

Abstract
We suggest here and evaluate a methodology for the measurement of specific interatomic distances from a combination of theoretical calculations and experimentally measured 13C kinetic isotope effects. This process takes advantage of a broad diversity of transition structures available for the epoxidation of 2-methyl-2-butene with oxaziridines. From the isotope effects calculated for these transition structures, a theory-independent relationship between the C-O bond distances of the newly forming bonds and the isotope effects is established. Within the precision of the measurement, this relationship in combination with the experimental isotope effects provides a highly accurate picture of the C-O bonds forming at the transition state. The diversity of transition structures also allows an evaluation of the Schramm process for defining transition state geometries based on calculations at non-stationary points, and the methodology is found to be reasonably accurate.
Introduction
Spectroscopic methods can be used to delineate ground-state structures, even when very short-lived, but only very rarely can spectroscopy be applied to define transition state geometries. Rather, transition states are characterized indirectly, invariably by some form of kinetics experiment. This is very limiting, but not infinitely so – many forms of kinetics observations resolve qualitative features of transition state structures. The quantitative experimental characterization of transition state geometries remains largely out of reach.1
Kinetic isotope effects (KIEs) have long been used as a qualitative measure of transition state geometry or as a semi-quantitative measure of transition state bond orders.2 The potential for a more detailed geometrical interpretation arose with Hess and Schaad’s first comparisons of experimental and ab initio-predicted isotope effects,3 though this was hampered by doubt in the predictions.3a,4 An advance came when Houk and Singleton found that conventional transition state theory with a one-dimensional tunneling correction could predict KIEs very well,5 particularly heavy atom KIEs, provided that the calculated transition state geometry is accurate and that the reaction does not involve a hydrogen transfer.6 The prediction of KIEs can be complicated by the substantial involvement of dynamics, multi-dimensional tunneling, variational transition state effects, and other specialized effects; however, these effects can often be accounted for using more complex theoretical treatments. 7 Complications also arise when solvation plays a significant role in the reaction, for example in reactions involving charge separation where solvent effects can be difficult to model accurately. Despite expected limitations, a close correspondence of predicted and experimental KIEs has been observed in many cases, and this has been interpreted as supporting the accuracy of the calculated transition structures.5,6,8,9
Schramm has developed a method for defining the transition states of enzymatic reactions based upon KIE measurements. In this method, a systematic search of constrained geometries is undertaken to identify structures that lead to correct predictions of the experimental KIEs.10 The use of such constrained geometries notably compromises the frequency calculations on which the KIE predictions rely, so the theoretical rigor of the Schramm process is questionable. Nonetheless, it is extraordinarily successful, having been used to design some of the most powerful enzyme inhibitors known.11
The precise interpretation of a KIE in terms of transition state geometry requires that the KIE, in fact, be directly related to a transition state geometrical parameter. Theory does not obviously require this; KIEs are a function of transition state and starting material vibrational frequencies that are themselves a complex function of the geometry. Several studies in the literature have probed the relationship between transition state geometry and KIEs.12 In an incisive series of studies, Jensen explored the relationship between transition state geometries and H/D KIEs in SN2 and E2 reactions.12b-g Jensen notably observed a smooth relationship between bond orders and primary isotope effects after compensation for complicating factors, though the relationship was flat for a broad range of bond orders, largely precluding a specific geometrical interpretation of primary isotope effects. The lore in the isotope effect area is that secondary H/D KIEs are closely related to transition state geometry, but Jensen’s studies show that the correlation of the two is complex when it exists at all. In four transition structures at varying calculational levels for the Claisen rearrangement, there was no trend in geometry versus predicted heavy-atom KIEs, and only geometrically extreme transition states could be completely excluded.6 These observations suggest a limited “resolution” for KIEs in determining transition state geometry. In contrast, we show here that a precise and detailed geometrical interpretation of KIEs is possible, and we identify an error to be avoided in relating KIEs to geometry.
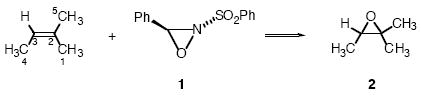
To probe the relationship of the 13C isotope effects to particular features of the transition state geometry, we needed a series of transition structures with varying geometries. Epoxidations of alkenes with oxaziridines (eq 1) provided a unique opportunity in this regard. Experimental studies of these reactions have been interpreted as favoring both planar and spiro transition states.13,14,15 A concerted mechanism has largely been assumed based upon the stereospecifity of the epoxidation, though stepwise mechanisms have also been proposed in special cases.16,17 Early theoretical studies on the epoxidation of alkenes with oxaziridines (RHF/STO-3G) by Bach predicted a planar transition structure.18 Later MP2 calculations predicted a late, spiro, synchronous transition structure,19 and the most recent B3LYP calculations by Houk20 predict a spiro but highly asynchronous transition state geometry. In each case, the calculational model was highly simplified compared to experimentally successful reactions, and we will see here that none of the calculations accurately describe a typical experimental reaction. For our purposes, the complexities inherent to the epoxidation of alkenes by oxaziridines afford a diverse array of structures that arise from varying substitutions, conformations, and orientations of the oxaziridine. The analysis of KIEs for these structures provides the basis for their geometric interpretation. This study also allows a detailed probe of the Schramm process for defining transition state geometries.
| (1) |
Results and Discussion
Experimental Isotope Effects
A qualitative question in additions to alkenes is whether there is synchronous or asynchronous bond formation to the olefinic carbons. Since unsymmetrically substituted alkenes necessarily react via unsymmetrical transition states, the question of synchroneity is only strictly meaningful with symmetrically-substituted alkenes. However, symmetrical alkenes must exhibit symmetrical isotope effects due to averaging, so probes of transition-state synchroneity have focused on “slightly unsymmetrical” reactants.8,21 Here, the epoxidation of 2-methyl-2-butene was chosen for study. The epoxidation of 2-methyl-2-butene with 1 proceeds slowly at room temperature and affords epoxide 2 quantitatively when an excess of 1 is used.
The 13C KIEs for this reaction were determined combinatorially by NMR methodology at natural abundance.22 Oxidations of 2-methyl-2-butene on a 0.15 mol scale with limiting 1 in chloroform at room temperature were taken to 77±3% and 78±3% conversion. The unreacted alkene was reisolated by successive distillations, and then analyzed by 13C NMR compared to a standard not subjected to the reaction conditions. The changes in isotopic composition were calculated using the C5 methyl carbon as an “internal standard” with the assumption that its isotopic composition does not change during the reaction.23 From the changes in isotopic composition, the KIEs were calculated as previously described.22 The results are summarized in Figure 1.
Figure 1.
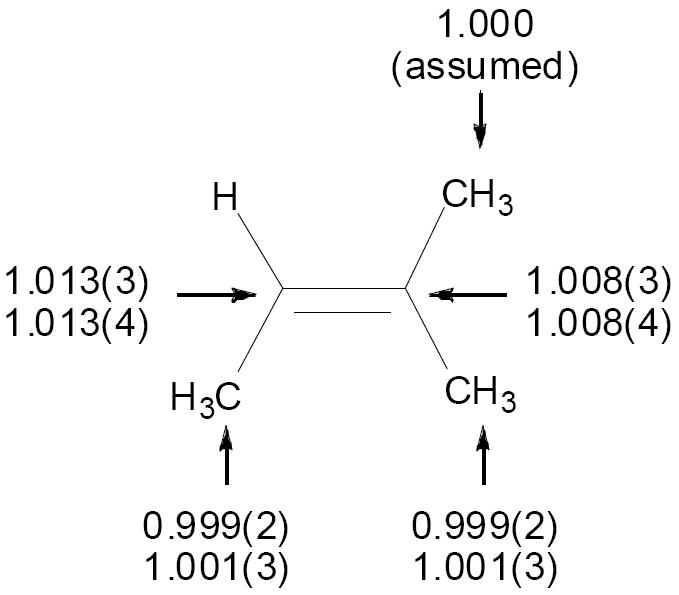
Experimental 13C KIEs (k12C/k13C, 25 °C) for the epoxidation of 2-methyl-2-butene with 1. The two sets of KIEs refer to two independent experiments, with each result based on the average of six measurements. Standard deviations in the last digit are shown in parentheses.
Theoretical Structures
As will be described, a variety of calculational methods were explored in these reactions, but the majority of the theoretical structures were derived from B3LYP calculations using a 6-31+G** basis set.24 This combination was supported by comparison of the transition structure geometry with a fully-optimized CCSD(T)/6-31+G** structure for the parent reaction (see the Supporting Information) along with the prediction of a reasonably accurate barrier versus experimental observations. This methodology is also ultimately supported by the isotope effect observations.
From a systematic search on the reaction of 2-methyl-2-butene with 2-(methylsulfonyl)oxaziridine, followed by more selective searches of reactions with trans- and cis-3-phenyl-2-methanesulfonyloxaziridine, a total of 18 transition structures were located in B3LYP calculations using a 6-31+G** basis set. All geometries were reoptimized using a PCM implicit solvent model to explore the effects of solvent in the stabilization of the transition state. Eight 2-(methylsulfonyl)oxaziridine-based transition structures were also reoptimized using an Onsager solvent model, leading to a total of 44 transition structures for the calculation of isotope effects below. (See the Supporting Information.)
The four lowest-energy transition structures located with 2-(methylsulfonyl)oxaziridine were 3-6; the rest are shown in Supporting Information. The multiplicity of structures arises from the possibility for, firstly, syn versus anti arrangement of the methylene group of the oxaziridine and the cis methyl groups of the alkene; secondly, orientation of the methanesulfonyl group toward either the more or less substituted end of the alkene; and thirdly, variation in rotational orientation of the methanesulfonyl group.
In the gas-phase structures, the oxygens of the methanesulfonyl group are partially associated with hydrogens of the alkene methyl groups. Since this charge-charge interaction would be greatly overemphasized relative to the solution chemistry, 3-6 were reoptimized using both the Onsager and PCM solvation models. This leads to significant changes in the structures: the sulfonyl oxygen – methyl hydrogen distances increase and there is a general shift toward an earlier transition structure with more synchronous formation of the epoxide C–O bonds. Solvent has a greater effect on the energetics of transition structures with two sulfonyl oxygen – methyl interactions (see structures 3, 5, and Supporting Information structures), resulting in an increase in the relative energies of these isomers. Solvent has previously been shown to have a substantial effect on the energetics of a fluorine–hydrogen interaction in epoxidations with fluorinated dioxiranes.25 We would suggest in general that great care should be taken in interpreting heteroatom – H–C interactions from gas-phase calculations.
The free energies of all eight of the solvent optimized transition structures are within 2.2 kcal/mol (PCM solvent model) of the lowest-energy structure 3. The multitude of similar-energy structures makes obvious the difficulty of obtaining high asymmetric induction in these reactions. The free energy barrier calculated by treating the experimental concentrations as the standard state is 22.2-24.4 kcal/mol (15.3-17.4 kcal/mol enthalpic barrier), in line with reactions that proceed slowly at room temperature.
The four transition structures obtained for the reaction of trans-3-phenyl-2-methanesulfonyloxaziridine, depicted in Figure 3 below as 7-10, are the analogs of isomers 3-6 (see Supporting Information for complete structures with stereo views). The presence of the phenyl group is predicted to notably increase the free energy barrier for the reaction in solvent by 4.7 kcal/mol. The free energies of all the trans-3-phenyl-2-methanesulfonyloxaziridine isomers are within 1.5 kcal/mol of lowest-energy structure 7. Transition structures 11 and 12 were located for the reaction of cis-3-phenyl-2-methanesulfonyloxaziridine – these structures are analogs of 3 and 4, respectively (see the Supporting Information for additional structures). The starting cis-oxaziridine is predicted to be less stable than the trans isomer by 6.7 kcal/mol. This higher energy is not expected to afford greater reactivity; transition structure 12, the lowest free energy cis structure, is 7.2 kcal/mol higher in energy than best trans structure 7, making the free energy barrier for the reaction of the cis oxaziridine 0.5 kcal/mol (0.8 kcal/mol activation energy) higher in energy than the trans. Yang has proposed that the cis oxaziridine acts as the oxidant in the epoxidation of alkenes with the in situ generated 3-phenyl-2-methanesulfonyloxaziridine.17 However, the current study suggests that the cis isomers play only a minor role in the reaction, since these transition structures have on average much higher barriers than the trans congeners (1.5 kcal/mol on average).
Figure 3.
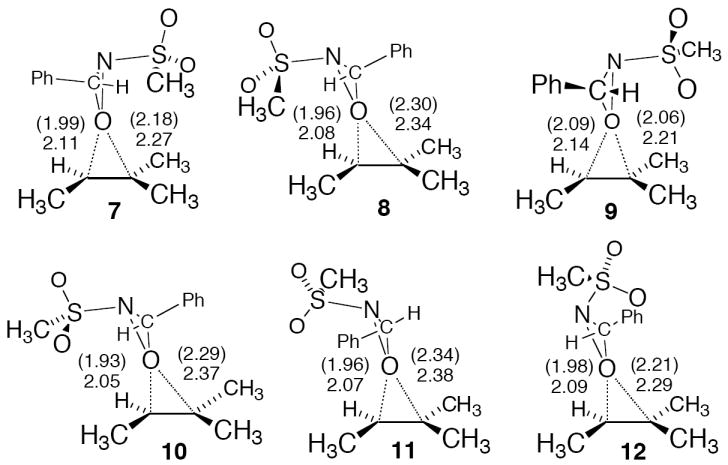
The B3LYP/6-31+G** transition structures for the epoxidation of 2-methyl-2-butene with trans-3-phenyl-2-methanesulfonyloxaziridine (7-10) and cis-3-phenyl-2-methanesulfonyloxaziridine (11 and 12). Distances (Å) in parentheses are from gas-phase optimizations, and distances without parentheses are from optimizations using a PCM implicit solvent model.
All of the epoxidation transition structures are best described as spiro; however, the asynchroneity of the two newly forming C–O bonds varies considerably. In the absence of obvious steric interactions, a nearly synchronous transition structure seems to be preferred, as shown in 7 and 9. Steric interactions between the phenyl group of the oxaziridine and the methyl groups of the alkene, in structures 8 and 10, cause the geometry to distort to highly asynchronous C–O bond formation. Unsurprisingly, the sterically encumbered cis structures distort readily to minimize steric interactions (compare 7 versus 11 and structures in the Supporting Information).
Predicted Isotope Effects
The isotope effects for each of the 44 transition structures were calculated from conventional transition state theory by the method of Bigeleisen and Mayer26 from the scaled theoretical vibrational frequencies,27 and tunneling corrections were applied using a one-dimensional infinite parabolic barrier model.28 Such KIE predictions have proven highly accurate in reactions not involving hydrogen transfer, so long as the calculation accurately depicts the mechanism and transition state geometry.5,6,8 The 88 predicted isotope effects for the olefinic carbons are summarized in the Supporting Information.
In analogy with previous studies, a comparison with experimental values to prune the theoretical structures and evaluate the accuracy of the best structures was applied. The predicted KIEs for 3-10 are shown in Figure 4. Though most of the structures calculated without using the implicit solvation model (displayed in parentheses) lead to isotope effects that do not match well with experimental values in Figure 1, all eight structures calculated using the implicit PCM solvation model give KIEs within error of the experimental measurements. Structures involving 2-(methylsulfonyl)oxaziridine (3-6) correctly lead to excellent preditions of the experimental KIEs, despite the simplicity of the calculational model. For the trans-3-phenyl-2-methanesulfonyloxaziridine series of structures (7-10), more closely resembling the experimental system, the lowest-energy transition structure 7 gives particularly good agreement with experiment. The predicted KIEs for structures 8 – 10 lie on the edge of experimental error; however, after allowing for two independent experimental measurements the 95% confidence range does not include the predicted KIEs for 8-10. Highly asynchronous structure 8 is similar in energy to 7, but the relative energies are sufficiently unreliable that the calculations do not exclude 7 as the predominant contributor to the transition state ensemble as suggested by the KIEs.
Figure 4.
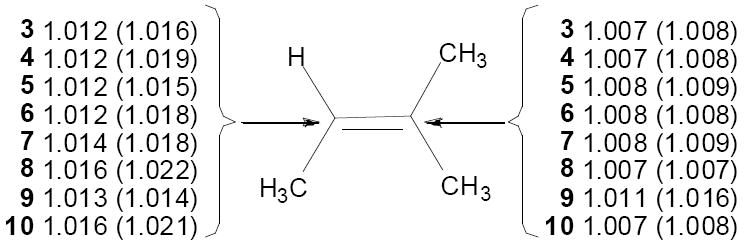
Predicted 13C KIEs (k12C / k13C) are shown for the olefinic carbons of transition state structures 3-10 for the epoxidation of 2-methyl-2-butene catalyzed by 2-(methylsulfonyl)oxaziridine (3–6) and trans-3-phenyl-2-methanesulfonyloxaziridine (7-10). Predicted KIEs from gas-phase optimizations are shown in parentheses, and KIEs from optimizations with an implicit PCM solvent model are shown without parentheses.
The arguably best theoretical structure 7 is consistent with the spiro transition state proposed by Bach; however, the model is much earlier and more synchronous than previously predicted by Houk for the reaction of ethylene with oxaziridine.20 The current study demonstrates the importance of including all alkene substituents in developing an accurate theoretical model.29 A solvent model is also necessary in systems involving oxygen-H-C interactions, which are overestimated in the gas-phase.
Geometries from Isotope Effects
From the diverse set of theoretical structures, we can examine in detail the connection between transition state geometry and KIEs. This analysis also provides a unique opportunity to evaluate the accuracy of the Schramm process in predicting transition state geometries.
To obtain a quantitative geometrical interpretation of the isotope effects, the relationship of the predicted KIEs to the C–O bond distances for the new C–O bond formation was examined for the various transition structures. Figure 5 shows a plot of the predicted 13C olefinic KIEs for each of the calculated structures versus the C–O bond distances. The significant observation in this plot is the close relationship of the predicted KIEs with the C–O distance across a wide variety of structures.
Figure 5.
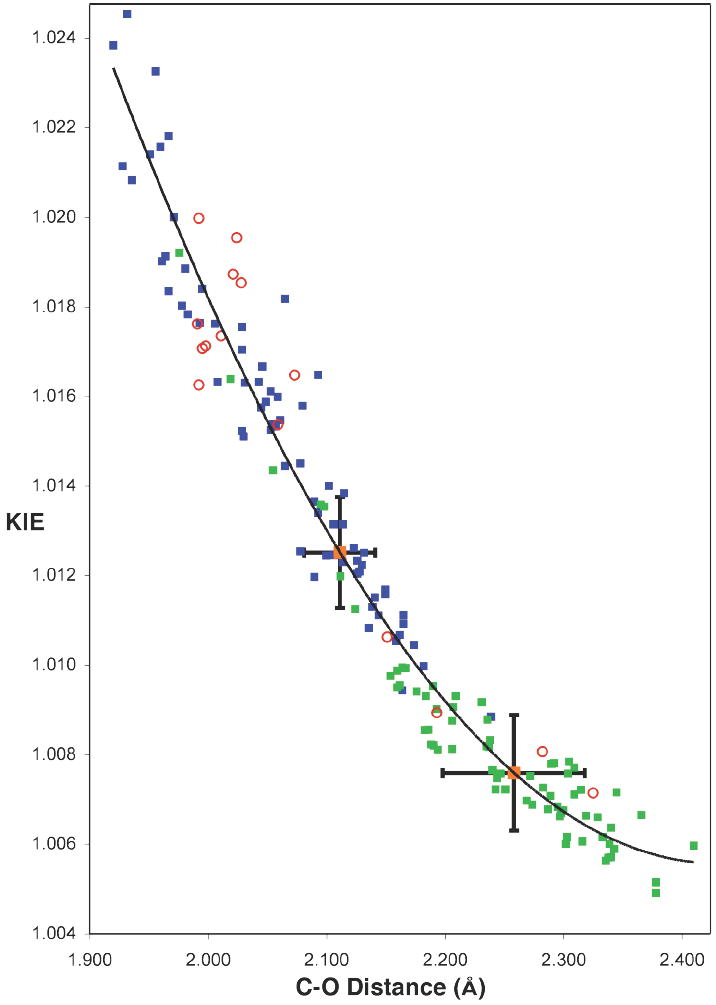
Relationship between 13C KIEs (k12C / k13C) and C–O bond distance of the newly forming olefinic bonds in the oxaziridination of 2-methyl-2-butene. Predictions were calculated from a variety of B3LYP optimized structures for C3 (blue squares) and C2 (green squares). A quadratic line is fit to all B3LYP points. Predictions from other theoretical methods with reasonable energetic barriers (BPW91, B3PW91, BP86, MPW3LYP, MPWLYP1M, MPW1B95, TPSSKCIS, PBE1KCIS) are also depicted (red open circles). Absolute experimental KIEs of 1.0125 and 1.0075 are shown (orange squares), with the uncertainties from a combination of experimental measurements. The absolute experimental KIEs (rather than the relative experimental KIEs in Figure 1) are represented here to facilitate comparison with the absolute predicted values. Absolute experimental values were determined as the product of each experimental measurement and the average of all predicted KIEs at C5 (0.9994).
This is profound. All 70 structures30 have 3N-6 vibrational frequencies that factor into the isotope effect calculation, and the 70 structures differ geometrically in a variety of ways, but the 13C isotope effect depends almost exclusively on the new C–O bond-forming distance. All of the KIEs are within 0.004 of a quadratic trend-line fitting the points; the absolute average deviation is 0.0007.
From this graph, the absolute experimental isotope effects correspond to C–O bond lengths of 2.11 Å and 2.26 Å. These bond lengths are in effect experimental transition state geometries, though they will be subject to both random and systematic error. Random error arises from both the uncertainty in the experimental measurements and the imperfection of the C–O bond distance / KIE relationship. Using KIE uncertainties from a combination of the two sets of experimental values gives C–O bond lengths of 2.11 ± 0.03 Å and 2.26 ± 0.06 Å.
Systematic error is more difficult to deal with in any measurement, and could arise here either because the method for calculating isotope effects and tunneling corrections is systematically inaccurate, or the theoretical method systematically produces an incorrect relationship between the C-O distance and the 13C KIE. Although there is no way to estimate the systematic error, there are two reasons to believe that it is not large. The first is that B3LYP calculations perform very well in modeling the oxidations of alkenes such that there is a striking agreement between experimental and predicted KIEs for the epoxidation of 1-pentene with peracids,8a the asymmetric dihydroxylation of t-butylethylene,8b and the Shi epoxidation of trans-β-methyl styrene.8d The second, more notable reason is that the KIE / C-O bond distance relationship is theory-independent within limits to be discussed.
To examine the dependence of the C-O bond distance / KIE relationship on the calculational method, a diverse combination of functionals (RHF, B3LYP, BPW91, B3PW91, BP86, MPW1K, MPW3LYP, MPWLYP1M, BB1K, MPWB1K, MPW1B95, TPSS1KCIS, PBE1KCIS, PBE1W, PBELYP1W, TPSSLYP1W), basis sets (6-31G*, 6-31+G*, 6-31+G**, 6-311+G**), and solvent models (PCM and Onsager) were used to generate 60 distinct transition structures based on the previously proposed models (see Supporting Information for complete structures). As shown in Figure 5, the C-O bond distance / KIE relationship remains valid across a variety of calculational methods, as long as the predicted activation barrier is reasonable (B3LYP, BPW91, B3PW91, BP86, MPW3LYP, MPWLYP1M, MPW1B95, TPSSKCIS, PBE1KCIS). Within the requirement that the calculation predicts the experimental rate within four orders of magnitude, the correlation between C-O bond distance and KIE is essentially independent of the basis set, solvent model, and functional.
Theoretical methods that gave calculated rates greater than four orders in magnitude above or below experimental observations – based upon free energy barriers with the experimental conditions considered as standard state – did not follow the relationship. For example, methods like MPW1K that overestimate the barrier of the reaction, lead to a shift of the bond distance / KIE relationship to the left of the trend-line (see plot in Supporting Information). The results here suggest that some caution should be taken with KIE calculations based on transition structures that do not correspond to an accurate barrier, and that a good correlation of KIEs with structure should not be expected across theoretical methods that predict widely varying barriers.6 The effect of barrier height may also be related to the lack of correlation between KIEs and geometrical parameters in literature studies that attempt to make correlations while significantly changing the electronics of the reaction.12b,c
We have previously emphasized the importance of applying tunneling corrections to the prediction of heavy atom KIEs.6 If an analogous relationship to that in Figure 5 were created without applying a tunneling correction to the prediction, the geometrical interpretation of a given KIE would correlate to a much shorter C-O bond distance. The error in bond distance increases with larger KIEs. For the experimental C2 KIE the error in the bond distance would be 0.05 Å; this error increases to a maximum of 0.14 Å if the KIE is extrapolated to 1.040. This emphasizes the importance of applying tunneling corrections to heavy atom KIEs, specifically in cases where KIEs are greater than 1%.
This relationship allows a unique geometrical interpretation of the KIEs. The actual reaction is likely to involve an entire ensemble of low-energy transition structures and the experimental KIEs reflect this ensemble. Therefore, a description of the transition state is not accurately represented by lowest-energy structure 7 alone. A more accurate description is derived from the average transition state geometry of the ensemble using the C-O bond distance / KIE relationship from several structures.
To explore whether distance / KIE relationships can be established and used in another system, previously published results from a mechanistic study on the Shi epoxidation of β-methyl styrene were also examined.8d The Shi epoxidation (eq 2) is a good candidate for this process because a variety of transition structures arise from conformational isomerism in the catalyst in combination with multiple orientations for approach of alkene to the intermediate dioxirane.
 |
(2) |
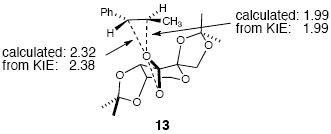
The transition state for the Shi epoxidation is spiro and asynchronous, as shown in lowest-energy transition structure 13. The previously calculated olefinic bond distances for the newly forming C-O bonds in structure 13 are 1.99 Å and 2.32 Å, for the Cβ-O and Cα-O bonds respectively, and the predicted KIEs based on this structure were in excellent agreement with experimentally measured values. A comparison of C-O bond distances to KIEs was examined for 18 calculated transition structures for the Shi epoxidation using B3LYP/6-31G* (see Supporting Information for plot).8d As in the epoxidations with oxaziridines, a correlation between the C–O bond distances and KIEs also exists for the Shi epoxidation. All of the KIEs are within 0.005 of a quadratic trend-line fitting the points; the absolute average deviation is 0.0018. From an average of the experimental KIE measurements, an average transition state geometry of 1.99 Å and 2.38 Å for Cβ-O and Cα-O bond distances respectively can be derived from the relationship. The agreement between the best transition structure 13 and the average transition state geometry derived from the relationship is remarkable, with errors of 0.00 Å and 0.06 Å for the Cβ-O and Cα-O bond distances respectively.
Analysis of KIE Predictions from Non-Stationary Points
In the Schramm process, the inability of geometrically unconstrained optimizations from gas-phase calculations to mimic interactions with the enzyme necessitates the restriction of geometries in the frequency calculations; therefore, this method involves the prediction of KIEs from non-stationary points on the potential energy surface. On its face this methodology appears dubious, since frequencies on which the isotope effect predictions depend are subject to intrinsic errors when calculated from non-stationary points.
To evaluate the Schramm process, we examined how well it could define the C-O bond forming distances in the transition structures for epoxidations with oxaziridines. This was done by first generating a grid of the predicted KIEs at C2 and C3 versus the C-O bond forming distances from B3LYP/6-31G* calculations in the gas-phase. This grid was based on structural analogs of 3 in which the C2-O and C3-O distances were fixed at 1.9 – 2.5 Å in 0.1 Å increments. The tunneling corrected KIE predictions for 38 non-stationary point structures in this range were obtained based on the Gaussian03-calculated frequencies, and interpolation was used to predict the KIEs for a total of 2800 geometries. The resulting grid was then used to translate the predicted KIEs based on 33 B3LYP stationary points (including both gas-phase and PCM-model structures) into the geometries expected for these structures. The deviation of the C-O distances obtained in this way versus the actual C-O distances in the stationary points is a measure of the error induced by the Schramm process. In the event, the absolute average deviation for the 66 total distances is 0.05 Å, and the maximum error is 0.13 Å. It must be judged that the Schramm process is strikingly successful.
As expected, the largest deviations occur at longer C-O bond lengths (>2.3 Å) where very slight changes in the KIEs have a large effect on bond distances. An average deviation of 0.04 Å reflects a systematic error that over predicts C–O bond distances. This systematic error is dominated by deviations of the fixed distance geometries from stationary point structures optimized with a PCM solvent model; this error could perhaps be reduced by exploring other theoretical models as the parent structure for the fixed parameter method. Using the grid to generate an estimate of the olefinic C–O bond lengths based upon the experimental KIEs for the reaction results in C-O bond distances within 0.03 Å and 0.08 Å (for C3 and C2 respectively).
Why does the Schramm process work so well? When distances associated with bond-forming or bond-breaking are constrained at non-stationary points, the greatest errors in the resulting calculated frequencies should tend to occur in low-energy modes. Because the zero-point energy associated with such modes is small, their direct contribution to the KIEs is small. In addition, the frequencies calculated for isotopologues as part of the KIE-prediction process are based on an identical set of (likely flawed) force constants. As a result, errors in the calculated frequencies for isotopologues are related and tend to cancel out in the isotope effect prediction.31
The ability of non-stationary points to empirically predict transition structures is plausible since the contribution of low-frequencies, which are incorrect in these constrained geometries, is small. Another possibility is that zero point energies associated with low-frequencies tend to cancel out the effect of the low-frequencies in the predictions. We cannot exclude the possibility that KIE predictions from non-stationary points may periodically contain errors associated with constrained motions at the transition state (see the Supporting Information for a discussion); however, these errors should be detectable in a grid of constrained transition structure calculations.
Conclusions
Since KIEs are only significant when substantial bonding changes occur, the KIEs can never provide information about many aspects of the geometry at the transition state. However, they do provide information about the most important aspects of the geometry where bonds are being formed and broken. The work here supports the idea that the isotope effects can be used in a quantitative manner to define interatomic distances at transition states. The general procedure to be followed is to use calculations of model reactions to establish a relationship between KIEs and geometry, and the results here suggest that with care, such relationships can be independent of the theoretical method. Having established such a relationship, the experimental KIEs provide a specific measurement of the geometry. As with all measurements there will be some associated uncertainty; despite this limitation, the match between these experimentally-based measurements and theoretical calculations can be remarkable. The precision in this methodology is sufficient to provide quantitative geometric insight into reaction mechanisms, and the methodology provides a quantitative picture that is not present in the usual qualitative interpretation of an experimental study.
The Schramm process accomplishes a similar quantitation of the transition state geometry. The advantage of the Schramm methodology is that it can be applied to complex reactions where the energy surface provides no stationary point models. The disadvantage is that the use of non-stationary points must inevitably introduce some degree of inaccuracy, but results here suggest that the inaccuracies in the derived geometries are small, supporting its use in defining approximate transition state geometries.
Experimental Section
Epoxidation of 2-methyl-2-butene
To an ice-cooled solution of 31.3 g (120 mmol) of trans-2-(phenylsulfonyl)-3-phenyloxaziridine32 in 70 mL of CDCl3 was added slowly a solution of 10.5 g (150 mmol) of 2-methyl-2-butene in 10 mL of CDCl3. After 48 h at room temperature the reaction was found to be 77±3% complete by 1H-NMR analysis of aliquots. The volatiles were then distilled from the reaction mixture using a 10-cm Vigreaux column until the head temperature reached 60 °C. The distillate was then redistilled fractionally to afford 1.1 g of the unreacted 2-methyl-2-butene (bp 35-38 °C, >99% pure by 1H-NMR). An analogous reaction under identical conditions was taken to 78±3% conversion.
NMR Measurements
NMR measurements were taken on 1.1 g samples of 2-methyl-2-butene in 10 mm NMR tubes diluted with CDCl3 to a constant height of 5 cm. A T1 determination by the inversion-recovery method was carried out for each NMR sample, and the T1 for each NMR signal remained constant within experimental error from sample to sample. The 13C spectra were recorded at 100.58 MHz with inverse gated decoupling, using 150 s delays between calibrated 45° pulses. An acquisition time of 7.301s was used and 262144 points were collected. Integrations were determined numerically using a constant region for each peak that was ≈5 times the peak width at half height on either side of the peak. A zeroth order baseline correction was generally applied, but in no case was a first order (tilt) correction applied. The results for all reactions are summarized in the Supporting Information.
Supplementary Material
Energies and full geometries of all calculated structures, NMR integration results for all reactions, table of KIEs versus C-O distances, plot of KIEs versus distance for the Shi epoxidation, a technical note on frequency calculations and KIE predictions, and complete reference 24a. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 2.
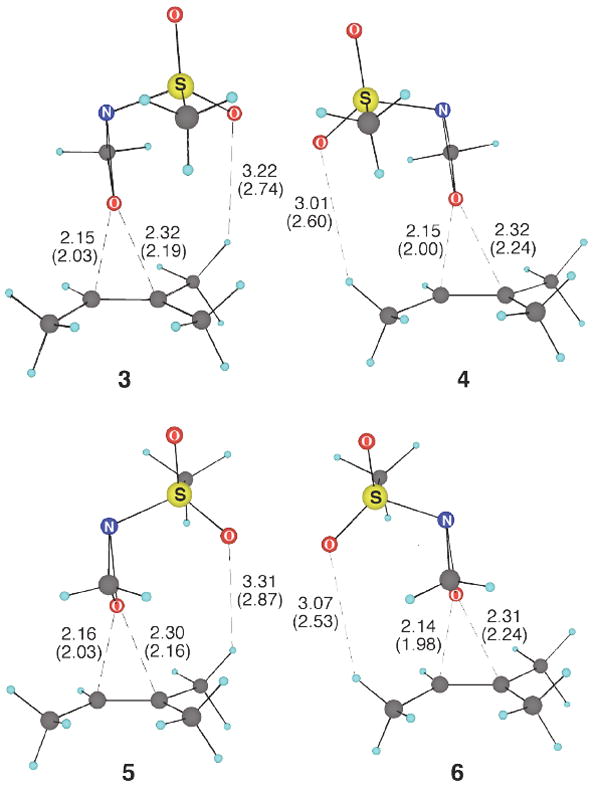
The four lowest-energy B3LYP/6-31+G** transition structures for the epoxidation of 2-methyl-2-butene with 2-(methylsulfonyl)oxaziridine. Distances (Å) in parentheses are from gas-phase optimizations, and distances without parentheses are from optimizations using a PCM implicit solvation model.
Acknowledgments
We thank NIH grant No. GM-45617, NSF-CRIF CHE-0541587, and The Robert A. Welch Foundation for support of this research.
References
- 1.For an exception, see: Wenthold PG, Hrovat DA, Borden WT, Lineberger WC. Science. 1996;272:1456–1459. doi: 10.1126/science.272.5267.1456.
- 2.(a) Bigeleisen J, Wolfsberg M. Advances in chemical physics. Vol. 1. Interscience; New York: 1958. pp. 16–76. [Google Scholar]; (b) Melander L, Saunders WH., Jr . Isotope effects on reaction rates. Wiley-Interscience; New York: 1980. [Google Scholar]; (c) More O’ Ferrall RA. J Chem Soc B. 1970:785–790. [Google Scholar]; (d) Katz AM, Saunders WH., Jr J Am Chem Soc. 1969;91:4469–4472. [Google Scholar]; (e) Streitwieser A, Jr, Jagow RH, Suzuki S. J Am Chem Soc. 1958;80:2326–2332. [Google Scholar]
- 3.(a) Hess BA, Jr, Schaad LJ, Pancir J. J Am Chem Soc. 1985;107:149–154. [Google Scholar]; (b) Baldwin JE, Reddy VP, Hess BA, Jr, Schaad LJ. J Am Chem Soc. 1988;110:8554–8555. [Google Scholar]
- 4.Lu D-h, Maurice D, Truhlar DG. J Am Chem Soc. 1990;112:6206–6214. [Google Scholar]
- 5.Beno BR, Houk KN, Singleton DA. J Am Chem Soc. 1996;118:9984–9985. [Google Scholar]
- 6.Meyer MP, DelMonte AJ, Singleton DA. J Am Chem Soc. 1999;121:10865–10874. [Google Scholar]
- 7.(a) Singleton DA, Hang C, Szymanski MJ, Greenwald EE. J Am Chem Soc. 2003;125:1176–1177. doi: 10.1021/ja027221k. [DOI] [PubMed] [Google Scholar]; (b) Ussing BR, Hang C, Singleton DA. J Am Chem Soc. 2006;128:7594–7607. doi: 10.1021/ja0606024. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nowlan DT, III, Singleton DA. J Am Chem Soc. 2005;127:6190–6191. doi: 10.1021/ja0504441. [DOI] [PubMed] [Google Scholar]; (e) Lin H, Zhao Y, Ellingson BA, Pu J, Truhlar DG. J Am Chem Soc. 2005;127:2830–2831. doi: 10.1021/ja0434026. [DOI] [PubMed] [Google Scholar]; (f) Ellingson BA, Pu J, Lin H, Zhao Y, Truhlar DG. J Phys Chem A. 2007;111:11706–11717. doi: 10.1021/jp072843j. [DOI] [PubMed] [Google Scholar]; (g) Ellingson BA, Truhlar DG. J Am Chem Soc. 2007;129:12765–12771. doi: 10.1021/ja072538b. [DOI] [PubMed] [Google Scholar]; (h) Garrett BC, Truhlar DG, Bowman JM, Wagner AF, Robie D, Arepalli S, Presser N, Gordon RJ. J Am Chem Soc. 1986;108:3515–3516. [Google Scholar]
- 8.For some other examples, see: Singleton DA, Merrigan SR, Liu J, Houk KN. J Am Chem Soc. 1997;119:3385–3386.DelMonte AJ, Haller J, Houk KN, Sharpless KB, Singleton DA, Strassner T, Thomas AA. J Am Chem Soc. 1997;119:9907–9908.Keating AE, Merrigan SR, Singleton DA, Houk KN. J Am Chem Soc. 1999;121:3933–3938.Singleton DA, Wang Z. J Am Chem Soc. 2005;127:6679–6685. doi: 10.1021/ja0435788.
- 9.For an example highlighting a limitation of this process, see: Singleton DA, Hang C. J Org Chem. 2000;65:7554–7560. doi: 10.1021/jo000922k.
- 10.Horenstein BA, Parkin DW, Estupinan B, Schramm VL. Biochemistry. 1991;30:10788–10795. doi: 10.1021/bi00108a026.Horenstein BA, Schramm VL. Biochemistry. 1993;32:7089–7097. doi: 10.1021/bi00079a004.(c) See also ref 12b, which employs a similar process.
- 11.(a) Singh V, Lee JE, Nunez S, Howell PL, Schramm VL. Biochemistry. 2005;44:11647–11659. doi: 10.1021/bi050863a. [DOI] [PubMed] [Google Scholar]; (b) Miles RW, Tyler PC, Furneaux RH, Bagdassarian CK, Schramm VL. Biochemistry. 1998;37:8615–8621. doi: 10.1021/bi980658d. [DOI] [PubMed] [Google Scholar]; (c) Taylor EA, Clinch K, Kelly PM, Li L, Evans GB, Tyler PC, Schramm VL. J Am Chem Soc. 2007;129:6984–6985. doi: 10.1021/ja071087s. [DOI] [PubMed] [Google Scholar]
- 12.(a) Owczarek E, Kwiatkowski W, Lemieszewski M, Mazur A, Rostkowski M, Paneth P. J Org Chem. 2003;68:9302–9310. doi: 10.1021/jo034799j. [DOI] [PubMed] [Google Scholar]; (b) Glad SS, Jensen F. J Am Chem Soc. 1997;119:227–232. [Google Scholar]; (c) Glad SS, Jensen F. J Org Chem. 1997;62:253–260. doi: 10.1021/jo9618379. [DOI] [PubMed] [Google Scholar]; (d) Nielsen PA, Glad SS, Jensen F. J Am Chem Soc. 1996;118:10577–10583. [Google Scholar]; (e) Jensen F. J Am Chem Soc. 1995;117:7487–7492. [Google Scholar]; (f) Wiest O, Houk KN, Black KA, Thomas B., IV J Am Chem Soc. 1995;117:8594–8599. [Google Scholar]; (g) Glad SS, Jensen F. J Am Chem Soc. 1994;116:9302–9310. [Google Scholar]; (h) Poirier RA, Wang Y, Westaway KC. J Am Chem Soc. 1994;116:2526–2533. [Google Scholar]; (i) Glad SS, Jensen F. J Chem Soc Perkin Trans 2. 1994;4:871–876. [Google Scholar]; (j) Wilkie J, Williams IH. J Am Chem Soc. 1992;114:5423–5425. [Google Scholar]; (k) Wolfe S, Kim C-K. J Am Chem Soc. 1991;113:8056–8061. [Google Scholar]
- 13.Davis FA, Harakal ME, Awad SB. J Am Chem Soc. 1983;105:3123–3126. [Google Scholar]
- 14.Baumstark AL, McCloskey CJ. Tetrahedron Lett. 1987;28:3311–3314. [Google Scholar]; Tu Y, Wang Z-X, Shi Y. J Am Chem Soc. 1996;118:9806–9807. [Google Scholar]
- 15.Anderson DR, Woods KW, Beak P. Org Lett. 1999;1:1415–1417. [Google Scholar]
- 16.Mithani S, Drew DM, Rydberg EH, Taylor NJ, Mooibroek S, Dmitrienko GI. J Am Chem Soc. 1997;119:1159–1160. [Google Scholar]
- 17.Yang D, Zhang C, Wang X-C. J Am Chem Soc. 2000;122:4039–4043. [Google Scholar]
- 18.Bach RD, Wolber GJ. J Am Chem Soc. 1984;106:1410–1415. [Google Scholar]
- 19.Bach RD, Andres J. J Org Chem. 1992;57:613–618. [Google Scholar]
- 20.Houk KN, Liu J, DeMello NC, Condroski KR. J Am Chem Soc. 1997;119:10147–10152. [Google Scholar]
- 21.Gajewski JJ, Peterson KB, Kagel JR, Huang YCJ. J Am Chem Soc. 1989;112:9078–9084. [Google Scholar]
- 22.Singleton DA, Thomas AA. J Am Chem Soc. 1995;117:9357–9358. [Google Scholar]
- 23.The predicted KIE for the methyl carbons ranged from 0.998 – 1.000 through 69 different transition structures calculated using B3LYP with various basis sets in gas-phase and with various solvent models.
- 24.Frisch MJ, et al. Gaussian 03. Gaussian, Inc.; Wallingford CT: 2004. Revision C.02.(b) See the Supporting Information for full details on the calculational methods employed.
- 25.Armstrong A, Washington I, Houk KN. J Am Chem Soc. 2000;122:6297–6298. [Google Scholar]
- 26.Bigeleisen J, Mayer MG. J Chem Phys. 1947;15:261–267.Wolfsberg M. Acc Chem Res. 1972;5:225–233.(c) It should be noted that the Bigeleisen method allows fully for free energy but is less subject to errors in low frequencies than a direct calculation of KIEs from activation free energies (see ref 31 and the Supporting Information).
- 27.The calculations used the program QUIVER( Saunders M, Laidig KE, Wolfsberg M. J Am Chem Soc. 1989;111:8989–8994.) with B3LYP frequencies scaled by 0.9614.
- 28.Bell RP. The Tunnel Effect in Chemistry. Chapman & Hall; London: 1980. pp. 60–63. [Google Scholar]
- 29.An optimized transition structure of the reaction of ethylene with oxazaridine using a PCM solvent model, results in a highly asynchronous structure with bond distances of 1.78Å and 2.22Å, and predicted 13C KIEs of 1.023 and 1.010 respectively, inconsistent with experimental observations.
- 30.Structures include all possible transition state explored for the epoxidation of 2-methyl-2-butene catalyzed by 2-(methylsulfonyl)oxaziridine, trans-3-phenyl-2-methanesulfonyloxaziridine, and cis-3-phenyl-2-methanesulfonyloxaziridine calculated using B3LYP/6-31G*, B3LYP/6-31+G**, and B3LYP/6-311+G** in gas-phase, using an implicit Onsager solvent model, or using an implicit PCM solvent model.
- 31.Schaad LJ, Bytautas L, Houk KN. Can J Chem. 1999;77:875–878. [Google Scholar]
- 32.Davis FA, Chattopadhyay S, Towson JC, Lai S, Reddy T. J Org Chem. 1988;53:2087–2089. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Energies and full geometries of all calculated structures, NMR integration results for all reactions, table of KIEs versus C-O distances, plot of KIEs versus distance for the Shi epoxidation, a technical note on frequency calculations and KIE predictions, and complete reference 24a. This material is available free of charge via the Internet at http://pubs.acs.org.