

Abstract
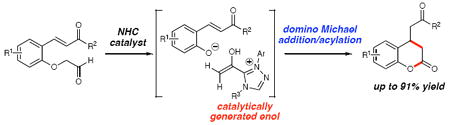
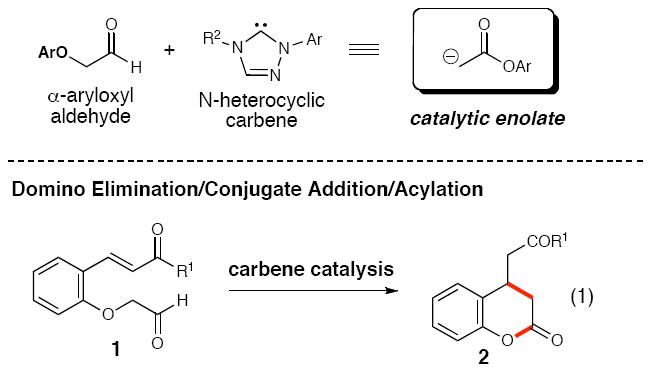
N-Heterocyclic carbenes (NHCs) catalyze a domino Michael addition/acylation reaction to form 3,4-dihydrocoumarins. The reaction proceeds through addition of the NHC to an aryloxyaldehyde followed by elimination of a phenoxide leaving group, generating an enol intermediate. This transient nucleophile generated in situ performs a 1,4-addition onto a conjugate acceptor and the carbene catalyst is regenerated upon acylation of the phenoxide anion resulting in formation of 3,4-dihydrocoumarins.
Enolates are essential intermediates in chemistry and biology due to their high utility in carbon-carbon bond-forming reactions.1 The catalytic, in situ generation of enolates or enols has become increasingly important due to their efficiency in synthesis and emerging environmental concerns. Consequently, significant effort in this area been directed toward metal-catalyzed processes2 that allow for the addition of the corresponding metalloenolate to various electrophiles.3 More recently, attention has turned toward the formation of enolates using only organic molecules as catalysts.4 Numerous groups5678 have employed secondary amines to catalyze enantioselective aldol, Michael, and Mannich reactions through the generation of a reactive enamine intermediate.9 We report here that N-heterocyclic carbenes (NHCs) catalyze the formation of enolates/enols through an elimination process of α-aryloxy aldehydes. The capture of these nucleophiles leads to substituted coumarins by a domino Michael addition/acylation process (eq 1).
In our recent carbene catalysis studies to access enols by the protonation of homoenolates,10 we became interested in whether new carbon-carbon bond-forming reactions were possible using reactive acetate-type enol-NHC intermediates from α-aryloxyacetaldehydes. This concept is distinctive since the phenol in the α-position serves an important dual purpose: first to facilitate generation of the enol equivalent, and second to liberate the carbene catalyst through an acylation event.11 We hypothesized that this process could be evaluated with an aldehyde containing a tethered conjugate acceptor in the ortho-position.12 The likely pathway involves initial addition of the NHC to aldehyde 1 (Scheme 2).13 A formal 1,2-shift of the aldehyde proton results in elimination of phenoxide/phenol derivative III and concomitant formation of enol equivalent II. A Michael addition could then occur which, when followed by intramolecular O-acylation, promotes regeneration of the carbene catalyst. A delicate balance of leaving group ability and nucleophilicity of the α-position substituent is crucial. A catalytic process is untenable if these factors are not optimal in this new type of “rebound” process. For example, a strong leaving group (e.g. chloride or sulfonate) would potentially initiate the reaction faster (I to II and III), but would fail to turn over the catalyst due to weak nucleophilicity (IV to 2).
Scheme 2.
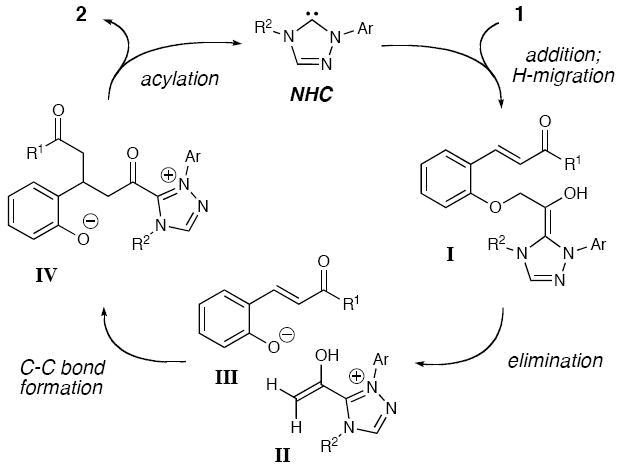
Proposed Reaction Pathway
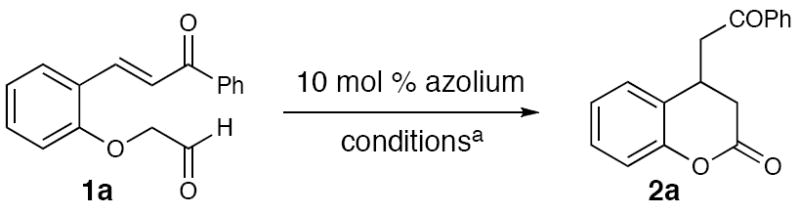
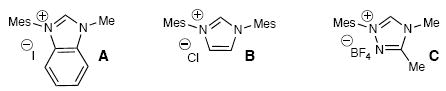
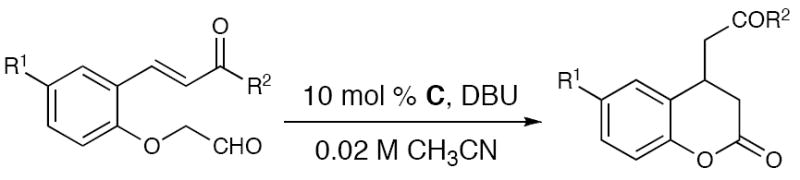
Enone 1a was chosen to explore this carbene-catalyzed enolate formation strategy and leads to the formation of 3,4-dihydrocoumarin products.14 Substituted coumarins and related compounds are biologically active15 and access to this particular substitution pattern by Michael additions to the parent coumarin remains challenging.16 For example, to the best of our knowledge, there are no general and high-yielding reports of conjugate additions of enolates to coumarins. While combining enone 1a with imidazolium or benzimidazolium salts in the presence of base produced a minor amount of the desired product (entries 1 and 2, Table 1), a large increase in yield was observed when triazolium pre-catalyst C17 was used with DBU (entry 5). Further optimization revealed that acetonitrile was the best solvent (60% yield, entry 6). In this particular case, the moderate yield was due to decomposition from prolonged exposure to silica gel. Indeed, quick filtration through SiO2 provided an improved 83% yield of pure desired product 2a (entry 7).
Table 1.
Optimization of Conditions
 | |||
|---|---|---|---|
| entry | azolium salt | Conditions | yieldb |
| 1 | A | 0.1 M CH2Cl2, 1.0 equiv i-Pr2EtN | <5 |
| 2 | B | 0.1 M CH2Cl2, 1.0 equiv i-Pr2EtN | <5 |
| 3 | B | 0.1 M CH2Cl2, 10 mol % DBU | <5 |
| 4 | C | 0.1 M CH2Cl2, 1.0 equiv i-Pr2EtN | <5 |
| 5 | C | 0.05 M CH2Cl2, 10 mol % DBU | 39 |
| 6 | C | 0.05 M CH3CN, 10 mol % DBU | 60 |
| 7 | C | 0.02 M CH3CN, 10 mol % DBU | 83 |
 | |||
All reactinos performed on 0.2 mmol scale.
Isolated yields.
With azolium salt C identified as the most efficient pre-catalyst to promote this well orchestrated domino process, we investigated the scope of the reaction. Electron-withdrawing (entry 3) and electron-donating aryl ketones (entries 4 and 5), as well as naphthyl ketones (entry 6), are accommodated by the reaction conditions. Electron-withdrawing and -donating groups (entries 7-12) can also be placed on the aromatic ring in a variety of patterns. The 3,4-dihydrocoumarin derived from substrate 7 (entry 11) is formed in good yield, although the diastereoselectivity with achiral NHCs is marginal. Additionally, catalyst loading can be decreased to 5 mol % without diminishing the yield (entry 2). Currently, substitution at the α-position of the aldehyde is not accommodated under the reaction conditions. The use of saturated substrates, which replace the phenoxide leaving group with an alkoxide, result in no reaction. From this data, it seems that normal alcohols are not sufficiently stabilized to undergo the first step of the catalytic cycle, i.e., elimination from the initial carbene•aldehyde adduct I. An extensive survey of known chiral azolium salts as catalysts in this reaction provide products in moderate yields but low levels of enantioselecitivity (<10% ee). Importantly, the discovery of this new process provides a strong impetus to develop new chiral catalysts to provide optically active substituted coumarins.
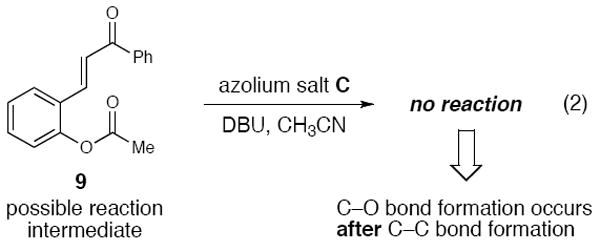
Our investigations of the proposed pathway have focused on probing alternative pathways to the one depicted in Scheme 1. The possibility of the acylation of the phenol by II occurring prior to the Michael addition was strongly discounted by subjecting 9 to reaction conditions. No reaction was observed, thereby supporting that intermediate II, a catalytic enolate equivalent, undergoes C–C bond formation prior to C–O bond formation (Scheme 3).
Scheme 1.
General Concept
Scheme 3.
Reaction Pathway Investigation
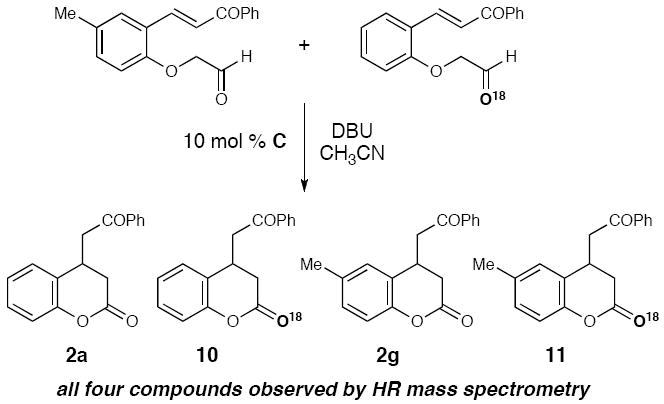
A cross-over experiment was performed (Scheme 4) to determine the fate of the enol (II) that is potentiallly formed in situ. When compounds 1g and 18O-labeled 1j were subjected to the optimized reaction conditions in a single flask, a mixture of compounds 2a, 2g, 10, and 11 was obtained (as observed by HRMS).18 The randomized distribution of the labelled oxygen and methyl group on the aryl ring lends additional support to a pathway involving a bimolecular Michael addition in which the α-aryloxy acetaldehyde fragments to form a reactive enol.
Scheme 4.
Cross-over Experiment
In conclusion, we have developed a new carbene-catalyzed domino process to generate substituted coumarins. A reactive enol is generated catalytically through the addition of an N-heterocyclic carbene to an α-aryloxy aldehyde followed by elimination of a phenoxide ion. The resulting enol intercepts a conjugate acceptor, thereby forming a 3,4-dihydrocoumarin after acylation of the phenoxide/phenol by the acyl azolium intermediate. This new concept of leaving group “rebound” has been developed in the context of carbene catalysis to access enolate reactivity in situ and yield an efficient synthesis of important oxygenated heterocycles. One can envision incorporating additional groups beyond phenols with this rebound capacity that will undoubtedly broaden the scope and utility of this process. Our mechanistic investigations employing both the subjection of potential intermediates to the reaction conditions and cross over experiments using isotopically-labelled substrates support a sequential elimination/C–C formation/C–O acylation pathway. There are many promising potential uses of enols derived from a carbene-catalyzed elimination process and our continued investigations in this area will be reported in due course.
Supplementary Material
Experimental procedures and spectral data for all new compounds, (PDF). This material is available free of charge via the internet at http://pubs.acs.org.
Table 2.
Substrate Scope
 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | product | yield (%)a | |
| 1 | 1a | H | Ph | 2a | 83 |
| 2 | 1a | H | Ph | 2a | 79b |
| 3 | 1b | H | 4-Br-C6H4 | 2b | 77 |
| 4 | 1c | H | 4-Me-C6H4 | 2c | 84 |
| 5 | 1d | H | 4-OMe-C6H4 | 2d | 80 |
| 6 | 1e | H | 2-Naphthyl | 2e | 90 |
| 7 | 1f | OMe | Ph | 2f | 68 |
| 8 | 1g | Me | Ph | 2g | 80 |
| 9 | 1h | Br | Ph | 2h | 77 |
| 10 | 1i | F | Ph | 2i | 91 |
| 11 | 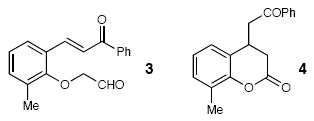 |
66 | |||
| 12 | 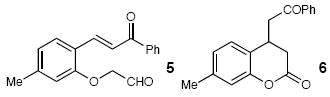 |
66 | |||
| 13 | 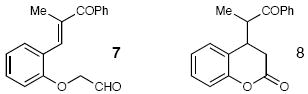 |
70c | |||
Isolated yields.
5 mol % of C and DBU. 1.3:1 as observed by 500 MHz 1H NMR spectroscopy.
Acknowledgments
Financial support for this work has been provided by the NIH (NIGMS, GM73072). We thank GlaxoSmithKline, AstraZeneca, and Boerhinger-Ingelheim for generous research support and Wacker Chemical Corp. and FMCLithium for reagent support. K.A.S. is a Sloan Foundation Fellow. Funding for the NU Integrated Molecular Structure Education and Research Center (IMSERC) has been furnished in part by the NSF (CHE-9871268). E.M.P. is a recipient of a 2008-2009 ACS Division of Organic Chemistry fellowship sponsored by Organic Reactions. We thank Prof. Regan Thomson (NU) for helpful discussions.
References
- 1.(a) Evans DA. Aldrichimica Acta. 1982;15:23. [Google Scholar]; (b) Mukaiyama T, Kobayashi S. Org React (NY) 1994;46:1–103. [Google Scholar]; (c) Arya P, Qin HP. Tetrahedron. 2000;56:917–947. [Google Scholar]
- 2.(a) Yoshikawa N, Yamada YMA, Das J, Sasai H, Shibasaki M. J Am Chem Soc. 1999;121:4168–4178. [Google Scholar]; (b) Trost BM, Ito H. J Am Chem Soc. 2000;122:12003–12004. [Google Scholar]; (c) Hamashima Y, Hotta D, Sodeoka M. J Am Chem Soc. 2002;124:11240–11241. doi: 10.1021/ja027075i. [DOI] [PubMed] [Google Scholar]; (d) Evans DA, Thomson RJ. J Am Chem Soc. 2005;127:10506–10507. doi: 10.1021/ja053386s. [DOI] [PubMed] [Google Scholar]; (e) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew Chem Int Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]; (f) Trost BM, Bream RN, Xu J. Angew Chem Int Ed. 2006;45:3109–3112. doi: 10.1002/anie.200504421. [DOI] [PubMed] [Google Scholar]
- 3.Sodeoka M, Hamashima Y. Bull Chem Soc. 2005;78:941–956. [Google Scholar]
- 4.Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]
- 5.(a) List B, Lerner RA, Barbas CF. J Am Chem Soc. 2000;122:2395–2396. [Google Scholar]; (b) Fonseca MTH, List B. Angew Chem Int Ed. 2004;43:3958–3960. doi: 10.1002/anie.200460578. [DOI] [PubMed] [Google Scholar]
- 6.Brown SP, Brochu MP, Sinz CJ, MacMillan DWC. J Am Chem Soc. 2003;125:10808–10809. doi: 10.1021/ja037096s. [DOI] [PubMed] [Google Scholar]
- 7.Hayashi Y, Yamaguchi J, Sumiya T, Shoji M. Angew Chem Int Ed. 2004;43:1112–1115. doi: 10.1002/anie.200353085. [DOI] [PubMed] [Google Scholar]
- 8.(a) Momiyama N, Torii H, Saito S, Yamamoto H. Proc Natl Acad Sci U S A. 2004;101:5374–5378. doi: 10.1073/pnas.0307785101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yamamoto Y, Momiyama N, Yamamoto H. J Am Chem Soc. 2004;126:5962–5963. doi: 10.1021/ja049741g. [DOI] [PubMed] [Google Scholar]
- 9.List B. Acc Chem Res. 2004;37:548–557. doi: 10.1021/ar0300571. [DOI] [PubMed] [Google Scholar]
- 10. Phillips EM, Wadamoto M, Chan A, Scheidt KA. Angew Chem Int Ed. 2007;46:3107–3110. doi: 10.1002/anie.200605235.Wadamoto M, Phillips EM, Reynolds TE, Scheidt KA. J Am Chem Soc. 2007;129:10098–10099. doi: 10.1021/ja073987e.. For excellent reviews on carbene catalysis, see: Enders D, Niemeier O, Henseler A. Chem Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z.Marion N, Diez-Gonzalez S, Nolan IP. Angew Chem Int Ed. 2007;46:2988–3000. doi: 10.1002/anie.200603380.
- 11.For a review on domino reactions, see: Tietze LF. Chem Rev. 1996;96:115–136. doi: 10.1021/cr950027e.. For NHC reactions using unstable α-chloroaldehydes in which the leaving group (chloride) is not involved in catalysts turnover, see: Reynolds NT, Rovis T. J Am Chem Soc. 2005;127:16406–16407. doi: 10.1021/ja055918a.He M, Uc GJ, Bode JW. J Am Chem Soc. 2006;128:15088–15089. doi: 10.1021/ja066380r.
- 12.(a) Yang JW, Chandler C, Stadler M, Kampen D, List B. Nature. 2008;452:453–455. doi: 10.1038/nature06740. [DOI] [PubMed] [Google Scholar]; (b) Hayashi Y, Itoh T, Ohkubo M, Ishikawa H. Angew Chem Int Ed. 2008;47:4722–4724. doi: 10.1002/anie.200801130. [DOI] [PubMed] [Google Scholar]
- 13.Reynolds TE, Stern CA, Scheidt KA. Org Lett. 2007;9:2581–2584. doi: 10.1021/ol0710515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Borges F, Roleira F, Milhazes N, Santana L, Uriarte E. Curr Med Chem. 2005;12:887–916. doi: 10.2174/0929867053507315. [DOI] [PubMed] [Google Scholar]; (b) Murray RDH. Nat Prod Rep. 1995;12:477–505. [Google Scholar]
- 15.(a) Okuda T, Kimura Y, Yoshida T, Hatano T, Okuda H, Arichi S. Chem Pharm Bull. 1983;31:1625–1631. doi: 10.1248/cpb.31.1625. [DOI] [PubMed] [Google Scholar]; (b) Demir AS, Gross RS, Dunlap NK, Bashirhashemi A, Watt DS. Tetrahedron Lett. 1986;27:5567–5570. [Google Scholar]; (c) Posakony J, Hirao M, Stevens S, Simon JA, Bedalov A. J Med Chem. 2004;47:2635–2644. doi: 10.1021/jm030473r. [DOI] [PubMed] [Google Scholar]
- 16.(a) Connor R, McClellan WR. J Org Chem. 1939;3:570–577. [Google Scholar]; (b) Dixon DJ, Horan RAJ, Monck NJT, Berg P. Org Lett. 2004;6:4427–4429. doi: 10.1021/ol048568q. [DOI] [PubMed] [Google Scholar]; (c) Matsuda T, Shigeno M, Murakami M. J Am Chem Soc. 2007;129:12086–12087. doi: 10.1021/ja075141g. [DOI] [PubMed] [Google Scholar]
- 17.Chan A, Scheidt KA. J Am Chem Soc. 2008;130:2740–2741. doi: 10.1021/ja711130p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and spectral data for all new compounds, (PDF). This material is available free of charge via the internet at http://pubs.acs.org.