

Abstract
HMG I(Y) proteins bind to double-stranded A+T oligonucleotides longer than three base pairs. Such motifs form part of numerous NF-AT-binding sites of lymphokine promoters, including the interleukin 4 (IL-4) promoter. NF-AT factors share short homologous peptide sequences in their DNA-binding domain with NF-κB factors and bind to certain NF-κB sites. It has been shown that HMG I(Y) proteins enhance NF-κB binding to the interferon β promoter and virus-mediated interferon β promoter induction. We show that HMG I(Y) proteins exert an opposite effect on the DNA binding of NF-AT factors and the induction of the IL-4 promoter in T lymphocytes. Introduction of mutations into a high-affinity HMG I(Y)-binding site of the IL-4 promoter, which decreased HMG I(Y)-binding to a NF-AT-binding sequence, the Pu-bB (or P) site, distinctly increased the induction of the IL-4 promoter in Jurkat T leukemia cells. High concentrations of HMG I(Y) proteins are able to displace NF-ATp from its binding to the Pu-bB site. High HMG I(Y) concentrations are typical for Jurkat cells and peripheral blood T lymphocytes, whereas El4 T lymphoma cells and certain T helper type 2 cell clones contain relatively low HMG I(Y) concentrations. Our results indicate that HMG I(Y) proteins do not cooperate, but instead compete with NF-AT factors for the binding to DNA even though NF-AT factors share some DNA-binding properties with NF-kB factors. This competition between HMG I(Y) and NF-AT proteins for DNA binding might be due to common contacts with minor groove nucleotides of DNA and may be one mechanism contributing to the selective IL-4 expression in certain T lymphocyte populations, such as T helper type 2 cells.
Keywords: T-cell activation, T helper type 2 cells
Numerous, if not all, lymphokine genes are strongly expressed after stimulation of T lymphocytes by signals that lead to T-cell activation and proliferation. The prototype of a lymphokine promoter, which is rapidly induced during T-cell activation, is the interleukin 2 (IL-2) promoter/enhancer (see ref. 1 for review). A further lymphokine promoter whose induction is similar, but not identical, to the IL-2 promoter is the interleukin 4 (IL-4) promoter. A number of signals that lead to the induction of the IL-2 promoter in T lymphoma cells, such as phorbol esters, Ca2+ ionophores, and lectins, also induce the IL-4 promoter, whereas low doses of the immunosuppressants cyclosporin A (CsA) and FK506, which prevent T-cell activation, suppress the induction of both the IL-2 and IL-4 promoters. Therefore, several transcription factors, which were found to induce the IL-2 promoter in T cells, also contribute to the induction of the IL-4 promoter in these cells (2–6).
One prominent class of transcription factors that control both the induction of IL-2 and IL-4 promoters in T lymphocytes are the NF-AT factors. There are two high-affinity-binding sites within the murine and human IL-2 and four (or five) NF-AT-binding sites within the murine and human IL-4 promoters (see ref. 7 for review). Mutations within the NF-AT-binding sites of the murine IL-4 promoter, which suppress NF-AT binding, had a deleterious effect on promoter induction (2, 3). One NF-AT-binding site, the mutation of which led to a drastic decrease of the IL-4 promoter activity, is the so-called Pu-bB site (2) [also designated as the “P site” (8, 9); upstream promoter site, IL-4 UPS (10); activation responsive element, ARE (5); and CS1 element (11) in other studies]. This is a composite factor-binding site, which was found to span about 35 nt in DNase I footprint experiments (2). In addition to NF-AT, several other factors such as AP-1, octamer, NF-κB, and C/EBP were described to bind to this site (2, 5, 8, 10, 12, 13). Due to a continuous stretch of 12 A+T nt, this site also provides a high-affinity-binding site for HMG I(Y) proteins.
HMG I(Y) proteins are abundant chromosomal proteins of low molecular weight, which were found to bind to the DNA minor groove of A+T stretches longer than 3 bp (14, 15). HMG I(Y) proteins are unable to stimulate (or inhibit) promoter activities on their own but they are able to interact with other DNA-binding proteins to modify promoter activities (16–19). This has been studied in greatest detail for the virus-mediated induction of the human interferon β (IFN-β) promoter, where HMG I(Y) proteins bind to several sites of the promoter, interact with NF-κB and ATF-2 factors, and thereby enhance the viral induction of the IFN-β promoter (16, 17, 20).
In an earlier report on the activity of the murine IL-4 promoter in T cells, we reported that HMG I(Y) exerts a suppressive effect on IL-4 promoter induction (2). However, it remained to be shown how this suppression by HMG I(Y) occurs. We show here that HMG I(Y) interferes with the binding of NF-AT factors to Pu-bB, a high-affinity NF-AT-binding site of the IL-4 promoter. The interference between HMG I(Y) and NF-AT factors seems to be one molecular mechanism for the selective activity of the IL-4 promoter in certain T lymphocyte subsets, such as in T helper type 2 (Th2) cells.
MATERIALS AND METHODS
Cell Culture, DNA Transfection, and Luciferase Assays.
Human Jurkat T leukemic cells and murine El4 T lymphoma cells were grown in RPMI medium, and HeLa cells were grown in DMEM medium containing 5% fetal calf serum. Usually, 4 × 107 T cells were transfected with 20 μg of DNA using the DEAE dextran transfection protocol. HeLa cells were transfected using the calcium phosphate protocol. The cells were cultured for 20 h after transfection and then divided. One portion of the cells was left as an uninduced control, one portion was induced by 10 ng phorbol 12-O-tetradecanoylphorbol-13-acetate (TPA) per ml and 0.5 μM ionomycin and one portion by TPA + ionomycin and CsA (100 ng/ml). Transfection experiments were repeated at least three times and standard deviations calculated as shown in the figures.
DNA Cloning.
The construction of numerous IL-4 promoter/chloramphenicol acetyltransferase (CAT) plasmids has been described (2). In all thymidine kinase (tk)-CAT constructs, the CAT reporter gene was removed by BglII/KpnI digestion and replaced by the BamHI/KpnI fragment from the luciferase vector ptkLuc containing the luciferase gene of plasmid pXPL (22). Trimers of the Pu-bB-HM1 site (see below, but with two SalI sites) were introduced into the SalI site in front of the tk promoter, the two distal copies in the direction of transcription, the proximal copy in the opposite orientation. Trimers of the Pu-bB-NM site were cloned between the SalI/BamHI sites, the most proximal and distal copies in the direction of transcription, the second copy in the opposite direction.
HMG I(Y) site mutations were introduced into the wild-type IL-4 promoter (construct IL4-270; ref. 2) and the promoter containing HMG I(Y) site mutations within Box II (2), using an oligonucleotide-directed mutagenesis system (Amersham) and a Pu-bB-HM1 primer similar to the oligonucleotide shown below. The pRSV-NF-ATc expression vector contains the complete cDNA of NF-ATc (23) and 195 additional 5′ untranslated mRNA nucleotides, which were cloned from a human B-cell cDNA library (S.C., unpublished work). The structure of the HMG I(Y) expression vector has been described (2).
Preparation of Bacterially Expressed Glutathione S-Transferase (GST) Proteins.
The GST-NF-ATp vector was constructed by the insertion of the NaeI/SmaI fragment of murine NF-ATp cDNA containing the Rel-like DNA-binding domain (24) into the GST vector pGEX-2 (25). Chimeric GST proteins were prepared as described (10).
Preparation of Nuclear Protein Extracts and DNA/Protein-Binding Studies.
Nuclear proteins from Jurkat cells, El4 cells, and human peripheral T lymphocytes were prepared (26) and used in electrophoretic mobility shift assays (EMSAs) as described previously (2). For the detection of HMG I(Y) binding, the EMSAs were carried out in the presence of 200 ng of sonicated herring sperm DNA per assay or, in a few cases (Fig. 1B, lanes 9–12), poly(dG·dC) instead of poly(dI·dC) as nonspecific inhibitor. In “supershift” EMSAs, 1 μl of α-NF-ATp (raised against peptide 67.1) (24) or α-HMG I(Y) (16) was added after the addition of nuclear proteins.
Figure 1.
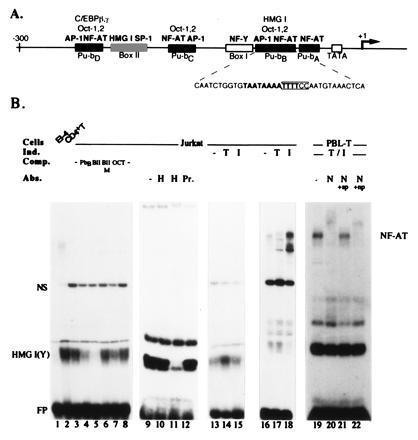
The Pu-bB (P site) of the murine IL-4 promoter is a strong HMG I(Y)-binding site. (A) Scheme of the murine IL-4 promoter. The factor binding sites were determined in DNase I footprint protection experiments (2) and are shown as solid boxes. The binding of C/EBP factors to Pu-bD has been demonstrated for the human IL-4 promoter (6, 27). Due to sequence homologies with the Pu-bB, the Pu boxes C and D should harbor weak AP-1 and octamer sites, although this has not been demonstrated so far (2, 12). The sequence of Pu-bB is shown below the promoter scheme. The “core” of NF-AT recognition sequence is indicated, and the HMG I(Y) site is shown in boldface letters. (B) Detection of HMG I(Y) binding to the Pu-bB in EMSAs. Four microgram nuclear proteins from El4 T lymphoma cells (lane 1), peripheral human CD4+ T lymphocytes (lane 2), Jurkat T leukemic cells (lanes 3–18), and peripheral human T lymphocytes (lanes 19–22) were incubated with a radioactively labeled Pu-bB probe in the presence of 200 ng herring sperm DNA (lanes 1–8 and 13–15), 2 μg poly(dG·dC) (lanes 9–12), and 700 ng (lanes 16–18) or 2 μg poly(dI·dC) (lanes 19–22) as nonspecific competitors. In lanes 14 and 17, nuclear proteins were isolated from T cells treated by TPA for 3 h (T), in lanes 15 and 18 by ionomycin for 3 h (I), and in lanes 19–22 by TPA and ionomycin for 10 h. In all the other lanes, nuclear proteins from uninduced cells were used. For specific competition (lanes 4–7), 50 ng of DNA of Pu-bB, Box II, mutated Box II, or an octamer consenus site were added as indicated. In lanes 10–12, 0.5 μl or 1 μl of a HMG I(Y)-specific antibody (H) (16) or 1 μl of pre-immune serum (Pr.) was added to the EMSAs for the detection of specific HMG I(Y) binding. In lanes 20–22, 1 μl of an antibody raised against a NF-ATp peptide (N) (24, 28) was added. In lanes 21 and 22, 50 ng of the immunogenic 67.1 peptide (ref. 28; N+sp) or an unrelated peptide (N+np) were added. The positions are indicated for the free probe (FP), the HMG I(Y), and NF-AT complexes. NS indicates an unspecific complex generated with nuclear proteins from Jurkat cells.
The following chemically synthesized oligonucleotides were used in EMSAs as probes or for competition:
Pu-bB:(−92)tcgaCAATCTGGTGTAATAAAATTTTCCAATGTAAACTCAg (−57) GTTAGACCACATTATTTTAAAAGGTTACATTTGAGTcctag Pu-bB-NM:(−92)tcgaCAATCTGGTGTAATAAAATTTTAAAATGTAAACTCAg (−57) GTTAGACCACATTATTTTAAAATTTTACATTTGAGTCctagPu-bB-OM:(−92)tcgaCAATCTGGTGTAATAAAATTTTCCAATGTGGACTCAg (−57) GTTAGACCACATTATTTTAAAAGGTTACACCTGAGTcctag Pu-bB-HM1:(−92)tcgaCAATCTGGTGTAATCAAATTTTCCAATGTAAACTCAg (−57) GTTAGACCACATTAGTTTAAAAGGTTACATTTGAGTcagct Pu-bB-HM2:(−92)tcgaCAATCTGGTGTAATCAAACTTTCCAATGTAAACTCAg (−57) GTTAGACCACATTAGTTTGAAAGGTTACACCTGAGTcagct Pu-bB-HM3:(−92)tcgaCAATCTGGTGTCATCAAACTTTCCAATGTAAACTCAg (−57) GTTAGACCACAGTAGTTTGAAAGGTTACACCTGAGTcagct Pu-bB-HOM3: (−92)tcgaCAATCTGGTGTCATCAAACTTTCCAATGTGGACTCAg (−57) GTTAGACCACAGTAGTTTGAAAGGTTACACCTGAGTcagctBoxII(−225)tcgaCACTTTGTAGATTTAAAAAAAAAAGGGGGGGGGAGGGGTGTTTCg (−182) GTGAAACATCTAAATTTTTTTTTTCCCCCCCCCTCCCCACAAAGcctagBoxIIM (−225)tcgaCACTTTGTAGATTTCAACAACAAAGGGGGGGGGG (−194) GTGAAACATCTAAAGTTGTTGAAACCCCCCCCCCctag Oct consensus site: ctagaGCAGAAATGCAAATTATACCCG CGTCTTTACGTTTAATATGGGCttcga
Cu2+/phenanthroline footprint assays, G methylation and KMnO4 T oxidation interference assays were done according to standard protocols as described (10).
RESULTS
Pu-bB, a NF-AT Site from the Murine IL-4 Promoter Is a Strong HMG I(Y)-Binding Site.
The murine IL-4 promoter contains two A+T stretches longer than 10 bp. In DNase I footprint protection experiments using protein extracts from El4 T lymphoma cells and P815 mast cells, these long A+T stretches were found to be part of two large protected DNA regions, the so-called Box II and the Pu-bB (2), also designated as P site (Fig. 1A). Both sites are composite factor-binding sites. Box II consists of two types of DNA segments, a G+C-rich half to which Sp-1-like factors bind with high affinity (S.K.-H., unpublished results), and an A+T-rich half, which is a binding site for HMG I(Y) proteins (2). The Pu-bB harbors one of the multiple NF-AT-binding motifs of the IL-4 promoter (Figs. 1 and 2) and also several other binding sites for nuclear proteins. One of them is HMG I(Y). This is shown in Fig. 1B, where nuclear proteins from El4 T lymphoma cells (lane 1), peripheral human CD4+ T lymphocytes (lane 2), and human Jurkat T leukemic cells (lanes 3–15) were investigated in EMSAs under conditions allowing the efficient binding of HMG I(Y) proteins to Pu-bB DNA. The specificity of HMG I(Y) binding is shown by competition of HMG I(Y) complex formation with an excess of other HMG I(Y)-binding sites and an antibody raised against HMG I(Y). Competition with a 250-fold molar excess of Box II, a strong HMG I(Y)-binding site, resulted in a significant reduction of HMG I(Y) complex formation (Fig. 1B, lane 5), whereas the same molar excess of a mutated Box IIM oligonucleotide was unable to compete (lane 6). A very weak competition was observed with an octamer consensus site, a poor HMG I(Y)-binding site (lane 7). HMG I(Y), but no other complex formation was also suppressed by an HMG I(Y)-specific antibody used in the EMSA incubations (Fig. 1B, lanes 9–12).
Figure 2.
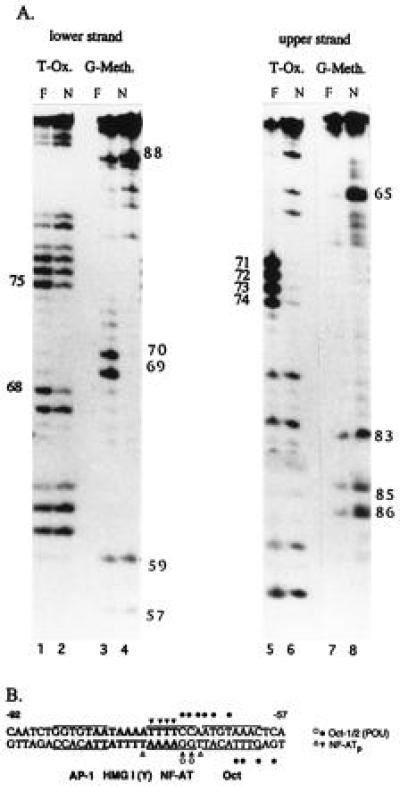
Binding of NF-ATp to the Pu-bB DNA. (A) KMnO4 T oxidation (T-Ox.) and G-methylation (G-Meth.) interference assays. In the T-oxidation assays, the T residues of single-stranded Pu-bB DNA labeled at their 5′ ends were oxidized by KMnO4 treatment, annnealed with cold complementary Pu-bB DNA, and incubated with bacterially expressed GST-NF-ATp, followed by a preparative EMSA as described (10). The free probes (F) and NF-ATp complexes (N) were electroblotted, isolated, cleaved by piperidine, and fractionated on a 12% polyacrylamide gel, along with a T-specific sequence reaction (not shown). In the G-methylation interference assays, labeled Pu-bB DNAs were methylated by dimethyl sulfate treatment, annealed, and processed as the oxidized probes. (B) Compilation of contacts of NF-ATp with the Pu-bB DNA. Strong interactions of NF-ATp are indicated by solid triangles, weak interactions by open triangles. The HMG I(Y)-binding motif is shown in boldface letters. The core motifs for the binding sites of NF-AT, AP-1, and octamer factors are boxed. The contact points of octamer factors shown as solid and open circles have been determined in T-oxidation and G-methylation interference studies (A.H., unpublished data) using bacterially expressed GST-Oct1-POU and GST-Oct2-POU domain proteins (see ref. 10).
The results of the EMSAs shown in Fig. 1B indicate that the concentrations of HMG I(Y) proteins vary conspicuously among different T cells. High concentrations of HMG I(Y) were found in Jurkat T leukemia cells and, in particular, in human peripheral T lymphocytes, whereas relatively low concentrations were found in murine El4 T lymphoma cells. Moreover, phorbol ester treatment of Jurkat T cells led to a significant increase of HMG I(Y) binding, similar to that published previously for human HUT-78 T lymphoma cells and K562 erythroleukemia cells (29). However, treatment with the Ca2+ ionophore ionomycin alone did not affect HMG I(Y) binding (Fig. 1B, lanes 13–15) but stimulated strongly the generation of NF-AT/Pu-bB complexes (lanes 16–18). This is consistent with the conditions for the induction of NF-AT/DNA complexes (7). Since in certain T cells, such as Th2 cells, the IL-4 promoter is efficiently stimulated by ionomycin alone (4), those differences in the induction conditions for HMG I(Y) and NF-AT complex formation might be of importance for the IL-4 promoter induction in these cells.
NF-AT and HMG I(Y) Proteins Overlap and Compete in Their Binding to the Pu-bB DNA.
In activated peripheral human T lymphocytes, which contain high concentrations of HMG I(Y) (Fig. 1B, lane 2), NF-ATp appears to be one of the major NF-AT proteins binding to the Pu-bB. EMSAs with an antibody raised against a NF-ATp peptide led to the “supershift” of almost all proteins in NF-AT/Pu-bB complexes (see Fig. 1B, lane 20). The supershift was abolished by the addition of an excess of the immunogenic NF-ATp peptide (+sp, lane 21), whereas addition of the same excess of an unrelated peptide was without effect on the supershift (+np, lane 22).
To investigate the binding of NF-ATp to the Pu-bB DNA in more detail, recombinant NF-ATp protein was used in G-methylation and T-oxidation interference studies with a Pu-bB DNA probe. In the methylation interference studies, the two G residues of positions −69 and −70, and in the oxidation experiments the T residues of positions −71, −72, −73, and −74 were found to be contacted by NF-ATp (Fig. 2A). Weaker contacts of NF-ATp were also observed for the T residues −68 and −75 on the opposite DNA strand. These two nucleotides flank the NF-AT “core” motif GGAAAA (see Fig. 2B). Probably due to the binding of HMG I(Y) to the minor groove of DNA (15), we were unable to detect contacts between HMG I(Y) proteins and Pu-bB DNA in parallel assays.
To determine the binding of HMG I(Y) to Pu-bB DNA we introduced mutations within the NF-AT, octamer, and putative HMG I(Y)-binding site of Pu-bB DNA and tested their protein binding in EMSAs. Substitution of two G residues at the positions −69 and −70 of Pu-bB by A residues in mutation NM, which destroy the NF-AT-binding motif and enlarge the A+T stretch, led to a distinct increase of HMG I(Y) binding (Figs. 3B, lane 8 and 5A, lanes 3 and 6). The octamer mutation, OM, did not affect HMG I(Y) binding (Fig. 3A, lane 2), whereas all mutations within the putative HMG I(Y) site had a deleterious effect on HMG I(Y) binding. Mutation HM1, which left intact an A+T stretch of 7 bp, had a relatively moderate reducing effect (see Fig. 3A, lane 3, but see also Figs. 3B, lane 4 and 5A, lanes 2 and 5), whereas the mutations HM2, HM3, and HOM3, which contain additional mutations in the flanking A/T bp of the proposed HMG I(Y) site (and HOM3 in the octamer site) abolished any HMG I(Y) binding. This is shown directly in Fig. 3 (lanes 4–6) using HM2, HM3, and HOM3 as probes, or as competitors in EMSAs in which HM2, HM3, and HOM3 were unable to compete for HMG I(Y) complex formation (Fig. 3B, lanes 5–7). These results demonstrate that the nucleotides from position −74 to −81 of Pu-bB overlapping the NF-AT-binding site correspond to a strong HMG I(Y)-binding site.
Figure 3.
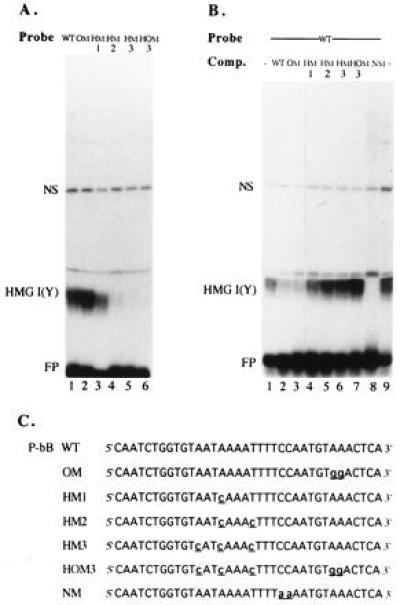
Binding of HMG I(Y) to Pu-bB mutated in the NF-AT, octamer, and HMG I(Y) motifs. (A) EMSAs. Four micrograms of nuclear proteins from Jurkat cells were incubated with a Pu-bB wild-type probe (WT, lane 1) or probes mutated in the octamer motif (OM, lane 2), the HMG I(Y)-binding site (HM1, HM2, and HM3, lanes 3–5) or in both the octamer and HMG I(Y) motifs (HOM3, lane 6). The sequence of mutated Pu-bB probes is shown below in C. (B) EMSA competitions. Nuclear proteins from Jurkat cells were incubated with a Pu-bB wild-type probe in the absence (lanes 1 and 9) or presence of 50 ng of the Pu-bB wild-type DNA (lane 2) or oligonucleotides containing mutations in the octamer motif (OM, lane 3), the HMG I(Y) motif (HM1, HM2, and HM3, lanes 4–6), the octamer and HMG I(Y) motifs (HOM3, lane 7), or the NF-AT motif (NM, lane 8). (C) Sequences of the mutated Pu-bB oligonucleotides used in the EMSAs in A and B.
Figure 5.
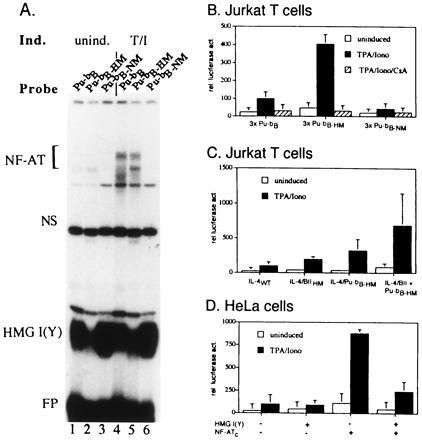
The binding of HMG I(Y) to Pu-bB suppresses transcriptional activity of Pu-bB and of the IL-4 promoter. (A) Interruption of the continuous A+T stretch of Pu-bB by mutation Pu-bB-HM1 impairs the binding of HMG I(Y) to Pu-bB, whereas mutation of the NF-AT-binding site, Pu-bB-NM, enhances HMG I(Y) but abolishes NF-AT binding. A wild-type Pu-bB probe (lanes 1 and 4) and Pu-bB probes bearing mutations within the HMG I(Y) (Pu-bB-HM1, lanes 2 and 5) or NF-AT-binding sites (Pu-bB-NM, lanes 3 and 6) were incubated with 3 μg nuclear proteins from uninduced Jurkat cells (lanes 1–3) or Jurkat cells induced by TPA/ionomycin for 3 h (lanes 4–6) in the presence of 200 ng herring sperm DNA. (B) Trimers of Pu-bB-HM1 bearing mutated HMG I(Y)-binding sites show an increased constitutive and inducible activity in comparison to trimers of wild-type Pu-bB in Jurkat cells. tk-luciferase constructs carrying trimers of Pu-bB, Pu-bB-HM1, or Pu-bB-NM in front of the tk promoter were transfected into Jurkat cells. Twenty hours later, one-third of the cells were induced by TPA and ionomycin for 8 h in the absence of CsA, one-third in the presence of CsA, and one-third were left as uninduced control. (C) Introduction of HMG I(Y) mutations into the IL-4 promoter enhances the promoter activity. tk-luciferase constructs containing the IL-4 wild-type promoter (IL-4WT), the promoter bearing a mutated HMG I(Y) site within Box II (IL-4/BIIHM), a mutated HMG I(Y) site, HM1 within Pu-bB (IL-4/Pu-bB-HM) or mutations in both sites (IL-4/BII+Pu-bBHM) were transfected into Jurkat cells. Twenty hours later, one-half of the cells were treated with TPA + ionomycin for 8 h, and one-half were left uninduced. (D) Overexpression of HMG I(Y) in HeLa cells suppresses NF-ATc-mediated transactivation of a Pu-bB controlled reporter gene in HeLa cells. A tk-luciferase reporter gene containing trimers of Pu-bB in front of the tk promoter was cotransfected with NF-ATc and/or HMG I(Y) expression vectors. Twenty hours later, the cells were induced by TPA/ionomycin. After an additional 18 h, cells were harvested, protein extracts prepared, and used for luciferase assays.
Figure 4.
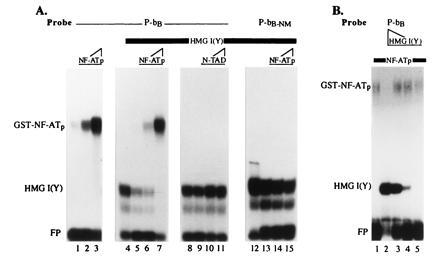
NF-ATp and HMG I(Y) compete for the binding to Pu-bB DNA. (A) A Pu-bB probe was incubated with increasing concentrations of (≈10, 20, and 100 ng) bacterially expressed GST-NF-ATp in the absence (lanes 1–3) or presence (lanes 5–7) of a constant amount of HMG I(Y) purified from Ehrlich ascites cells [see ref. 2 for the isolation of HMG I(Y)]. In lane 4, Pu-bB DNA was incubated with HMG I(Y) alone. In lanes 8–11, Pu-bB DNA was incubated with a constant amount of HMG I(Y) in the absence (lane 8) or presence (lanes 9–11) of increasing concentrations of (≈10, 20, and 100 ng) of GST-NFATc-TAD B, i.e., a GST-NF-ATc fusion protein containing the transacting domain B of NF-ATc (S.C., unpublished work). In lanes 12–15, a constant amount of HMG I(Y) was incubated in the absence (lane 12) or presence (lanes 13–15) of increasing concentrations of (10, 20, and 100 ng) GST-NF-ATp with the Pu-bB-NM motif mutated in the NF-AT-binding site of Pu-bB (see Fig. 3C). Note the inhibition of HMG I(Y) binding to Pu-bB by high concentrations of NF-ATp (lanes 6 and 7). No competition of HMG I(Y) binding by NF-ATp was observed to a Pu-bB-NM motif (lanes 12–15) to which NF-ATp is unable to bind. Likewise, an NF-AT protein lacking the DNA-binding domain was unable to displace HMG I(Y) from the binding to Pu-bB (lanes 8–11). (B) Increasing concentrations of purified HMG I(Y) were incubated with a low, constant amount of GST-NF-ATp (lanes 2–5). In lane 1, NF-ATp protein alone was incubated. Note the inhibition of NF-ATp binding by high concentrations of HMG I(Y) (lane 2).
The overlapping binding of NF-AT and HMG I(Y) proteins to the Pu-bB could have different consequences for factor binding such as cooperation between the two factors, as has been described for NF-κB and HMG I(Y) proteins (20), or in contrast, competition between the factors, as we detected for NF-AT and octamer factors (10). To determine whether NF-ATp and HMG I(Y) proteins cooperate or compete with each other at the Pu-bB, recombinant NF-ATp and HMG I(Y) proteins purified from mammalian cells were titrated in EMSAs. As shown in Fig. 4A (lanes 5–7), high concentrations of GST-NF-ATp protein were able to suppress the binding of HMG I(Y) to Pu-bB. Vice versa, high concentrations of HMG I(Y) were also able to suppress the binding of GST-NF-ATp to Pu-bB DNA (Fig. 4B, lanes 2–5). Under the same conditions high concentrations of an unrelated GST fusion protein, i.e., GST-NF-ATc-TAD coding for the transacting domain of NF-ATc, were unable to displace HMG I(Y) from Pu-bB DNA (Fig. 4A, lanes 9–11). Likewise, GST-NF-ATp protein was unable to displace HMG I(Y) from a site, i.e., Pu-bB-NM, to which NF-AT was unable to bind (Fig. 4A, lanes 13–15). These results demonstrate that HMG I(Y) and NF-AT do not cooperate but compete in their binding to the Pu-bB of the IL-4 promoter.
Inhibition of HMG I(Y) Binding to the Pu-bB Enhances the Activity of the IL-4 Promoter in T Cells.
To determine whether the binding of HMG I(Y) to the Pu-bB has any effect on the induction of the IL-4 promoter, we investigated the effect of mutations within the HMG I(Y)-binding site on the Pu-bB activity in T cells. In a first set of experiments, trimers of “wild-type” Pu-bB, or trimers of Pu-bB containing mutations within the HMG I(Y) or NF-AT motifs, were inserted in front of a tk-luciferase reporter gene and transfected into Jurkat cells. The mutations HM1 and NM either reduced the binding of HMG I(Y) (Fig. 5A, lanes 2 and 5) or NF-AT (lane 6) to Pu-bB. As shown in Fig. 5B, suppression of HMG I(Y) binding to the Pu-bB had an enhancing effect on both the constitutive and inducible activity of the reporter gene. In contrast, suppression of NF-AT binding resulted in a distinct decrease of reporter gene activity. Since the increase of the inducible activity of 3xPu-bB-HM construct could be inhibited by CsA, one may conclude that the increase in the Pu-bB activity may be mediated by NF-AT, the activity of which is inhibited by low doses of CsA (7).
A similar positive effect on the reporter gene activity was also observed when mutations were introduced into the HMG I(Y)-binding sites in the context of the entire IL-4 promoter. Mutation HM1 resulted in a 3-fold increase of IL-4 promoter activity in Jurkat cells, whereas double mutations within the Pu-bB as well as Box II resulted in a 7-fold increase of inducible promoter activity (Fig. 5C). These data support our earlier conclusion that HMG I(Y) proteins exert a negative effect on the IL-4 promoter (2).
To test the mutual functional interference between HMG I(Y) and NF-AT proteins in another system, we cotransfected HMG I(Y) and NF-ATc expression vectors, along with a 3xPu-bB controlled reporter gene into HeLa cells, which do not express NF-AT factors (30). NF-ATc is a strong transactivator and its DNA-binding properties are similar if not identical to NF-ATp (31). Overexpression of NF-ATc in HeLa cells led to a 6-fold increase of constitutive and a 9-fold increase of inducible Pu-bB activity. Coexpression of HMG I(Y) in the same cells resulted in a distinct decrease in NF-ATc-mediated Pu-bB activity (Fig. 5D). Again, these results indicate a functional interference between HMG I(Y) and NF-AT proteins in the control of IL-4 promoter activity.
DISCUSSION
The results of this study show that HMG I(Y) and NF-AT factors compete for binding to the NF-AT Pu-bB site of the murine IL-4 promoter. The results also indicate that the competition between these types of factors for DNA binding plays an important role in the IL-4 promoter induction upon T-cell activation. In peripheral T lymphocytes and Jurkat T cells, which contain high HMG I(Y) concentrations, the IL-4 promoter might be more strictly controlled than in T cells with lower HMG I(Y) concentrations, such as in El4 cells (see Fig. 1B). Similar low HMG I(Y) concentrations were also found in the TH2 cell lines D10 and ATOP (S.K.-H., unpublished work).
The interference between HMG I(Y) and NF-AT factors is in striking contrast to the synergistic binding of HMG I(Y) and NF-κB to the κB site of the IFN-β promoter, which enhances the viral induction of this promoter (16, 20). In addition to the reported direct protein–protein interactions between the HMG I(Y) and NF-κB proteins, the synergistic binding of NF-κB and HMG I(Y) to the IFN-β promoter might also be facilitated by the binding of NF-κB to the major groove and HMG I(Y) to the minor groove of DNA (16). In contrast to NF-κB factors, which share several properties with NF-AT factors, NF-AT also contacts several nucleotides via the minor groove of DNA. Several lines of evidence indicate that NF-ATp contacts some of the GGAAAA base pairs of its core recognition sequence in the minor DNA groove (ref. 21; G. Verdine, personal communication), which overlaps with the HMG I(Y)-binding site of Pu-bB. It has been shown that a minor groove binding agent is able to displace NF-AT but not other factors, including NF-κB, from their binding sequence (32).
Numerous, if not all, lymphokine promoters are controlled by multiple NF-AT-binding sites. Among the four NF-AT sites of the murine IL-4 promoter that we detected in DNase I footprint protection studies (2), similar to Pu-bB, the Pu-bC spans a long stretch of eight A+T base pairs and might also be controlled by HMG I(Y) proteins. The NF-AT/HMG I(Y) site of Pu-bC is also bound by STAT6, an IL-4 induced transcription factor implicated in the development of Th2 cells (33).
However, the interference between HMG I(Y) and NF-AT factors does not seem to be the only mechanism by which HMG I(Y) proteins suppress the IL-4 promoter. Introduction of mutations into the HMG I(Y) site of Box II of the IL-4 promoter also resulted in an increase of IL-4 promoter induction (Fig. 5B), although the binding of HMG I(Y) to this site does not interfere with the binding of Sp-1 or similar factors (S.K.-H., unpublished results). One explanation might be an effect of HMG I(Y) binding to this site on the architecture of the IL-4 promoter. Because one HMG I(Y) molecule contains three so-called A+T-hook-binding motifs (34) it is possible that HMG I(Y) proteins bind simultaneously to two (and three) promoter sites, thereby altering the promoter structure. Long contiguous A+T stretches are also part of the murine IL-5 promoter (35) and could be of similar functional relevance.
The IL-4 promoter is not the only promoter that is suppressed by HMG I(Y) proteins in lymphoid cells. In a recent report, a negative effect of HMG I(Y) on the Ig heavy chain ɛ germ-line RNA promoter has been described, which seems to be mediated by the binding of HMG I(Y) to a binding site for NF-BRE, a STAT-like factor induced by IL-4 (36).
The interference between HMG I(Y) and NF-AT factors in the control of the IL-4 promoter might have implications for the different expression of lymphokines in certain T lymphocyte subsets, in particular in Th2 cells that express IL-4 and IL-5 but not IL-2 and IFN-γ (37). For Jurkat cells we observed a distinct increase of DNA binding for HMG I(Y) after TPA but not ionomycin treatment, whereas the opposite was found for NF-AT. NF-AT binding was strongly enhanced by ionomycin but not TPA treatment (Fig. 1B). It has been reported for several T-cell lines that ionomycin alone is sufficient to induce the IL-4 promoter but not the IL-2 promoter, which needs both TPA and ionomycin for induction (4, 9, 38). Such a situation seems to be typical for Th2 cells in which the predominant increase of free cellular Ca2+ might lead to a preferential induction of the IL-4 but not IL-2 promoter. In Jurkat cells, TPA treatment even lowered the induction of the human IL-4 promoter, compared with the promoter induction by ionomycin alone. This suppression was attributed to the binding of RelA/NF-κB to the Pu-bB of the human IL-4 promoter (13), which differs in 1 bp from the murine Pu-bB. Thus, the equilibrium between HMG I(Y), NF-κb, and other transcription factors induced by TPA-activated protein kinase cascades and NF-AT factors (including c-Maf interacting with NF-AT; ref. 39) induced by enhanced Ca2+ levels might be important for the selective expression of lymphokines in Th1 and Th2 cells.
Acknowledgments
We wish to thank Elke Leibold and Ilona Pietrowski for excellent technical support. We are indebted to Drs. I. Johnston and Anneliese Schimpl for critical reading of the manuscript. For gifts of reagents we are indebted to Drs. J. Altschmied, R. Gerwig, F. Grummt, T. Maniatis, A. Rao, and D. Thanos. We are particularly indebted to Michael Wegner for his generous gifts of purified HMG I(Y). For communicating unpublished results we wish to thank Greg Verdine. This study was supported by the Sonderforschungsbereich 165 (Würzburg) of the Deutsche Forschungsgemeinschaft and the Wilhelm Sander-Stiftung.
Footnotes
Abbreviations: IL-2, interleukin 2; IL-4, interleukin 4; CsA, cyclosporin A; Th2, T helper type 2; IFN-β, interferon β; TPA, 12-O-tetradecanoylphorbol-13-acetate; tk, thymidine kinase; GST, glutathione S-transferase; EMSA, electrophoretic mobility shift assay.
References
- 1.Serfling E, Avots A, Neumann M. Biochim Biophys Acta. 1995;1263:181–200. doi: 10.1016/0167-4781(95)00112-t. [DOI] [PubMed] [Google Scholar]
- 2.Chuvpilo S, Schomberg C, Gerwig R, Heinfling A, Reeves R, Grummt F, Serfling E. Nucleic Acids Res. 1993;21:5694–5704. doi: 10.1093/nar/21.24.5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szabo S J, Gold J S, Murphy T L, Murphy K M. Mol Cell Biol. 1993;13:4793–4805. doi: 10.1128/mcb.13.8.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rooney J W, Hodge M R, McCaffrey P G, Rao A, Glimcher L H. EMBO J. 1994;13:625–633. doi: 10.1002/j.1460-2075.1994.tb06300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tara D, Weiss D L, Brown M A. J Immunol. 1995;154:4592–4602. [PubMed] [Google Scholar]
- 6.Davydov I V, Krammer P H, Li-Weber M. J Immunol. 1995;155:5273–5279. [PubMed] [Google Scholar]
- 7.Rao A. Immunol Today. 1994;15:274–281. doi: 10.1016/0167-5699(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 8.Matsuda I, Masuda E S, Tsuboi A, Behnam S, Arai N, Arai K. Biochem Biophys Res Commun. 1994;199:439–446. doi: 10.1006/bbrc.1994.1248. [DOI] [PubMed] [Google Scholar]
- 9.Kubo M, Kincaid R L, Ransom J T. J Biol Chem. 1994;269:19441–19446. [PubMed] [Google Scholar]
- 10.Pfeuffer I, Klein-Hessling S, Heinfling A, Chuvpilo S, Escher C, Brabletz T, Hentsch B, Schwarzenbach H, Matthias P, Serfling E. J Immunol. 1994;153:5572–5585. [PubMed] [Google Scholar]
- 11.Bruhn K W, Nelms K, Boulay J-L, Paul W E, Lenardo J. Proc Natl Acad Sci USA. 1993;90:9707–9711. doi: 10.1073/pnas.90.20.9707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rooney J W, Hoey T, Glimcher L H. Immunity. 1995;2:473–483. doi: 10.1016/1074-7613(95)90028-4. [DOI] [PubMed] [Google Scholar]
- 13.Casolaro V, Georas S N, Song Z, Zubkoff I D, Abdulkadir S A, Thanos D, Ono S J. Proc Natl Acad Sci USA. 1995;92:11623–11627. doi: 10.1073/pnas.92.25.11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Solomon M J, Strauss F, Varshavsky A. Proc Natl Acad Sci USA. 1986;83:1276–1280. doi: 10.1073/pnas.83.5.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geierstanger B H, Volkman B F, Kremer W, Wemmer D E. Biochemistry. 1994;33:5347–5355. doi: 10.1021/bi00183a043. [DOI] [PubMed] [Google Scholar]
- 16.Thanos D, Maniatis T. Cell. 1992;71:777–789. doi: 10.1016/0092-8674(92)90554-p. [DOI] [PubMed] [Google Scholar]
- 17.Du W, Thanos D, Maniatis T. Cell. 1993;74:887–898. doi: 10.1016/0092-8674(93)90468-6. [DOI] [PubMed] [Google Scholar]
- 18.Leger H, Sock E, Renner K, Grummt F, Wegner M. Mol Cell Biol. 1995;15:3738–3747. doi: 10.1128/mcb.15.7.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.John S, Reeves R B, Lin J-X, Child R, Leiden J M, Thompson C B, Leonard W J. Mol Cell Biol. 1995;15:1786–1796. doi: 10.1128/mcb.15.3.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thanos D, Maniatis T. Cell. 1995;83:1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- 21.Jain J, Loh C, Rao A. Curr Opin Immunol. 1995;7:333–343. doi: 10.1016/0952-7915(95)80107-3. [DOI] [PubMed] [Google Scholar]
- 22.Nordeen S K. BioTechniques. 1988;6:454. [PubMed] [Google Scholar]
- 23.Northrop J P, Ho S N, Chen L, Thomas D J, Timmerman L A, Nolan G P, Admon A, Crabtree G R. Nature (London) 1994;369:497–502. doi: 10.1038/369497a0. [DOI] [PubMed] [Google Scholar]
- 24.McCaffrey P G, Luo C, Kerppola T K, Jain J, Badalian T M, Ho A M, Burgeon E, Lane W S, Lambert J N, Curran T, Verdine G L, Rao A, Hogan P G. Science. 1993;262:750–754. doi: 10.1126/science.8235597. [DOI] [PubMed] [Google Scholar]
- 25.Smith D B, Johnson K S. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 26.Schreiber E, Matthias P, Müller M M, Schaffner W. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davydov I V, Bohmann D, Krammer P H, Li-Weber M. Gene. 1995;161:271–275. doi: 10.1016/0378-1119(95)00271-7. [DOI] [PubMed] [Google Scholar]
- 28.Wang D Z, McCaffrey P G, Rao A. Ann NY Acad Sci. 1995;766:182–194. doi: 10.1111/j.1749-6632.1995.tb26661.x. [DOI] [PubMed] [Google Scholar]
- 29.Friedmann M, Holth L T, Zoghbi H Y, Reeves R. Nucleic Acids Res. 1993;21:4259–4267. doi: 10.1093/nar/21.18.4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brabletz T, Pietrowski I, Serfling E. Nucleic Acids Res. 1991;19:61–67. doi: 10.1093/nar/19.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoey T, Sun Y-L, Williamson K, Xu X. Immunity. 1995;2:461–472. doi: 10.1016/1074-7613(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 32.Ho S N, Boyer S H, Schreiber S L, Danishifevsky S J, Crabtree G R. Proc Natl Acad Sci USA. 1994;91:9203–9207. doi: 10.1073/pnas.91.20.9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lederer J A, Perez V L, desRoches L, Kim S M, Abbas A K, Lichtman A H. J Exp Med. 1996;184:397–406. doi: 10.1084/jem.184.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maher J F, Nathans D. Proc Natl Acad Sci USA. 1996;93:6716–6720. doi: 10.1073/pnas.93.13.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee H, Masuda E S, Arai N, Arai K-I, Yokota T. J Biol Chem. 1995;270:17541–17550. doi: 10.1074/jbc.270.29.17541. [DOI] [PubMed] [Google Scholar]
- 36.Kim J, Reeves R, Rothman P, Boothby M. Eur J Immunol. 1995;25:798–808. doi: 10.1002/eji.1830250326. [DOI] [PubMed] [Google Scholar]
- 37.Mosmann T R, Moore K W. Immunol Today. 1991;12:A49–A53. doi: 10.1016/S0167-5699(05)80015-5. [DOI] [PubMed] [Google Scholar]
- 38.Kubo M, Kincaid R L, Webb D R, Ransom J T. Int Immunol. 1994;6:179–188. doi: 10.1093/intimm/6.2.179. [DOI] [PubMed] [Google Scholar]
- 39.Ho I, Hodge M R, Rooney J W, Glimcher L H. Cell. 1996;85:973–983. doi: 10.1016/s0092-8674(00)81299-4. [DOI] [PubMed] [Google Scholar]