at room temperature in the same buffer. Sections were rinsed in the same buffer and incubated with DAB reagent according to the manufacturer's instructions (Vector Laboratories) for 2–10 min. After DAB incubation, sections were washed briefly with distilled water and dehydrated in graded alcohols, cleared in xylene, and mounted using xylene-based mounting medium. Images were captured on an Axiophot-2 visible/fluorescence microscope using an AxioVision4Ac software system (Carl Zeiss).
Double/triple immunofluorescence staining.
Coronal sections were incubated with 10% normal donkey serum for 1 h at room temperature in PBS containing 0.1% Triton X-100 followed by incubation with appropriate primary antibodies overnight at 4°C in the same buffer. The following primary antibodies were used in different combinations: anti-NeuN (1:500; Millipore), anti-Dkk1 (1:50; Santa Cruz Biotechnology), anti-phospho-c-Jun (anti-p-c-Jun; 1:50; Santa Cruz Biotechnology), anti-β-catenin (1:100; Abcam), and anti-survivin (1:100; Cell Signaling Technology). After primary antibody incubation, sections were washed for 4 × 10 min at room temperature followed by incubation with Alexa Fluor594/647 donkey anti-mouse/rabbit, Alexa-Fluor488 donkey anti-rabbit/mouse, and Alexa-Fluor488 donkey anti-goat secondary antibody (1:500; Invitrogen) for 1 h at room temperature. Sections were then washed with PBS containing 0.1% Triton X-100 four times for 10 min each followed by three times for 5 min each with PBS and briefly with water, and then mounted with water-based mounting medium containing antifading agents (Biomeda; Thermo Fischer Scientific). A simultaneous examination of negative controls (omission of primary antibody) confirmed the absence of nonspecific immunofluorescent staining, cross-immunostaining, or fluorescence bleed-through.
Confocal microscopy and image analysis.
All of the double- and triple-labeled images were captured on an LSM510 Meta confocal laser microscope (Carl Zeiss) using either a 5× or 40× oil-immersion Neofluor objective (numerical aperture, 1.3) with the image size set at 1024 × 1024 pixels. The following excitation laser/emission filter settings were used for various chromophores: an argon/2 laser was used for Alexa Fluor 488, with maximum excitation at 490 nm and emission in the range of 505–530 nm, an HeNe1 laser was used for Alexa Fluor 594 with maximum excitation at 543 nm and emission in the range of 568–615 nm, and an HeNe2 laser was used for Alexa Fluor 647 with maximum excitation at 633 nm and emission in the range of 650–800 nm. The captured images were viewed and analyzed using LSM510 Meta imaging software.
Brain homogenates and subcellular fractionations.
For brain tissue preparation, rats were killed under anesthesia at the time points stated in the experiments. Whole brains were removed and the hippocampal CA1 region microdissected from both sides of the hippocampal fissure and immediately frozen in liquid nitrogen. Tissues were homogenized in ice-cold homogenization medium consisting of (in mm) 50 HEPES, pH 7.4, 150 NaCl, 12 β-glycerophosphate, 3 dithiotheitol (DTT), 2 sodium orthovanadate (Na3VO4), 1 EGTA, 1 NaF, 1 phenylmethylsulfonyl fluoride (PMSF), 1% Triton X-100, and inhibitors of proteases and enzymes (0.5 mm PMSF, 10 μg/ml each of aprotinin, leupeptin, and pepstatin A) with a Teflon-glass homogenizer. The homogenates were centrifuged at 15,000 × g for 30 min at 4°C; supernatants were collected and stored at −80°C for use. When necessary, cytosol fractions and nuclear fractions were extracted with some modifications to a previously described procedure (Ogita and Yoneda, 1994). Briefly, tissues were homogenized in ice-cold buffer A containing (in mm) 10 HEPES, pH 7.9, 1 DTT, 1 Na3VO4, 1 4-nitrophenyl phosphate (PNPP), and inhibitors of proteases and enzymes. The homogenates were allowed to swell on ice for 10 min. Then, tubes were vigorously vortexed for 30 s and centrifuged at 800 × g for 10 min after the addition of NP-40 (0.6% of total solution). Supernatants were centrifuged at 15,000 × g for 30 min at 4°C. The nuclear pellets were washed three times with buffer A and resuspended in buffer B [(in mm) 20 HEPES, pH 7.9, 400 NaCl, 20% glycerine, 1 DTT, 1 Na3VO4, 1 PNPP] with inhibitors of proteases and enzymes, and then the tubes were vigorously rocked at 4°C for 30 min. After centrifugation at 12,000 × g for 15 min, the nuclear extracts were aliquoted and frozen in liquid nitrogen and stored at −80°C until use. The protein concentrations were determined by a Lowry protein assay kit with bovine serum albumin as standard.
Western blot analysis.
For Western blot analysis, samples were mixed with loading buffer and boiled for 5 min. An aliquot of 20–50 μg of protein was separated by 4–20% SDS-PAGE. Proteins were transferred to nitrocellulose membrane, blocked for 3 h, and incubated with 1° antibodies against Dkk1 (1:200; Santa Cruz Biotechnology), Wnt3 (1:200; Santa Cruz Biotechnology), p-β-catenin (1:1000; Cell Signaling Technology), β-catenin (1:1000; Abcam), survivin (1:1000; Cell Signaling Technology), p-JNK(1:200; Santa Cruz Biotechnology), p-c-Jun (1:200; Santa Cruz Biotechnology), and PHF1 (1:5000; gift from Dr. Peter Davies) at 4°C overnight. β-actin and NeuN were used as loading controls for the total cell protein and nuclear protein, respectively. The membrane was then washed with TBS containing Tween 20 to remove unbound antibody, followed by incubation with 2° HRP-conjugated or Alexa Fluor 680 goat anti-rabbit/mouse IgG for 1–2 h at room temperature.
Bound proteins were visualized using the Odyssey Imaging System (LI-COR Bioscience) and semiquantitative analysis of the bands were performed with the ImageJ analysis software (version 1.30v; Wayne Rasband, National Institutes of Health, Bethesda, MD). To quantitate hippocampal protein abundance, band densities for the indicated phosphoprotein were corrected for variations in loading and normalized to the corresponding band densities for total protein signals, respectively, and the indicated total proteins were expressed relative to actin or NeuN signals. Normalized means were then expressed as fold changes of the corresponding value for control (sham operated) animals. A mean ± SE was calculated from the data from all of the animals for graphical presentation and statistical comparison.
Administration of drugs.
The recombinant human Dkk1 protein (rhDkk1; R&D Systems) was dissolved in sterile PBS at a concentration of 1 μg/μl. Five microliters of the solutions were administrated to both of the cerebral ventricles at 12 h after ischemia through intracerebroventricular injection. Infusion of sterile PBS served as vehicle control. For intracerebroventricular injection, the rats were placed on ear bars of a stereotaxic instrument under anesthesia. Drug infusion was performed using a Hamilton microsyringe at a rate of 1 μl/min to the cerebral ventricle (from the bregma: anteroposterior, −0.8 mm; lateral, 1.5 mm; depth, 3.5 mm). Anthra(1–9-cd) pyrazol-6(2H)-one (SP600125) was dissolved in PPCES vehicle (30% polyethylene glycol–400/20% polypropylene glycol/15% cremophor EL/5% ethanol/30% saline) as described previously (Bennett et al., 2001), and was treated by tail-vein injection 15 min before ischemia at a dose of 10 mg/kg.
Statistical analysis.
Statistical analysis was performed using two-way ANOVA followed by Student–Newman–Keuls test. When only two groups were compared, a Student's t test was used. Statistical significance was accepted at the 95% confidence level (p < 0.05). Data are expressed as mean ± SE.
Results
E2 protects the hippocampus CA1 region from global cerebral ischemia-induced neuronal damage and cell death
We first examined the neuroprotective effect of low serum levels of E2 in the hippocampus CA1 after global cerebral ischemia (Fig. 1 A,B). Measurement of serum E2 by radioimmunoassay revealed that the subcutaneous E2 pellets produced low physiological serum levels of E2 (10.0 ± 3.6 pg/ml), which are equivalent to the low E2 serum levels observed in intact young adult rats on diestrus I of the ovulatory cycle (Gore et al., 2000). As shown in Figure 1, A and B, representative NeuN staining of hippocampal sections at 7 d after global cerebral ischemia demonstrates a profound protective effect of E2 on hippocampus CA1 (Fig. 1 A), an effect that was shown to be highly statistically significant (p < 0.01 vs placebo) (Fig. 1 B). Histochemical staining for Fluoro-Jade B, a marker of neuronal degeneration, revealed that cerebral ischemia induced significant Fluoro-Jade B staining in hippocampus CA1 compared with sham at 7 d after cerebral ischemia, suggesting that global cerebral ischemia induced marked neuronal degeneration in the CA1 region (Fig. 1 A). E2 treatment essentially abolished Fluoro-Jade B staining in the CA1 region, suggesting that E2 strongly attenuates cerebral ischemia-induced neuronal degeneration.
Figure 1.
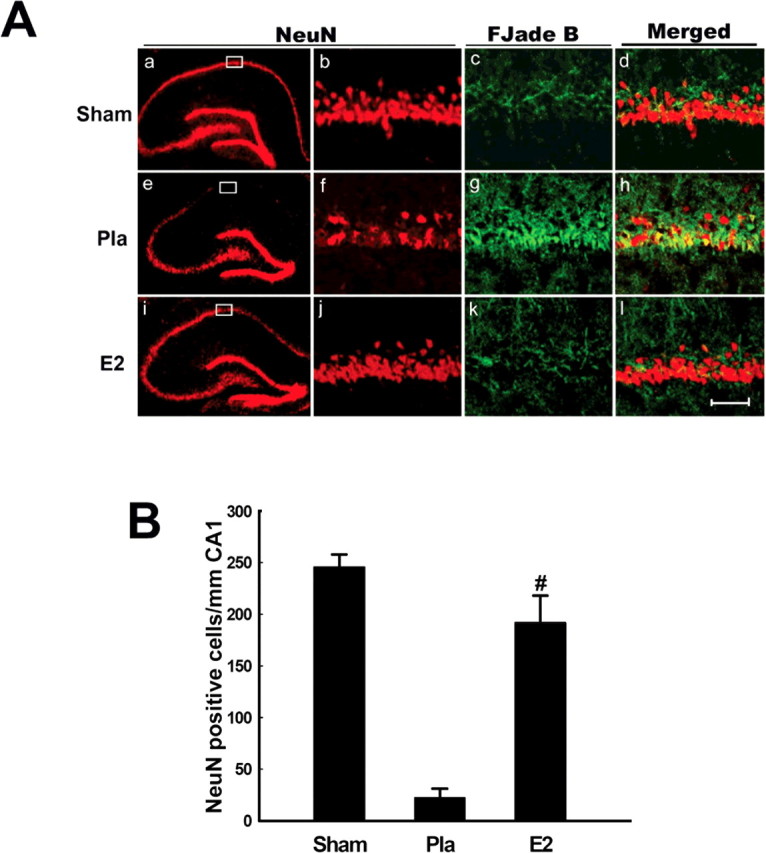
Aa–Al, NeuN immunostaining and Fluoro-Jade B staining of representative coronal brain sections at the level of the dorsal hippocampus at 7 d after reperfusion from sham, placebo (Pla)- and E2-treated female ovariectomized rats subjected to global cerebral ischemia. Global ischemia induced significant neuronal degeneration and neuronal cell loss in the CA1 pyramidal cell layer, with little or no cell loss apparent in CA3 or DG. E2 treatment afforded nearly complete protection of the hippocampus CA1 from global cerebral ischemia-induced neuronal degeneration and neuronal cell loss. Boxed areas in the left column are shown at higher magnification in right column (magnification, 40×). Scale bar, 50 μm. B, Neuronal counts of NeuN-positive neurons. Counts refer to CA1 neurons of sham-, placebo-, and 17β-estradiol-treated ovariectomized rats. Note the significant neuroprotection by E2. Values are mean ± SEM of determinations from seven individual rats. # p < 0.01 versus placebo.
17β-Estradiol attenuates cerebral ischemia-induced elevation of Dkk1 in hippocampus CA1 region
We next examined the regulation of the Wnt/β-catenin antagonist Dkk1 after cerebral ischemia. Figure 2 A shows representative Western blot analysis results, B shows quantification and statistical analysis of the Western blot results from all animals, and C shows immunohistochemical results from representative animals that confirms the Western blot data and illustrate that changes in Dkk1 occur in pyramidal neurons of the CA1. As shown in Figure 2 A–C, Dkk1 protein levels were extremely low in the hippocampal CA1 region of Sham control animals as indicated by Western blot analysis (Fig. 2 A,B) and by immunohistochemistry (Fig. 2 C). In contrast, global cerebral ischemia (placebo) induced a robust elevation of Dkk1 in the hippocampal CA1 region at 24 and 48 h after cerebral ischemia compared with the sham controls and earlier placebo time points (Fig. 2 A–C). As shown in Figure 2 C, double immunohistochemistry showed that cerebral ischemia induction of Dkk1 was in neurons in the CA1 region, because Dkk1 and NeuN signals were strongly colocalized. Of significant interest, E2 treatment dramatically attenuated the cerebral ischemia-induced elevation of Dkk1 at 24 and 48 h (Fig. 2 A–C). As shown in supplemental Figure 1, A and B (available at www.jneurosci.org as supplemental material), E2 had no effect on Dkk1 levels in the hippocampus CA1 of sham animals. This further confirms that E2 is acting specifically to suppress cerebral ischemia-induced Dkk1 elevation.
Figure 2.

A,B, Effect of 17β-estradiol on Dkk1 protein levels in hippocampus CA1 after global cerebral ischemia. Values are mean ± SEM of determinations from five to six individual rats and expressed as fold change versus sham control. Pla, Placebo; R, reperfusion. *p < 0.05 versus sham control; # p < 0.05 versus placebo treatment group. Ca–Ci, Confocal analysis of NeuN and Dkk1 immunostaining in hippocampus CA1 at 24 h after global cerebral ischemia (magnification, 40×). Scale bar, 50 μm.
It is well known that the hippocampal CA3 region and dentate gyrus (DG) are much less sensitive to cerebral ischemia damage as is the CA1 region. We thus examined whether Dkk1 induction in these regions was correspondingly less as to what we observed in the hippocampal CA1 region after cerebral ischemia. Immunohistochemical analysis showed that Dkk1 induction was very low in the hippocampal CA3 region and DG at 24 h after cerebral ischemia, a time point at which Dkk1 was strongly induced in the hippocampal CA1 region (supplemental Fig. 2A,B,D, available at www.jneurosci.org as supplemental material). Interestingly, Dkk1 was shown to be induced moderately in the cerebral cortex after global cerebral ischemia, where some cell death is known to occur after global cerebral ischemia (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material). Although induction of Dkk1 was less in cortex, the Dkk1 induction was still abolished by E2 treatment, suggesting that E2 protected cells in the cortex with similar efficacy as observed in the hippocampal CA1 region.
17β-Estradiol enhances Wnt3 levels in the hippocampus CA1 region after global cerebral ischemia
We next examined whether E2 regulates Wnt3, a ligand for the Wnt receptor that is highly expressed in the hippocampus CA1 region (Salinas and Nusse, 1992) after cerebral ischemia. As shown in Fig, 3 A–C, sham controls showed low Wnt3 levels in the hippocampus CA1, with cerebral ischemia (placebo) having no significant effect on Wnt3 levels. In contrast, E2 induced a highly significant increase in Wnt3 levels at 24 and 48 h after cerebral ischemia (Fig. 3 A,B). The E2 upregulation of Wnt3 levels was confirmed by immunohistochemistry, in which E2 strongly enhanced Wnt3 staining intensity in the hippocampus CA1 at 24 h after cerebral ischemia (Fig. 3 C). E2 only slightly increased Wnt3 in the CA1 of nonischemic sham control animals, an effect that was not statistically significant (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material).
Figure 3.

A, B, 17β-Estradiol enhances Wnt3 protein levels in hippocampus CA1 after global cerebral ischemia. Values are mean ± SEM of determinations from five to six individual rats expressed as fold change versus sham control. Pla, Placebo; R, reperfusion. # p < 0.05 versus the Pla group at the same time point. Ca–Cf, DAB immunostaining for Wnt3 in hippocampus CA1 at 24 h after global cerebral ischemia. Results are representative of staining observed in five (n = 5) individual animals per group. Ca–Cc, Magnification is 5×. Scale bar, 200 μm. Cd–Cf, Magnification is 40×. Scale bar, 50 μm.
17β-Estradiol prevents ischemia-induced downregulation of Wnt/β-catenin signaling in the hippocampus CA1 region after global cerebral ischemia
Because E2 decreased Dkk1 and enhanced Wnt3, we next examined whether Wnt/β-catenin signaling was significantly enhanced by E2 after cerebral ischemia. Phosphorylation of β-catenin targets it for degradation, an effect reversed by activation of the Wnt signaling pathway. Activation of Wnt signaling pathway thus leads to nuclear retention of β-catenin, allowing transcription of potential prosurvival genes. We thus examined levels of phosphorylated and nonphosphorylated total β-catenin levels in the nucleus after cerebral ischemia and determined the regulatory effect of E2. As shown in Figure 4 A–C, phospho-β-catenin levels in the nucleus remain stable at 30 min and 6 h after cerebral ischemia in placebo animals, but then show a strong increase at 24 and 48 h after cerebral ischemia. In contrast, E2 significantly attenuated the cerebral ischemia-induced elevation of nuclear p-β-catenin levels in the CA1 region at 24 and 48 h (Fig. 4 A–C), suggesting that E2 prevents degradation of β-catenin and preserves its retention in the nucleus. In support of this interpretation, we observed that total nuclear β-catenin protein levels in CA1 of cerebral ischemia (placebo) animals were similar to sham control at 30 min and 6 h after cerebral ischemia, but showed a marked and significant reduction at 24 and 48 h after cerebral ischemia (Fig. 4 D,E). Of significant interest, E2 treatment completely prevented the cerebral ischemia-induced reduction of nuclear β-catenin protein levels in CA1 at 24 and 48 h after cerebral ischemia (Fig. 4 D,E). The phosphorylated and total β-catenin levels in the cytoplasmic fractions were unchanged by cerebral ischemia and were not significantly affected by E2 (data not shown). Double immunohistochemistry for NeuN and β-catenin in hippocampal CA1 region at 24 h after cerebral ischemia confirmed the Western blot results by demonstrating that β-catenin was localized in nuclei of neurons as evidenced by colocalization with the neuronal marker NeuN (Fig. 4 F). It further confirmed that nuclear β-catenin immunoreactivity was markedly reduced by cerebral ischemia (placebo), an effect prevented by E2 (Fig. 4 F). E2 had no significant effect on p-β-catenin or nuclear β-catenin levels in the hippocampus CA1 of nonischemic sham control animals (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material), demonstrating that E2 specifically reversed ischemia-induced alterations of Wnt/β-catenin signaling in the hippocampus CA1.
Figure 4.

A, B, 17β-Estradiol prevents cerebral ischemia-induced elevation of p-β-catenin levels in nuclear fraction of hippocampus CA1 after global cerebral ischemia. Values are mean ± SEM of determinations from five to six individual rats expressed as fold change versus sham control. Pla, Placebo; R, reperfusion. Ca–Cf, DAB immunostaining for p-β-catenin in hippocampus CA1 at 24 h after global cerebral ischemia. Results are representative of staining observed in four individual animals per group. D, E, E2 prevents cerebral ischemia-induced reduction of nuclear β-catenin levels in hippocampus CA1 after global cerebral ischemia. Values are mean ± SEM of determinations from five to six individual rats expressed as fold change versus sham control. *p < 0.05 versus sham control; # p < 0.05 versus the Pla group at the same time point. Fa–Fi, Confocal analysis of NeuN and β-catenin immunostaining in hippocampus CA1 at 24 h after global cerebral ischemia. Results are representative of staining observed in five individual animals per group (magnification, 40×). Scale bars, 50 μm.
17β-Estradiol enhances survivin levels in the hippocampus CA1 region after global cerebral ischemia
We next examined regulation of a downstream β-catenin-induced gene, survivin, which is a putative prosurvival factor (Ambrosini et al., 1997). As shown in Figure 5 A–C, ischemia reduced survivin levels at 24–48 h compared with sham control. However, E2 markedly upregulated survivin in hippocampus CA1 at 24–48 h compared with placebo group (Fig. 5 A,B), which was further confirmed by DAB staining (Fig. 5 C). Double immunohistochemistry revealed that survivin was induced in neurons in the CA1 region, as survivin and NeuN signals were strongly colocalized in E2-treated animals (Fig. 5 C). Additionally, at 7 d after cerebral ischemia, we observed that survivin colocalized with NeuN-positive but not with Fluoro-Jade B-positive neurons, suggesting that neurons that are degenerating lose survivin expression, whereas surviving neurons maintain survivin expression (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). Furthermore, the more resistant CA3 and DG show strong survivin expression, which appeared to be less affected by cerebral ischemia compared with CA1 region (supplemental Fig. 3B,D, available at www.jneurosci.org as supplemental material). The cortex, which shows mild loss of neurons after the 10 min cerebral ischemia used in our studies, showed a mild to moderate reduction in NeuN and survivin staining, an effect antagonized by E2 treatment (supplemental Fig. 3C, available at www.jneurosci.org as supplemental material). E2 appeared to slightly increase survivin levels in the hippocampus CA1 of nonischemic sham control animals, but this effect was not statistically significant (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material). Thus, E2 appears to be preventing cerebral-ischemia induced downregulation of survivin.
Figure 5.

A, B, 17β-Estradiol enhances expression of the antiapoptotic protein survivin in hippocampus CA1 after global cerebral ischemia. Values are mean ± SEM of determinations from five to six individual rats expressed as fold change versus sham control. Pla, Placebo; R, reperfusion. *p < 0.05 versus sham control; # p < 0.05 versus the Pla group at the same time point. C, DAB and confocal analysis shows that survivin is induced in NeuN-positive neurons by E2 in hippocampus CA1 at 24 h after global cerebral ischemia. Results are representative of staining observed in five individual animals per group (magnifications, 40×). Scale bars, 50 μm.
17β-Estradiol attenuates cerebral ischemia-induced upregulation of Dkk1 levels in hippocampal CA1 region by reducing activation of the JNK/c-Jun signaling pathway
Previous work suggests that during development c-Jun can regulate induction of the Dkk1 gene (Grotewold and Rüther, 2002). We therefore determined whether E2 regulates JNK/c-Jun signaling in the hippocampus CA1 region after cerebral ischemia, and whether Dkk1 and p-c-Jun are induced in the same neurons after cerebral ischemia. As shown in Figure 6, A and B, p-JNK levels in placebo-treated animals were increased twofold to threefold over sham control at 30 min, and 24 and 48 h after cerebral ischemia. Similarly, p-c-Jun levels showed a twofold to fourfold elevation at all time points in the CA1 region of placebo-treated animals compared with sham control. Interestingly, E2 treatment blocked the p-JNK and p-c-Jun elevations after cerebral ischemia at all time points (Fig. 6 A,B). Figure 6 C shows the result of triple immunohistochemistry for NeuN, Dkk1, and p-c-Jun in hippocampal CA1 region at 24 h after cerebral ischemia. The results confirm the Western blot findings that Dkk1 and p-c-Jun levels in the CA1 region are low in sham animals, markedly elevated in placebo-treated animals at 24 h after cerebral ischemia, and that E2 treatment blocks the elevation of both Dkk1 and p-c-Jun in the CA1 region. Additionally, the results demonstrate that Dkk1 and p-c-Jun are induced in the same neurons in the CA1 region at 24 h after cerebral ischemia (Fig. 6 C), further supporting a potential role for c-Jun in the regulation of Dkk1. To confirm that JNK/c-Jun signaling is critical for induction of Dkk1 after cerebral ischemia, we administered a JNK inhibitor, SP600125 (10 mg/kg body weight), via intravenous injection 15 min before induction of cerebral ischemia and examined the effect on p-JNK, p-c-Jun and Dkk1 at 24 h after cerebral ischemia in placebo animals. As shown in Figure 6, D and E, SP600125 prevented the cerebral ischemia activation of JNK and c-Jun, an effect that was correlated with a significant attenuation of the induction of Dkk1. This finding suggests that JNK/c-Jun activation is critical for induction of Dkk1, and that E2 ability to attenuate JNK/c-Jun activation may be a critical mechanism for its ability to reduce Dkk1 induction after cerebral ischemia. Additionally, E2 did not significantly affect JNK activation in the hippocampus CA1 of nonischemic sham control animals (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material), demonstrating that its p-JNK regulatory action was specific for the ischemic hippocampus CA1.
Figure 6.

A, B, 17β-Estradiol prevents cerebral ischemia-induced elevation of p-JNK and p-c-Jun levels in hippocampus CA1 after global cerebral ischemia. Values are mean ± SEM of determinations from five to six individual rats, corrected by total JNK or c-Jun, and expressed as fold change versus sham control. Pla, Placebo; R, reperfusion. Ca–Cl, Confocal analysis demonstrates that p-c-Jun and Dkk1 are induced in the same neurons at 24 h after global cerebral ischemia. Results are representative of staining observed in five individual animals per group (magnification, 40×). Scale bar, 50 μm. D, E, Administration of the JNK inhibitor SP600125 (10 mg/kg, i.v.) prevents activation of JNK and c-Jun and prevents cerebral ischemia induction of Dkk1 in hippocampus CA1 at 24 h after global cerebral ischemia. Values are mean ± SEM of determinations from five to six individual rats expressed as fold change versus sham control. *p < 0.05 versus sham control; # p < 0.05 versus Pla group at the same time point.
17β-Estradiol attenuates hyperphosphorylation of tau after cerebral ischemia by attenuating JNK/c-Jun-Dkk1 signaling
Dkk1 has been demonstrated to induce hyperphosphorylation of tau and to play a role in neurodegeneration in Alzheimer's disease (Caricasole et al., 2004; Scali et al., 2006). E2, in contrast, has been reported to attenuate hyperphosphorylation of tau, protect against β-amyloid-induced neuronal cell death and to potentially delay onset of Alzheimer's disease (Wen et al., 2004; Alvarez-de-la-Rosa et al., 2005; Simpkins et al., 2005). We thus examined whether E2 attenuation of Dkk1 could be correlated with reduction of tau hyperphosphorylation after cerebral ischemia. We also determined the role of JNK/c-Jun/Dkk1 signaling in tau hyperphosphorylation by administering a JNK inhibitor, SP600125, and determining whether cerebral ischemia-induced hyperphosphorylation of tau is attenuated. As shown Figure 7 A–C, sham controls had low phospho-tau levels, whereas cerebral ischemia (placebo) induced a 1.5-fold to fourfold induction of phospho-tau in hippocampus CA1 compared with sham, with 24 h showing peak phospho-tau levels. E2 treatment completely blocked cerebral ischemia-induced hyperphosphorylation of tau at all time points examined (Fig. 7 A–C). As shown in Figure 7, D and E, intravenous administration of the JNK inhibitor SP600125 significantly attenuated cerebral ischemia-induced hyperphosphorylation of tau in CA1 region at 24 h after cerebral ischemia, suggesting that JNK/c-Jun activation, which is important for induction of Dkk1, is critical for hyperphosphorylation of tau after cerebral ischemia. Figure 7 F demonstrates that treatment with the JNK inhibitor SP600125 significantly protected the hippocampus CA1 from cerebral ischemia-induced neuronal cell death.
Figure 7.

A, C, 17β-Estradiol reduces tau hyperphosphorylation after global cerebral ischemia as determined by Western blot analysis with PHF-1 antibodies. Values are mean ± SEM of determinations from five to six individual rats, and expressed as fold change versus sham control. Pla, Placebo; R, reperfusion. Ba–Bf, DAB immunostaining showing tau hyperphosphorylation at 24 h after global cerebral ischemia in hippocampus CA1 and attenuation by E2. Results are representative of staining observed in five individual animals per group (magnification, 40×). Scale bar, 50 μm. D, E, Administration of the JNK inhibitor SP600125 abolishes global cerebral ischemia-induced tau hyperphosphorylation at 24 h after global cerebral ischemia. Values are means ± SEM of determinations from five to six individual rats expressed as fold change versus sham control. *p < 0.05 versus sham control, # p < 0.05 versus Pla group at the same time point. F, Administration of the JNK inhibitor SP600125 significantly attenuated hippocampal CA1 neuronal cell death as measured by counting of NeuN-positive cells at 7 d after global cerebral ischemia. Representative NeuN staining in hippocampus CA1 from vehicle- and SP600125-treated rats is shown in the inset. Values are mean ± SEM of determinations from five to six individual rats. # p < 0.01 versus vehicle and placebo.
Exogenous Dkk1 administration reverses 17β-estradiol neuroprotection and tau regulation in the hippocampal CA1 region after cerebral ischemia
Finally, we examined the ability of exogenous Dkk1 to reverse E2 attenuation of tau hyperphosphorylation and neuroprotection after cerebral ischemia. Exogenous recombinant Dkk1 (5 μg/μl) was injected into both lateral cerebral ventricles of E2-treated rats at 12 h after cerebral ischemia. As a control, Dkk1 was also injected into the lateral cerebral ventricles of E2-treated nonischemic sham control animals to demonstrate that Dkk1 was specifically reversing E2 neuroprotection in the ischemic situation. Figure 8 A shows representative photomicrographs of NeuN staining in the hippocampus CA1 region at 7 d after cerebral ischemia in E2-treated ovariectomized animals or in E2-treated sham controls. As shown in Figure 8, A and B, Dkk1 had no significant effect on CA1 neuronal cell density in E2-treated sham control animals, as demonstrated by NeuN staining and quantification of NeuN-positive cells in the CA1. As also shown in Figure 8, A and B, E2 treatment exerted significant neuroprotection in the CA1 region of animals after 10 min global cerebral ischemia, as evidenced by dense NeuN positive cells in the CA1 region. Intriguingly, Dkk1 administration in the lateral ventricle of E2-treated animals having undergone cerebral ischemia abolished E2 neuroprotection as evidenced by a dramatic decrease in NeuN-positive cells in the CA1 region. Figure 8, C and D, further demonstrates that exogenous Dkk1 administration reverses E2-induced attenuation of tau hyperphosphorylation and enhancement of nuclear β-catenin levels at 24 h after cerebral ischemia. Collectively, these findings suggest that the ability of E2 to prevent cerebral ischemia-induced elevation of Dkk1 in the hippocampal CA1 is an important mechanism underlying its neuroprotection and regulation of tau hyperphosphorylation.
Figure 8.

Exogenous Dkk1 administration reverses 17β-estradiol-induced neuroprotection and phospho-tau regulation after global cerebral ischemia. Dkk1 (5 μg/5 μl) was administered via intracerebroventricular injection into both lateral ventricles at 12 h after global cerebral ischemia. For a control, separate animals received vehicle into both lateral cerebral ventricles. Additionally, a nonischemic sham control also received Dkk1 into the lateral ventricles. Aa–Ad, High-power magnification of NeuN staining in hippocampus CA1 at 7 d in sham animals and after 7 d reperfusion after global cerebral ischemia in E2-treated rats that received vehicle or exogenous Dkk1 in both lateral ventricles. Magnification is 40×. Scale bar, 50 μm. Ae–Ah, Low-power magnifications of representative whole hippocampus sections showing NeuN staining in hippocampus CA1 at 7 d in sham animals and after 7 d reperfusion after global cerebral ischemia in E2-treated rats that received vehicle or exogenous Dkk1 in both lateral ventricles. Magnification is 5×. Scale bar, 200 μm. B, CA1 cell counts of NeuN-positive neurons in all animals show that exogenous Dkk1 had no significant effect on CA1 neuronal cell survival in nonischemic sham controls, although it significantly reversed E2 neuroprotection in ischemic animals. Values are mean ± SEM of determinations from five to six individual rats. # p < 0.01 versus vehicle. C, D, Exogenous Dkk1 injection also reversed E2 reduction of phospho-tau and elevation of nuclear β-catenin in the hippocampus CA1 at 24 h after global cerebral ischemia. Values are mean ± SEM of determinations from five to six individual rats expressed as fold change versus sham control. # p < 0.05 versus vehicle treatment group.
Discussion
Chronic treatment with low physiological doses of E2 is well established to exert neuroprotection in animal models of both focal and global cerebral ischemia, a finding further confirmed by this study (Toung et al., 1998; Rusa et al., 1999; Horsburgh et al., 2002; Brann et al., 2007). Although E2 clearly is neuroprotective in cerebral ischemia, the mechanisms underlying its protective effects remain poorly understood. The findings of this study enhance our understanding of the molecular mechanisms underlying E2 neuroprotection after cerebral ischemia by demonstrating that E2 prevents cerebral ischemia induction of the neurodegenerative factor Dkk1, an antagonist of the Wnt/β-catenin signaling pathway. E2 also acts in a parallel manner to enhance expression of Wnt3, an activator of the Wnt/β-catenin signaling pathway. These combined effects lead to activation of the Wnt/β-catenin signaling pathway and a profound protection of the hippocampal CA1 region from cerebral ischemia-induced neuronal death.
The mechanisms underlying Dkk1 induction in cerebral ischemia and other neurodegenerative disorders are not fully understood. The results of our study provide illumination to this issue by demonstrating that JNK/Jun signaling is critical for cerebral ischemia induction of Dkk1. This finding agrees with previous work in vertebrate limb development where Dkk1 induction was similarly demonstrated to be dependent on induction by c-Jun (Grotewold and Rüther, 2002). In our study, E2 markedly attenuated JNK/c-Jun activation after cerebral ischemia. This suggests that E2 reduction of Dkk1 is likely attributable to its ability to attenuate activation of JNK/c-Jun signaling in the hippocampus CA1 after cerebral ischemia. This conclusion is further supported by the fact that inhibition of JNK/c-Jun signaling via a different approach (e.g., administration of a JNK inhibitor) also blocked Dkk1 induction after cerebral ischemia. In addition to c-Jun induction, Dkk1 is also induced by the tumor-suppressing protein, p53, which is a sensor for DNA damage in cells (Wang et al., 2000; Cappuccio et al., 2005). Preliminary studies in our laboratory show that E2 decreases P53 after global cerebral ischemia (L. Raz, Q. G. Zhang, D. W. Brann, unpublished observation). Thus, E2 suppression of P53 induction could also play a role in the ability of E2 to prevent Dkk1 induction after cerebral ischemia.
An additional interesting observation from our data are that we noted a biphasic regulation of p-JNK and phospho-tau after cerebral ischemia, in which p-JNK and phospho-tau levels were elevated at 30 min and 24 h after cerebral ischemia, with a much smaller increase at 6 h. The precise reason for this pattern of regulation is unclear, but previous work by a member of our group showed that Akt activation had an inverse pattern to JNK activation at 3–12 h and 3 d after cerebral ischemia (Zhang et al., 2007). This suggests that the biphasic JNK activation may be attributable to Akt regulation. In support of this possibility, administration of antisense oligonucleotides to PTEN (phosphatase/tensin homolog on chromosome 10), a phosphatase that negatively regulates Akt activation, led to Akt phosphorylation and dephosphorylation of JNK after cerebral ischemia. This finding suggests a critical role for Akt cross talk in the control of JNK activation and may explain the biphasic JNK activation observed in our studies after cerebral ischemia. Because we provide data that JNK is critical in the phosphorylation of tau, the biphasic pattern of tau phosphorylation reported in our study after cerebral ischemia may be attributable to the biphasic changes in JNK activation.
An additional novel finding of our study was that E2 markedly elevated survivin levels in CA1 neurons after cerebral ischemia. Survivin, which is a β-catenin-regulated gene, is a member of the inhibitor of apoptosis protein family, which exerts anti-apoptotic effects in cells (Ambrosini et al., 1997; Jiang et al., 2005; Ma et al., 2005; Tapia et al., 2006). Thus, its induction further verifies activation of Wnt/β-catenin signaling by E2 after cerebral ischemia, and it also provides a potential mechanism for neuroprotection by E2 because of the well known anti-apoptotic actions of survivin. Survivin has been implicated to exert its antiapoptotic effects by blocking caspase activation and inhibiting activated caspases (Shin et al., 2001; Chiou et al., 2003). Along these lines, survivin has been shown to enhance degradation of p53 via its ability to inhibit caspase cleavage of Mdm2, a factor that regulates expression, function and degradation of p53 (Wang et al., 2004). Interestingly, we observed that survivin was induced in healthy but not degenerating CA1 pyramidal neurons, suggesting that the lack of survivin induction in degenerating neurons may play a role in their demise after cerebral ischemia. CA3 and DG showed strong survivin levels after cerebral ischemia with very few degenerating neurons compared with the CA1 region, suggesting that maintenance of survivin in CA3 and DG may aid in the relative resistance of these hippocampal regions to global cerebral ischemia compared with the CA1 region.
E2 has been shown previously to inhibit tau hyperphosphorylation, which may be important for its purported beneficial effects in delaying onset of Alzheimer's disease (Wen et al., 2004; Alvarez-de-la-Rosa et al., 2005; Simpkins et al., 2005). Aberrant aggregation of hyperphosphorylated tau protein has been implicated as a factor in formation of neurofibrillary tangles and neurodegeneration in Alzheimer's disease, as well as other neurodegenerative diseases, such as Pick's disease, corticobasal degeneration, and progressive supranuclear palsy, which collectively are known as tauopathies (Maccioni et al., 2001; Iqbal et al., 2005). Intriguingly, cerebral ischemia has been shown to also be a tauopathy, as it displays significant tau hyperphosphorylation and neurofibrillary tangle formation in the brain after cerebral ischemia (Wen et al., 2004). Our study sheds light on the potential mechanism underlying E2 suppression of tau hyperphosphorylation by demonstrating that E2 attenuation of cerebral ischemia-induced tau hyperphosphorylation is correlated with reduction of Dkk1, a known inducer of tau hyperphosphorylation. Furthermore, replacement with exogenous Dkk1 was able to reinstate tau hyperphosphorylation in E2-treated animals, further suggesting that a reduction of Dkk1 is important for E2 suppression of tau hyperphosphorylation in cerebral ischemia.
Our data also suggests an important role for modulation of JNK signaling in the E2 suppression of tau hyperphosphorylation in the hippocampus CA1 after cerebral ischemia. Previous work has demonstrated that formation of p-JNK granules in the hippocampus is an early event in Alzheimer disease (Lagalwar et al., 2006), and p-JNK has been shown to phosphorylate tau at many proline-directed serine/threonine residues (Okazawa and Estus, 2002). Our study supports a role for p-JNK in regulating tau hyperphosphorylation after cerebral ischemia as evidenced by the fact that administration of the JNK inhibitor SP600125 significantly reduced hyperphosphorylation of tau after cerebral ischemia. Because E2 attenuated JNK activation in our study, we propose that one of the central mechanisms for E2 attenuation of tau hyperphosphorylation involves the attenuation of JNK activation. Furthermore, because JNK enhances phosphorylation of tau by a direct mechanism, as well as by an indirect mechanism involving activation of c-Jun and induction of Dkk1, E2 attenuation of JNK activation thus provides an attractive dual mechanism for attenuating tau hyperphosphorylation after cerebral ischemia, e.g., by attenuating a direct phosphorylation of tau by JNK, as well as attenuating a JNK indirect pathway for tau phosphorylation, which involves JNK induction of Dkk1 with subsequent downstream activation of the tau phosphorylating kinase, GSK-3β. Because tau hyperphosphorylation is also a hallmark of Alzheimer's disease, and Dkk1 is induced in degenerating neurons in the disorder, these findings may also have importance and application to Alzheimer's disease.
In conclusion, as illustrated in the summary diagram provided in Figure 9, our study demonstrates that E2 neuroprotection after global cerebral ischemia involves two parallel mechanisms that lead to activation of Wnt/β-catenin signaling. First, E2 acts to prevent cerebral ischemia-induced downregulation of Wnt/β-catenin signaling in the hippocampus CA1 by inhibiting cerebral ischemia-induced activation of JNK, which prevents (1) induction of the Wnt/β-catenin antagonist Dkk1, (2) subsequent downstream phosphorylation of tau and β-catenin, and (3) delayed neuronal cell death. Second, E2 enhances expression of Wnt3, which further enhances Wnt/β-catenin signaling, leading to nuclear β-catenin accumulation, enhanced expression of survivin, inhibition of apoptosis, and increased neuronal survival. Because Dkk1 plays a role in neurodegeneration in both stroke and Alzheimer's disease, it is suggested that E2 regulation of Dkk1 and Wnt/β-catenin signaling may be a critical mechanism underlying its beneficial effects in both disorders.
Figure 9.

Summary diagram of E2 regulation of Wnt/β-catenin signaling pathway after cerebral ischemia and proposed role in E2 ischemic neuroprotection. E2 acts to inhibit cerebral ischemia activation of JNK/Jun/Dkk1 signaling, which prevents subsequent phosphorylation of tau and β-catenin and cell death, while it acts to enhances expression of Wnt3, leading to accumulation of nuclear β-catenin and enhanced expression of survivin, which inhibits apoptosis and enhances neuronal cell survival.
Footnotes
This work was supported by National Institutes of Health–National Institute of Neurological Diseases and Stroke Research Grant NS050730.
References
- Alkayed NJ, Goto S, Sugo N, Joh HD, Klaus J, Crain BJ, Bernard O, Traystman RJ, Hurn PD. Estrogen and Bcl-2: gene induction and effect of transgene in experimental stroke. J Neurosci. 2001;21:7543–7550. doi: 10.1523/JNEUROSCI.21-19-07543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-de-la-Rosa M, Silva I, Nilsen J, Pérez MM, García-Segura LM, Avila J, Naftolin F. Estradiol prevents neural tau hyperphosphorylation characteristic of Alzheimer's disease. Ann N Y Acad Sci. 2005;1052:210–224. doi: 10.1196/annals.1347.016. [DOI] [PubMed] [Google Scholar]
- Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–921. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brann DW, Dhandapani K, Wakade C, Mahesh VB, Khan MM. Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications. Steroids. 2007;72:381–405. doi: 10.1016/j.steroids.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busceti CL, Biagioni F, Aronica E, Riozzi B, Storto M, Battaglia G, Giorgi FS, Gradini R, Fornai F, Caricasole A, Nicoletti F, Bruno V. Induction of the Wnt inhibitor, Dickkopf-1, is associated with neurodegeneration related to temporal lobe epilepsy. Epilepsia. 2007;48:694–705. doi: 10.1111/j.1528-1167.2007.01055.x. [DOI] [PubMed] [Google Scholar]
- Cappuccio I, Calderone A, Busceti CL, Biagioni F, Pontarelli F, Bruno V, Storto M, Terstappen GT, Gaviraghi G, Fornai F, Battaglia G, Melchiorri D, Zukin RS, Nicoletti F, Caricasole A. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is required for the development of ischemic neuronal death. J Neurosci. 2005;25:2647–2657. doi: 10.1523/JNEUROSCI.5230-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caricasole A, Copani A, Caraci F, Aronica E, Rozemuller AJ, Caruso A, Storto M, Gaviraghi G, Terstappen GC, Nicoletti F. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer's brain. J Neurosci. 2004;24:6021–6027. doi: 10.1523/JNEUROSCI.1381-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou SK, Jones MK, Tarnawski AS. Survivin - an anti-apoptosis protein: its biological roles and implications for cancer and beyond. Med Sci Monit. 2003;9:PI25–PI29. [PubMed] [Google Scholar]
- Di Carlo A, Lamassa M, Baldereschi M, Pracucci G, Basile AM, Wolfe CD, Giroud M, Rudd A, Ghetti A, Inzitari D. Sex differences in the clinical presentation, resource use, and 3-month outcome of acute stroke in Europe: data from a multicenter multinational hospital-based registry. Stroke. 2003;34:1114–1119. doi: 10.1161/01.STR.0000068410.07397.D7. [DOI] [PubMed] [Google Scholar]
- Gore AC, Yeung G, Morrison JH, Oung T. Neuroendocrine aging in the female rat: the changing relationship of hypothalamic gonadotropin-releasing hormone neurons and N-methyl-D-aspartate receptors. Endocrinology. 2000;141:4757–4767. doi: 10.1210/endo.141.12.7841. [DOI] [PubMed] [Google Scholar]
- Grotewold L, Rüther U. The Wnt antagonist Dickkopf-1 is regulated by Bmp signaling and c-Jun and modulates programmed cell death. EMBO J. 2002;21:966–975. doi: 10.1093/emboj/21.5.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson VW. Estrogen-containing hormone therapy and Alzheimer's disease risk: understanding discrepant inferences from observational and experimental research. Neuroscience. 2006;138:1031–1039. doi: 10.1016/j.neuroscience.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Horsburgh K, Macrae IM, Carswell H. Estrogen is neuroprotective via an apolipoprotein E-dependent mechanism in a mouse model of global ischemia. J Cereb Blood Flow Metab. 2002;22:1189–1195. doi: 10.1097/01.wcb.0000037991.07114.4e. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Jiang Y, de Bruin A, Caldas H, Fangusaro J, Hayes J, Conway EM, Robinson ML, Altura RA. Essential role for survivin in early brain development. J Neurosci. 2005;25:6962–6970. doi: 10.1523/JNEUROSCI.1446-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jover-Mengual T, Zukin RS, Etgen AM. MAPK signaling is critical to estradiol protection of CA1 neurons in global ischemia. Endocrinology. 2007;148:1131–1143. doi: 10.1210/en.2006-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jover T, Tanaka H, Calderone A, Oguro K, Bennett MV, Etgen AM, Zukin RS. Estrogen protects against global ischemia-induced neuronal death and prevents activation of apoptotic signaling cascades in the hippocampal CA1. J Neurosci. 2002;22:2115–2124. doi: 10.1523/JNEUROSCI.22-06-02115.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagalwar S, Guillozet-Bongaarts AL, Berry RW, Binder LI. Formation of phospho-SAPK/JNK granules in the hippocampus is an early event in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:455–464. doi: 10.1097/01.jnen.0000229236.98124.d8. [DOI] [PubMed] [Google Scholar]
- Ma H, Nguyen C, Lee KS, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene. 2005;24:3619–3631. doi: 10.1038/sj.onc.1208433. [DOI] [PubMed] [Google Scholar]
- Maccioni RB, Muñoz JP, Barbeito L. The molecular bases of Alzheimer's disease and other neurodegenerative disorders. Arch Med Res. 2001;32:367–381. doi: 10.1016/s0188-4409(01)00316-2. [DOI] [PubMed] [Google Scholar]
- Miller NR, Jover T, Cohen HW, Zukin RS, Etgen AM. Estrogen can act via estrogen receptor alpha and beta to protect hippocampal neurons against global ischemia-induced cell death. Endocrinology. 2005;146:3070–3079. doi: 10.1210/en.2004-1515. [DOI] [PubMed] [Google Scholar]
- Murphy SJ, McCullough LD, Smith JM. Stroke in the female: role of biological sex and estrogen. ILAR J. 2004;45:147–159. doi: 10.1093/ilar.45.2.147. [DOI] [PubMed] [Google Scholar]
- Niewada M, Kobayashi A, Sandercock PA, Kamiński B, Czlonkowska A. Influence of gender on baseline features and clinical outcomes among 17,370 patients with confirmed ischaemic stroke in the international stroke trial. Neuroepidemiology. 2005;24:123–128. doi: 10.1159/000082999. [DOI] [PubMed] [Google Scholar]
- Ogita K, Yoneda Y. Selective potentiation of DNA binding activities of both activator protein 1 and cyclic AMP response element binding protein through in vivo activation of N-methyl-d-aspartate receptor complex in mouse brain. J Neurochem. 1994;63:525–534. doi: 10.1046/j.1471-4159.1994.63020525.x. [DOI] [PubMed] [Google Scholar]
- Okazawa H, Estus S. The JNK/c-Jun cascade and Alzheimer's disease. Am J Alzheimers Dis Other Demen. 2002;17:79–88. doi: 10.1177/153331750201700209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke. 1979;10:267–272. doi: 10.1161/01.str.10.3.267. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Buchan AM. The four-vessel occlusion rat model: method for complete occlusion of vertebral arteries and control of collateral circulation. Stroke. 1988;19:913–914. doi: 10.1161/01.str.19.7.913. [DOI] [PubMed] [Google Scholar]
- Roquer J, Campello AR, Gomis M. Sex differences in first-ever acute stroke. Stroke. 2003;34:1581–1585. doi: 10.1161/01.STR.0000078562.82918.F6. [DOI] [PubMed] [Google Scholar]
- Rusa R, Alkayed NJ, Crain BJ, Traystman RJ, Kimes AS, London ED, Klaus JA, Hurn PD. 17beta-Estradiol reduces stroke injury in estrogen-deficient female animals. Stroke. 1999;30:1665–1670. doi: 10.1161/01.str.30.8.1665. [DOI] [PubMed] [Google Scholar]
- Salinas PC, Nusse R. Regional expression of the Wnt-3 gene in the developing mouse forebrain in relationship to diencephalic neuromeres. Mech Dev. 1992;39:151–160. doi: 10.1016/0925-4773(92)90042-i. [DOI] [PubMed] [Google Scholar]
- Scali C, Caraci F, Gianfriddo M, Diodato E, Roncarati R, Pollio G, Gaviraghi G, Copani A, Nicoletti F, Terstappen GC, Caricasole A. Inhibition of Wnt signaling, modulation of Tau phosphorylation and induction of neuronal cell death by DKK1. Neurobiol Dis. 2006;24:254–265. doi: 10.1016/j.nbd.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Shin S, Sung BJ, Cho YS, Kim HJ, Ha NC, Hwang JI, Chung CW, Jung YK, Oh BH. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and -7. Biochemistry. 2001;40:1117–1123. doi: 10.1021/bi001603q. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Rajakumar G, Zhang YQ, Simpkins CE, Greenwald D, Yu CJ, Bodor N, Day AL. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg. 1997;87:724–730. doi: 10.3171/jns.1997.87.5.0724. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Wen Y, Perez E, Yang S, Wang X. Role of nonfeminizing estrogens in brain protection from cerebral ischemia: an animal model of Alzheimer's disease neuropathology. Ann N Y Acad Sci. 2005;1052:233–242. doi: 10.1196/annals.1347.019. [DOI] [PubMed] [Google Scholar]
- Tapia JC, Torres VA, Rodriguez DA, Leyton L, Quest AF. Casein kinase 2 (CK2) increases survivin expression via enhanced beta-catenin-T cell factor/lymphoid enhancer binding factor-dependent transcription. Proc Natl Acad Sci U S A. 2006;103:15079–15084. doi: 10.1073/pnas.0606845103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatemichi TK, Desmond DW, Mayeux R, Paik M, Stern Y, Sano M, Remien RH, Williams JB, Mohr JP, Hauser WA, Figueroa M. Dementia after stroke: baseline frequency, risks, and clinical features in a hospitalized cohort. Neurology. 1992;42:1185–1193. doi: 10.1212/wnl.42.6.1185. [DOI] [PubMed] [Google Scholar]
- Toung TJ, Traystman RJ, Hurn PD. Estrogen-mediated neuroprotection after experimental stroke in male rats. Stroke. 1998;29:1666–1670. doi: 10.1161/01.str.29.8.1666. [DOI] [PubMed] [Google Scholar]
- Wakade C, Khan MM, De Sevilla LM, Zhang QG, Mahesh VB, Brann DW. Tamoxifen neuroprotection in cerebral ischemia involves attenuation of kinase activation and superoxide production and potentiation of mitochondrial superoxide dismutase. Endocrinology. 2008;149:367–379. doi: 10.1210/en.2007-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Shou J, Chen X. Dickkopf-1, an inhibitor of the Wnt signaling pathway, is induced by p53. Oncogene. 2000;19:1843–1848. doi: 10.1038/sj.onc.1203503. [DOI] [PubMed] [Google Scholar]
- Wang Z, Fukuda S, Pelus LM. Survivin regulates the p53 tumor suppressor gene family. Oncogene. 2004;23:8146–8153. doi: 10.1038/sj.onc.1207992. [DOI] [PubMed] [Google Scholar]
- Wen Y, Yang S, Liu R, Brun-Zinkernagel AM, Koulen P, Simpkins JW. Transient cerebral ischemia induces aberrant neuronal cell cycle re-entry and Alzheimer's disease-like tauopathy in female rats. J Biol Chem. 2004;279:22684–22692. doi: 10.1074/jbc.M311768200. [DOI] [PubMed] [Google Scholar]
- Zhang QG, Tian H, Li HC, Zhang GY. Antioxidant N-acetylcysteine inhibits the activation of JNK3 mediated by the GluR6-PSD95-MLK3 signaling module during cerebral ischemia in rat hippocampus. Neurosci Lett. 2006a;408:159–164. doi: 10.1016/j.neulet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Zhang QG, Wang XT, Han D, Yin XH, Zhang GY, Xu TL. Akt inhibits MLK3/JNK3 signaling by inactivating Rac1: a protective mechanism against ischemic brain injury. J Neurochem. 2006b;98:1886–1898. doi: 10.1111/j.1471-4159.2006.04020.x. [DOI] [PubMed] [Google Scholar]
- Zhang QG, Wu DN, Han D, Zhang GY. Critical role of PTEN in the coupling between PI3K/Akt and JNK1/2 signaling in ischemic brain injury. FEBS Lett. 2007;581:495–505. doi: 10.1016/j.febslet.2006.12.055. [DOI] [PubMed] [Google Scholar]