

Abstract
The (X;1)(p11;q21) translocation is a recurrent chromosomal abnormality in a subset of human papillary renal cell carcinomas, and is sometimes the sole cytogenetic abnormality present. Via positional cloning, we were able to identify the genes involved. The translocation results in a fusion of the transcription factor TFE3 gene on the X chromosome to a novel gene, designated PRCC, on chromosome 1. Through this fusion, reciprocal translocation products are formed, which are both expressed in papillary renal cell carcinomas. PRCC is ubiquitously expressed in normal adult and fetal tissues and encodes a putative protein of 491 aa with a relatively high content of prolines. No relevant homologies with known sequences at either the DNA or the protein level were found.
It is by now well established that recurrent chromosomal translocations play a major role in the etiology of cancer (1). Based on translocation breakpoint studies in mostly hematopoietic tumors, two basic mechanisms have emerged, that can lead to (i) the altered expression of protooncogene products or (ii) the formation of fusion proteins. In solid tumors, the second mechanism seems to be the predominant one. Studies on tumor-specific translocations have led to the identification of several genes involved, among which are often those encoding transcription factors (1).
Chromosome 3 abnormalities are frequently found in sporadic and familial cases of clear cell nonpapillary renal cell carcinomas (RCCs; ref. 2). For instance, in patients with the Von Hippel–Lindau disease, mutations in the VHL gene on chromosome 3p were found to be predisposing to several types of cancer, including renal cancer (3). Mutations and loss of heterozygosity of the VHL gene have also been detected in sporadic renal cell carcinomas (4). Recently, another gene on chromosome 3 was identified (FHIT) that is disrupted in hereditary t(3;8)-positive clear cell nonpapillary renal cancer (5) and that appears to be a member of the histidine triad gene family. In contrast, chromosome 3 abnormalities appear to be absent in chromophilic RCCs with a papillary growth pattern, commonly referred to as papillary RCCs. Instead, a combination of gain of chromosomes 7 and 17 and loss of the Y chromosome are found (2, 6). In addition, a t(X;1)(p11;q21) or variants thereof have repeatedly been described in a subgroup of these tumors (7–13), sometimes as the sole cytogenetic anomaly present. Previously, positional cloning strategies were used to narrow down the translocation regions, thereby excluding several candidate genes in Xp11 and 1q21 (14, 15). We now report the identification of the genes that are involved in this specific translocation.
MATERIALS AND METHODS
Tumor Material, Somatic Cell Hybrids, and Tissues.
Two male-derived renal cell cultures (CL89-12117 and CL89-17872), one male-derived primary tumor (REN11-TT; ref. 9), and one female-derived primary tumor (RP-T) were used in this study. CL89-12117 and RP-T contain the t(X;1) as the only cytogenetic abnormality, whereas CL89-17872 also shows numerical aberrations next to the t(X;1) (7). The karyotype of REN11-TT is as follows: 49,Y,t(X;1)(p11;q21),+der(X)t(X;1)(p11;q21),+5,-16,+17,+18. REN11-N and RP-N were taken as controls and consist of normal tissue adjacent to the corresponding tumors. CL89-12117-derived somatic cell hybrids were obtained as described before (14, 16). Briefly, after fusion of A3 or Wg3h hamster cell lines and the t(X;1)-positive renal carcinoma cells, a panel of somatic cell hybrids was isolated in which the reciprocal translocation chromosomes segregate. As controls, two hybrid lines containing the normal X chromosome (17) or normal chromosome 1 (GM13.139; Coriell Cell Repositories, Camden, NJ) as the only human constituent and parental hamster (A3) and mouse (A9) cell lines were included. HL60 is a myeloid leukemia cell line. Rat organs that were used for Northern blot analysis were: spleen, liver, brain, heart, striated muscle, kidney, testes, thymus, seminal vesicles, lung, bladder, intestines, and stomach. Blots containing 2 μg of poly(A)+-selected RNA of fetal tissues (brain, lung, liver, and kidney) and adult tissues (heart, brain, placenta, lung, liver, skeletal muscle, kidney, and pancreas) were purchased from CLONTECH.
RNA Isolation, Northern Blot Analysis, and Reverse Transcription PCR (RT-PCR).
Total RNA was isolated as described by Auffray and Rougeon (18). Oligodeoxythymidine selections were performed using oligodeoxythymidine columns (type II; Collaborative Research). Total RNA (10 μg) was blotted on Hybond N-plus (Amersham) according to the procedure recommended by the manufacturer, after glyoxylation (19) and size fractionation on 1% agarose gels. RT-PCR was performed using 5 μg of total RNA and Superscript RT (Life Technologies, Grand Island, NY) using the conditions as described by the manufacturer. Priming was performed using a specific primer for PRCC (5′-GGGGAAGCAAGGCAGACAAACCA-3′) or TFE3 (TFE3b; 5′-GGTGGGGTGAGGGCCTGGGA-3′). PCR (30–35 cycles) was performed under standard conditions using 10 pmol of priming oligonucleotide and 10 pmol of a second TFE3-specific primer (TFE3a; 5′-CGCGGCTCCCTTCCCCAGTC-3′) according to the following protocol: denaturation at 94°C for 45 sec, annealing at 60°C (PRCC) or 62°C (TFE3) for 45 sec, and extension at 72°C for 1 min.
Construction of the CL89-12117 cDNA Library.
A CL89-12117-derived cDNA library was constructed from 5 μg of oligodeoxythymidine-selected RNA into Uni-ZAP XR using a unidirectional cDNA cloning kit (Stratagene). The size of the primary cDNA library was ≈3 × 106 recombinant clones. Clones (106) of the amplified library were used for screening.
Hybridization, DNA Probes, and Southern Blot Analysis.
Probes were labeled by random prime labeling and hybridized at 65°C in 0.5 M sodium phosphate buffer, 1 mM EDTA, and 7% SDS. Washes were performed in 40 mM sodium phosphate/0.1% SDS. Hybridizations of cDNA library filters were performed in the same manner with the addition of denatured herring sperm DNA to the hybridization mixture. Washes were performed stepwise, starting with 100 mM phosphate/0.5% SDS/1 mM EDTA, which was decreased to 50 mM phosphate/0.1% SDS. Northern blot hybridization and washes were performed likewise with the addition of 1% BSA to the hybridization mixture.
Genomic DNA was isolated using standard protocols by proteinase K/SDS treatment followed by phenol/chloroform extractions and ethanol precipitation. After digestion and size selection on an agarose gel, the DNA was blotted onto Hybond N-Plus (Amersham) or Genescreen Plus (DuPont/NEN) membranes.
DNA Sequencing and Computer Analysis.
A set of overlapping deletion clones was constructed using the Erase-a-Base system (Promega). DNA sequences were analyzed on an automated DNA sequencer (Applied Biosystems model no. 373A) using the Taq Dye Deoxy Terminator Cycle Sequencing kit (Applied Biosystems/Perkin–Elmer). Searches for known sequences were performed using the GenBank and European Molecular Biology Laboratory (EMBL) data bases. Searches for motifs, structure predictions, open reading frames, and alignments were performed using the cammsa programs, which are all part of the Wisconsin Package version 8.0.
RESULTS
Isolation of a t(X;1)(p11;q21) Breakpoint-Spanning RT-PCR Product.
In a previous study we have shown that the translocation (X;1)(p11;q21) breakpoint maps between the markers SYP and TFE3 on the X chromosome (15). When using a 2.3-kb subclone of a breakpoint-spanning TFE3 cosmid as a probe, shifted restriction fragments could be detected on Southern blots in t(X;1)-positive tumors as compared with normal cells. After isolation of a chimeric (X;1) genomic breakpoint fragment, which was also present on der(X), partial sequencing revealed a 150-bp stretch on the chromosome X part with a high degree of homology to mouse TFE3 and a 48-bp stretch on the chromosome 1 part corresponding to an exon identical to the 5′ part of a known expressed sequence tag (EST; GenBank data base accession no. R93849R93849; ref. 15). To confirm that the two corresponding genes are really involved in the translocation we performed an RT-PCR on RNA from three independent t(X;1)-positive RCCs and normal renal tissues, using one primer within the 150-bp stretch on one side of the breakpoint and a second primer just downstream of the 48-bp fragment based on the sequence of the EST as deposited in the data base. As shown in Fig. 1, a product of the expected size (209 bp including primers) could be detected in all three t(X;1)-positive RCCs, whereas this was not the case in the adjacent normal tissue or normal renal tissue from other persons. Subsequent cloning and sequencing of this RT-PCR product showed the expected fusion of TFE3 to a novel gene as represented by the EST (not shown).
Figure 1.
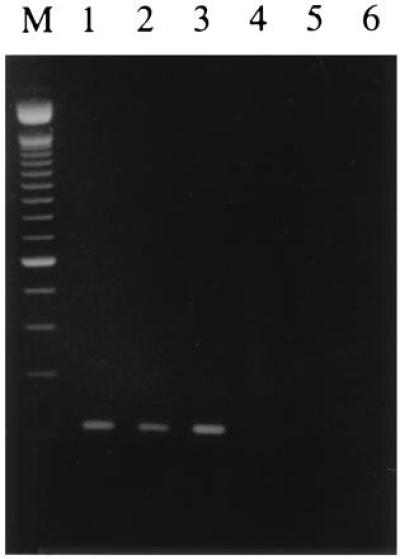
RT-PCR on three different t(X;1)-positive RCCs (lanes 1–3) and t(X;1)-negative controls (lanes 4–6). Lanes: 1, CL89-12117; 2, CL89-17872; 3, RP-T; 4, RP-N; 5, normal renal tissues pooled from several persons; and 6, normal renal tissue from a patient with RCC. The marker shown is the 100-bp ladder (Life Technologies). All three RT-PCR products showed the same sequence joining 150 bp of TFE3 to 48 bp of EST R93849R93849.
Isolation of PRCC and Chimeric TFE3/PRCC cDNAs.
The cloned 209-bp PCR breakpoint product was used as a probe to screen a cDNA library that was constructed from RNA of the t(X;1)-positive CL89-12117 tumor cell line. Several positive clones were isolated. Based on the restriction patterns, they could be divided into two main groups. When screening these clones with a genomic subfragment containing the 150-bp sequence homologous to mouse TFE3, one group was positive indicating that these cDNAs might represent the translocation product, whereas the other group was negative and thus might represent the normal mRNA transcribed from the normal chromosome 1. Since CL89-12117 tumor cells were taken from a male patient, no normal X chromosomal product is expected. The longest cDNAs of each group (clone C1:TFE3/PRCC and clone C12:PRCC, both ≈2 kb in size) were chosen for further characterization.
Involvement of TFE3/PRCC in t(X;1)(p11;q21).
To confirm that the cDNAs that we isolated indeed represent the genes involved in the translocation, C1 (TFE3/PRCC; TFE3-positive) was labeled and hybridized to a Southern blot containing three t(X;1)-positive tumors, adjacent normal tissue and other human cells, hybrid cell lines containing either der(X), der(1), or normal chromosomes X and 1, and the parental rodent lines A3 (hamster) and A9 (mouse) as controls. Normal X chromosome- and chromosome 1-derived fragments (Fig. 2, lanes X and 1) were present in all three t(X;1)-positive tumors, the CL89-12117-derived der(X)-containing somatic hybrid, and human control cells. In the der(1)-containing somatic hybrid, only hamster bands could be detected. Altered restriction fragments were present in two out of three t(X;1)-positive RCCs (CL89-12117 and CL89-17872) and in the der(X)-containing somatic cell hybrid (Fig. 2, arrows). These results indicate that the C1 cDNA indeed represents an (X;1) fusion gene. Mouse and hamster hybridizing fragments were readily detected, suggesting that the corresponding sequences are well conserved during evolution.
Figure 2.
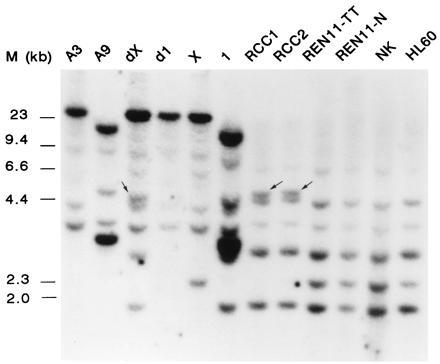
Southern blot analysis of somatic cell hybrids using C1 as a probe. A3 and A9 are the hamster and mouse controls, X and 1 the hybrids containing these chromosomes as the only human component, and dX and d1 the CL89-12117-derived somatic cell hybrids containing either der(X) or der(1). RCC1 (CL89-12117), RCC2 (CL89-17872), and REN11-TT are the t(X;1)-positive RCCs. Lane REN11-N, normal tissue adjacent to REN11-TT; and lane NK, normal kidney tissue. HL60 is a myeloid leukemia cell line. As a molecular size marker, λ HindIII DNA was used. DNA samples were cleaved with EcoRI.
Expression of PRCC, TFE3, and Chimeric Genes.
After hybridization of a Northern blot containing RNA from tumor cells and normal kidney tissue with the cloned RT-PCR breakpoint fragment or cDNA clone C1, an mRNA of ≈2 kb was detected (Fig. 3a). Only a very long exposure of the blot hybridized with the initial RT-PCR fragment showed a very weak 3.2-kb band in normal renal tissue (not shown). Hybridization with a human TFE3 probe containing 3′ sequences (1.9 kb) also revealed an mRNA of ≈3.2 kb, in both tumor and normal tissue (Fig. 3b). Apparently, the normal and fusion transcripts are about equal in size, and, therefore, the size of the exchanged parts of the mRNAs must also be similar. To confirm this and to discriminate between normal and fusion products, we rescreened the same blot using a probe from the 5′ region of C12 (positions 1–678; Fig. 4a), which is present in the normal PRCC mRNA (2 kb) and also in PRCC/TFE3 (3.2 kb; Fig. 3c). Now, mRNAs of different size can be detected in the tumor lanes, corresponding to PRCC/TFE3 and PRCC, whereas, as expected, in normal kidney, only the 2-kb mRNA (PRCC) is present. The PRCC/TFE3 fusion gene is expressed at approximately the same level as PRCC itself.
Figure 3.
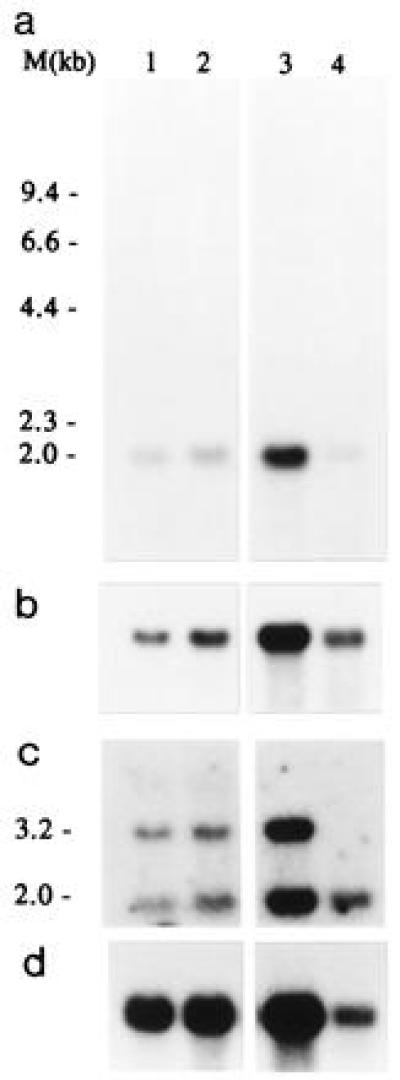
Northern blot analysis of t(X;1)-positive RCCs and normal kidney using C1-cDNA (a), TFE3-cDNA (b), or a 5′ fragment from C12 (positions 1–678) (c) as a probe. Lanes 1 and 2 contain total RNA from CL89-17872 and CL89-12117, respectively, lane 3 and 4 contain oligo(dT)-selected RNA from CL89-12117 (1.5 μg) and normal kidney tissue (cortex and medulla; 0.8 μg), respectively. An actin control hybridization is shown (d). As a molecular size marker glyoxylated λ HindIII DNA was used.
Figure 4.

Nucleotide sequences and deducted amino acid sequences of C12/PRCC [GenBank accession no. X99720X99720 (a) and the TFE3 sequence (b)] present in C1(TFE3/PRCC) (positions 1–639) and the additional 5′ part (accession no. X99721X99721) present upstream of the known TFE3 sequence (accession no. X51330X51330). Breakpoint locations are indicated by arrows (asterisks in the margins). The first 31 nt of the known human TFE3 sequence including the primer sequence are underlined.
Sequence Analysis of PRCC, TFE3/PRCC, and TFE3.
Sequence analysis of both C12 (PRCC) and C1 (TFE3/PRCC) (Fig. 4) demonstrated that the translocation results in a fusion of the two genes at positions 680–681 in the PRCC cDNA sequence. In C1 (TFE3/PRCC), this 5′ part is replaced by 639 bp of 5′ human TFE3. This exchange interrupts the predicted protein sequence of PRCC (Fig. 4, arrow). However, a novel open reading frame is formed resulting in a putative fusion protein of 513 aa. Screening of data bases for known sequences revealed no significant homologies with PRCC except for several ESTs, among which the one mentioned above. Searches for motifs in the predicted protein revealed 1 N-linked glycosylation site (position 189), 3 cAMP/GMP-dependent protein kinase phosphorylation sites, 15 casein kinase II phosphorylation sites, 8 protein kinase C phosphorylation sites distributed over the protein, and 1 tyrosine kinase phosphorylation site (position 337).
The reciprocal translocation product contains the first 680 bp of PRCC coupled to the 3′ part of TFE3 (position 640–poly(A)-tail). The 150-bp stretch that we originally identified showed an 87% homology to the first 150 bp of the mouse TFE3 (GenBank accession no. S76673S76673). Based on this homology, we assume that the 150-bp stretch represents an as yet unidentified 5′ sequence of the human TFE3 gene. When aligning the mouse and human TFE3 cDNAs as deposited in the GenBank data base (accession no. X51330X51330), the known human sequence starts at position 255 of the mouse sequence, suggesting that a stretch of ≈100 bp of human TFE3 was still missing (corresponding to positions 150–255 of the mouse sequence). To clone this missing part, we devised another RT-PCR using a second TFE3 primer located in the known human sequence to prime the RNA (from placenta and kidney) and as the second primer the same TFE3 upstream primer that we used before. Translation of this sequence (Fig. 4b) again revealed an open reading frame that is in line with the open reading frames of both the 5′ C1/TFE3 part and known TFE3 sequences downstream. This means that PRCC/TFE3 also encodes a putative fusion protein, using the first PRCC ATG at position 213 as a start codon (Fig. 4b). This fusion protein includes the activation, helix-loop-helix, and leucine-zipper domains of TFE3.
PRCC and TFE3 Expression in Fetal and Adult Tissues.
Expression of normal PRCC was examined in various human and rat tissues (Fig. 5) and was detected in every organ analyzed. The TFE3 gene is also ubiquitously expressed. Fetal brain, lung, liver, and kidney all showed expression of both PRCC and TFE3. In rat, PRCC expression was very low in bladder, and relatively high in brain. Rat kidney showed a relatively low expression of PRCC (not shown).
Figure 5.
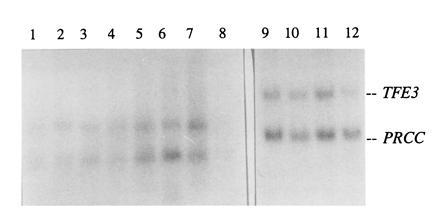
Northern blot analysis of human tissues using C12/PRCC and 3′ TFE3 as probes. Lanes 1–8 contain adult tissues, and lanes 9–12 contain fetal tissues. Lanes: 1, heart; 2, brain; 3, placenta; 4, lung; 5, liver; 6, skeletal muscle; 7, kidney; 8, pancreas; 9, kidney; 10, liver; 11, lung; and 12, brain.
DISCUSSION
Cloning of recurrent translocation breakpoints in tumors has revealed the existence of several genes important in the etiology of cancer. We have identified the genes involved in the papillary RCC-specific t(X;1)(p11;q21), which in some cases occurs as the sole cytogenetic anomaly present in these tumors. Via RT-PCR, we were able to demonstrate a fusion between the transcription factor TFE3 gene on the X chromosome and a novel gene, designated PRCC, on chromosome 1. Screening of a papillary RCC-derived cDNA library with this RT-PCR product resulted in the isolation of both the normal transcript (PRCC) and the translocation product (TFE3/PRCC). Southern blot analysis of three t(X;1)-positive RCCs using the TFE3/PRCC cDNA as a probe confirmed the notion that these genes are indeed involved in the translocation. Aberrant restriction fragments were detected in two of the tumors (CL89-12117 and CL89-17872). The third tumor (REN11-TT) did not show any shifted bands. Previously, a slightly different position of the breakpoint in this RCC was observed (15).
The first suggestion that TFE3, which is a leucine-zipper containing transcription factor of the helix-loop-helix family, might be involved in renal tumors with an Xp11 translocation came from mapping studies (20) and was based on the fact that transcription factors are more often involved in translocation breakpoints. Although previous reports excluded TFE3 as a candidate gene (16, 21), this can retrospectively be attributed to the incompleteness of the known 5′ human TFE3 sequence. Moreover, the sizes of the fusion mRNAs cannot be distinguished from those of their normal counterparts at the Northern blot level. Several ATGs were found in the additionally cloned 5′ TFE3 sequence of which the first one (position 106) more probably functions as a translation initiation site than the one described previously (that corresponds to position 850 of the presently known TFE3 sequence; Fig. 4b). Since the overall homology between the published human and murine TFE3 cDNAs is 80%, and the homology of the 150-bp stretch that we originally found in the genomic breakpoint fragment and subsequently in the RT-PCR breakpoint product is 87% to the mouse TFE3 (1), we assume that this novel sequence represents the 5′ part of the human TFE3 mRNA. In addition, we could demonstrate that a PCR product is formed when using primers corresponding to this 5′ novel sequence and the 3′ known part of human TFE3, which completely supports this assumption.
On the chromosome 1 side of the breakpoint, a novel gene, designated PRCC, was encountered. No significant homologies were found after data base searches. After submission of this manuscript, Sidhar et al. (22) also identified this gene as being involved in the t(X;1). Since no nonfrequent motifs were detected in the predicted PRCC protein sequence, suggestions about its function must remain speculative. The highest homology at the protein level was observed with mouse formin 4 (32.5% in 120 aa overlap; Swiss-Prot accession no. Q05859Q05859), which is a limb deformity protein. Since the mouse and chicken formin (accession no. Q05858Q05858) show a 59% homology over the whole length of the protein, it seems highly improbable that the PRCC gene encodes the human counterpart. The predicted protein is relatively acidic (isoelectric point of 4.85) and contains a relatively high percentage of prolines with several P-P-P-P motifs present. In the SH3 domain of CrkII, binding was observed to proteins containing the P-X-X-P motif (23). A consensus site for tyrosine kinases is also present, indicating that this predicted protein may function in a signaling cascade and, as such, may contribute to tumor formation as part of a fusion product.
Two translocation-related mechanisms for tumor formation have been reported: deregulation of the expression of a protooncogene without the formation of an altered protein and formation of a fusion protein that has transforming capacities. In the first case, which is predominantly encountered in B- and T-cell leukemias and lymphomas, T-cell receptor or immunoglobulin genes become juxtaposed to a protooncogene, often a transcription factor, thereby activating it (1). The second mechanism leads to the formation of fusion proteins. In t(X;1)-positive tumors, the open reading frames of the two genes involved, TFE3, a transcription factor which binds to the μE3 element of the immunoglobulin enhancer (20), and a novel gene, PRCC, are both interrupted, leading to the formation of two putative fusion proteins. Since both fusion genes are expressed, it remains to be determined if both are needed for the establishment of the malignant phenotype or if one of them is causally related to malignant transformation. In addition, an alternative mechanism that may lead to transformation cannot be ruled out. Since most patients carrying a t(X;1) reported to date are male, one can speculate that loss of function of a gene on the X chromosome might represent a crucial step in tumor formation. An indication in this direction comes from a male patient with papillary RCC without an (X;1) translocation but instead, a deletion in the Xp11 region (24). Only two female patients with a t(X;1) have been described so far. One of these patients, who already suffered from a kidney tumor at the age of 9 years, developed multiple tumors in the right kidney at 29 years of age (J. Couturier, personal communication), suggesting that a constitutional mutation may have been present on the X chromosome not involved in the translocation. A cell line which was developed from a second female patient did not contain a normal X chromosome next to der(X) (21). So, teleologically, disruption of the function of TFE3 may be sufficient for establishing the malignant phenotype. Further studies are needed to reveal which mechanism ultimately leads to the formation of this type of renal cell carcinomas. Now that the relevant genes have been identified, tools are available to determine the molecular events underlying the development of t(X;1)-positive papillary renal cell carcinomas.
Acknowledgments
The authors thank E. van den Berg, B. de Jong, G. Kappe, and M. van Asseldonk for advice and support. Dr. A. M. Meloni, Dr. Tomlinson, and Dr. J. Couturier are acknowledged for generously providing tumor and normal material (CL89-12117, CL89-17872, REN11-TT, REN11-N, RP-T, and RP-N). Use of the services and facilities of the Dutch National Expertise Center CAOS/CAMM is gratefully acknowledged. This work was supported by the Dutch Cancer Society Grant 94–733.
Footnotes
Abbreviations: RCC: renal cell carcinoma; RT-PCR, reverse transcription PCR; EST, expressed sequence tag.
References
- 1.Rabbitts T H. Nature (London) 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- 2.Meloni A M, Bridge J, Sandberg A A. J Urol. 1992;148:253–265. doi: 10.1016/s0022-5347(17)36565-5. [DOI] [PubMed] [Google Scholar]
- 3.Latif F, Tory K, Gnarra J, Yao M, Duh F-M, et al. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 4.Shuin T, Kondo K, Torigoe S, Kishida T, Kubota Y, Hosaka M, Nagashima Y, Kitamura H, Latif F, Zbar B, Lerman M I, Yao M. Cancer Res. 1994;54:2852–2855. [PubMed] [Google Scholar]
- 5.Ohta M, Inoue H, Cotticelli M G, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, Mccue P, Druck T, Croce C M, Huebner K. Cell. 1996;84:587–597. doi: 10.1016/s0092-8674(00)81034-x. [DOI] [PubMed] [Google Scholar]
- 6.van den Berg E, van der Hout A H, Oosterhuis J W, Stoerkel S, Dijkhuizen T, Dam A, Zweers H M M, Mensink H J A, Buys C H C M, de Jong B. Int J Cancer. 1993;55:223–227. doi: 10.1002/ijc.2910550210. [DOI] [PubMed] [Google Scholar]
- 7.Meloni A M, Dobbs R M, Jr, Pontes J E, Sandberg A A. Cancer Genet Cytogenet. 1993;65:1–6. doi: 10.1016/0165-4608(93)90050-v. [DOI] [PubMed] [Google Scholar]
- 8.de Jong B, Molenaar I M, Leeuw J A, Idenburg V I S, Oosterhuis J W. Cancer Genet Cytogenet. 1986;21:165–169. doi: 10.1016/0165-4608(86)90042-7. [DOI] [PubMed] [Google Scholar]
- 9.Tonk V, Wilson K S, Timmons C F, Schneider N R, Tomlinson G E. Cancer Genet Cytogenet. 1995;81:72–75. doi: 10.1016/s0165-4608(94)00195-2. [DOI] [PubMed] [Google Scholar]
- 10.Tomlinson G E, Nisen P D, Timmons C F, Schneider N R. Cancer Genet Cytogenet. 1991;57:11–17. doi: 10.1016/0165-4608(91)90184-v. [DOI] [PubMed] [Google Scholar]
- 11.Dijkhuizen T, van den Berg E, Wilbrink M, Weterman M, Geurts van Kessel A, Stoerkel S, Folkers R P, Braam A, de Jong B. Genes Chromosomes Cancer. 1995;14:43–50. doi: 10.1002/gcc.2870140108. [DOI] [PubMed] [Google Scholar]
- 12.Zhao W P, Gnarra J R, Liu S, Knutsen T, Linehan W M, Whang-Peng J. Cancer Genet Cytogenet. 1995;82:128–139. doi: 10.1016/0165-4608(95)00024-j. [DOI] [PubMed] [Google Scholar]
- 13.Hernandez-Marti M J, Orellana-Alonso C, Badia-Garrabou L, Verdeguer Miralles A, Paradis-Alos A. Cancer Genet Cytogenet. 1995;83:82–83. doi: 10.1016/0165-4608(94)00184-7. [DOI] [PubMed] [Google Scholar]
- 14.Weterman M A J, Wilbrink M, Dijkhuizen T, van den Berg E, Geurts van Kessel A. Hum Genet. 1996;98:16–21. doi: 10.1007/s004390050153. [DOI] [PubMed] [Google Scholar]
- 15.Weterman, M. A. J., Wilbrink, M., Janssen, I., Janssen, H. A. P., van den Berg, E., Fisher, S. E., Craig, I. & Geurts van Kessel, A. (1996) Cytogenet. Cell Genet., in press. [DOI] [PubMed]
- 16.Sinke R J, de Leeuw B, Janssen H A P, Olde Weghuis D, Suijkerbuijk R F, Meloni A M, Gilgenkrantz S, Berger W, Ropers H H, Sandberg A A, Geurts van Kessel A. Hum Genet. 1993;92:305–308. doi: 10.1007/BF00244478. [DOI] [PubMed] [Google Scholar]
- 17.Wieacker P, Davies K E, Cooke H J, Pearson P L, Williamson P L, Bhattacharya S, Zimmer J, Ropers H H. Am J Hum Genet. 1984;36:265–270. [PMC free article] [PubMed] [Google Scholar]
- 18.Auffray C, Rougeon F. Eur J Biochem. 1980;107:303–314. doi: 10.1111/j.1432-1033.1980.tb06030.x. [DOI] [PubMed] [Google Scholar]
- 19.McMaster G K, Carmichael G. Proc Natl Acad Sci USA. 1977;74:4835–4838. doi: 10.1073/pnas.74.11.4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henthorn P S, Stewart C C, Kadesch T, Puck J M. Genomics. 1991;11:374–378. doi: 10.1016/0888-7543(91)90145-5. [DOI] [PubMed] [Google Scholar]
- 21.Shipley J M, Birdsall S, Clark J, Crew J, Gill S, Linehan M, Gnarra J, Fisher S, Craig I W, Cooper C S. Cytogenet Cell Genet. 1995;71:280–284. doi: 10.1159/000134127. [DOI] [PubMed] [Google Scholar]
- 22.Sidhar S K, Clark J, Gill S, Hamoundi R, Crew A J, Gwilliam R, Ross M, Linehan W M, Birdsall S, Shipley J, Cooper C S. Hum Mol Genet. 1996;5:1333–1338. doi: 10.1093/hmg/5.9.1333. [DOI] [PubMed] [Google Scholar]
- 23.Meeting Review. Genes Dev. 1995;9:1289–1301. [Google Scholar]
- 24.Ohjima Y, Isawaki H, Ishiguro M, Hara H, Ohgami A, Kikuchi M, Kaneko Y. Cancer Genet Cytogenet. 1993;70:77–78. doi: 10.1016/0165-4608(93)90136-a. [DOI] [PubMed] [Google Scholar]